氧化还原液流电池电极用碳催化剂的制作方法
本发明涉及氧化还原液流电池的电极中使用的催化剂,特别涉及能够适用于钒系氧化还原液流电池的由碳质材料构成的氧化还原液流电池电极用碳催化剂。
背景技术:
近年来,为了实现低碳社会,推动着风力发电和光伏发电等可再生能源转换。然而,风力发电和光伏发电存在随着时间带和季节、天气导致的输出波动问题。为了维持大规模的电力需求,需要针对这种输出波动的电力需求的负载均衡化对策。
作为针对该电力负载均衡化的一个对策,蓄电池的蓄电被关注。在蓄电池中,尤其是氧化还原液流电池由于具有容易大型化、高安全性和稳定性、充放电循环寿命长等的优点,被认为有潜力用于大规模电力存储用电池。
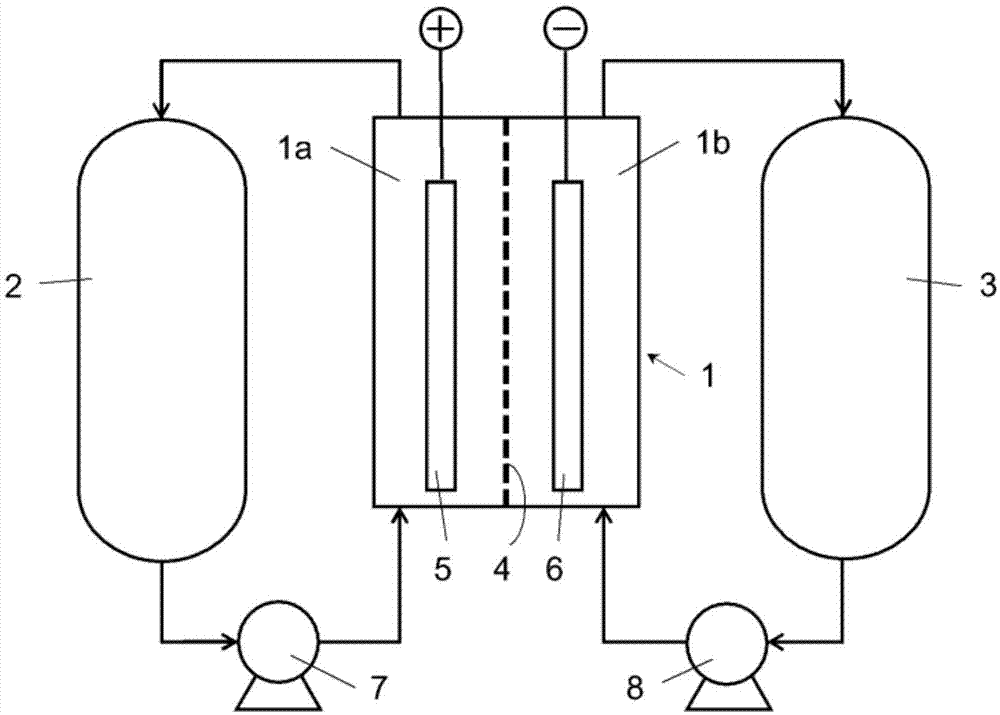
氧化还原液流电池是指通过泵使电解液循环而进行离子的氧化还原反应来进行充放电的液流电池。图1为示出了通常的氧化还原液流电池的结构的示意图。图1所示的氧化还原液流电池包括电池1、正极液罐2和负极液罐3。电池1被离子透过性隔膜4分隔为正极室1a和负极室1b,在正极室1a内设置正极5,在负极室1b内设置负极6。正极室1a与用于供给以及回收正极液的正极液罐2连接,并通过正极液泵7进行液体循环。负极室1b与用于供给以及回收负极液的负极液罐3连接,并通过负极液泵8进行液体循环。在氧化还原液流电池中,电解液(正极液和负极液)中含有的阳离子经由隔膜4在正极室1a和负极室1b之间移动,分别在正极5和负极6发生氧化还原反应,电解液中的离子价数变化从而反复充放电。
与其它蓄电池相比,上述那样的氧化还原液流电池有能量密度低的缺点。因此,正进行着用于提高能量密度的研究开发,并从促进氧化还原液流电池的电极反应的观点出发,进行着与电极相关的各种改良和提案。
例如,作为能够减小充放电循环的经时变化的碳质电极,专利文献1提出了一种由碳表面的结合氧原子为碳原子数的10-25%的碳质纤维构成的电极。
另外,作为钒氧化还原液流电池用碳电极材料,专利文献2中记载有含有平均纤维直径为0.05-0.3μm、平均长径比为10-500的气相法碳纤维的电极材料。
此外,还报道有通过经氧化处理的石墨烯、部分还原的氧化石墨、氮掺杂碳、金属负载碳材料等制造电极等。
另外,除了电极自身的改良以外,也有报道作为电极催化剂使用酞菁、铂催化剂等。另外,专利文献3中记载有,使金属酞菁热分解,对碳电极表面通过含有金属的碳薄膜进行修饰的电极催化剂,对于氧化还原液流电池的电极反应具有高催化剂活性。
现有技术文献
专利文献
专利文献1:日本特开平5-234612号公报
专利文献2:日本特开2006-156029号公报
专利文献3:日本特开2015-115158号公报
技术实现要素:
然而,上述专利文献1中记载的电极,由于通过空气氧化进行使得电极表面的结合氧原子数相对于碳原子数为10-25%的处理,因此存在电极的电阻率变高的问题。另外,难以提高碳的结晶性,特别是,电解液含有1.5mol/l以上的钒离子的氧化还原液流电池中,耐氧化性不充分,伴随着反复充放电的循环电池电阻增加,能量效率大大降低。
另外,上述专利文献2中记载的电极的氧化还原的可逆反应性不充分。
其它以往的电极在实用上制造困难,并且对于在金属负载碳材料等上具体的金属种类和负载方法等的研究也不能说是充分的。
另外,以往的电极催化剂从催化剂活性和耐久性不充分、价格高等的观点来看,实用性差,另外,上述专利文献3中记载的电极催化剂的催化活性也不能说是充分的。此外,专利文献3的实施例中,通过含有金属的碳薄膜覆盖在玻璃碳(GC)平板上,制造碳薄膜覆盖的GC平板电极,但是在适合用作氧化还原液流电池的电极的碳毡和碳纤维不织布中的实用性差。
因而,从氧化还原液流电池的实际实用化和普及的观点来看,需求能够使能量密度提高的电极,具体地,需求促进电极反应,并且耐久性优异的电极。
本发明是为了解决上述问题而完成,其目的是提供能够促进氧化还原液流电池中的电极反应、并且耐久性优异的氧化还原液流电池电极用碳催化剂。
本发明,在使氧化还原液流电池的能量密度提高的基础上,着眼于电极催化剂,发现具有规定的表面特性的碳催化剂能够促进电极反应,并且得到良好的耐久性,基于此完成了本发明。
即,本发明提供以下的[1]-[5]。
[1]一种氧化还原液流电池电极用碳催化剂,通过X射线光电子能谱法表面分析测定的氧原子数相对于碳原子数的比为0.05-0.20。
[2]根据上述[1]所述的碳催化剂,其中,通过所述表面分析测定的氮原子数相对于碳原子数的比为0.005-0.30。
[3]一种氧化还原液流电池用电极,该氧化还原液流电池用电极包括上述[1]或[2]所述的碳催化剂。
[4]一种氧化还原液流电池,该氧化还原液流电池包括上述[3]所述的电极。
[5]根据上述[4]所述的氧化还原液流电池,所述氧化还原液流电池为钒系氧化还原液流电池。
本发明的氧化还原液流电池电极用碳催化剂能够促进氧化还原液流电池中的电极反应,并且耐久性优异。
因而,通过使用本发明的氧化还原液流电池电极用碳催化剂,能够提供显示出良好的充放电循环特性的氧化还原液流电池用电极,进而,能够使氧化还原液流电池的能量密度提高。本发明的氧化还原液流电池电极用碳催化剂,特别适合用于钒系氧化还原液流电池。
附图说明
图1为示意性地示出氧化还原液流电池的一个例子的构成的示意图。
具体实施方式
以下,详细说明本发明。
[碳催化剂]
本发明的氧化还原液流电池电极用碳催化剂,其特征在于,通过X射线光电子能谱法(XPS:X-ray Photoelectron Spectroscopy)表面分析测定的氧原子数相对于碳原子数的比(O/C比)为0.05-0.20。
通过具有这样的表面特性的碳催化剂,促进氧化还原液流电池中的电极反应,提高充放电特性,得到充分的耐久性。
此外,通过XPS的表面分析,具体地,可以通过后述实施例中记载的方法进行。
本发明的碳催化剂能够适用于使用水系或非水系的电解液中任意的电解液的氧化还原液流电池,由于具有亲水性,因此优选适用于使用水系电解液的氧化还原液流电池。
氧化还原液流电池的电解液含有价态变化的金属离子,例如可举出铁-铬系、铁-钛系、钛-锰系、锰-铬系、铬系、钒系等。其中,从得到高电动势来看,优选为钒系(VO2+/VO2+(V4+/V5+)、V2+/V3+)。这些电解液由于是作为活性物质的各金属离子或者含氧金属离子稳定存在的溶液,因此优选为硫酸、盐酸、硝酸或磷酸等的酸性水溶液,更优选为硫酸水溶液。
即,本发明中的电解液优选为正极液为VO2+/VO2+(V4+/V5+)的硫酸水溶液、负极液为V2+/V3+的硫酸水溶液的钒系。
所述碳催化剂通过XPS表面分析测定的O/C比为0.05-0.20,优选为0.06-0.18,更优选为0.10-0.18。通过使O/C比在上述范围内,通过与电解液的润湿性的提高,能够实现催化剂活性的提高,促进电极反应。
O/C比不足0.05时,有与电解液的润湿性差,能量密度(电流密度)降低的倾向。另一方面,O/C比超过0.20时,碳容易氧化,耐久性差。
另外,优选所述碳催化剂通过XPS表面分析测定的氮原子数相对于碳原子数的比(N/C比)为0.005-0.30,更优选为0.010-0.10。通过使N/C比在上述范围内,能够在不降低碳催化剂的导电性的情况下,使催化剂活性进一步提高。
另外,优选地,所述碳催化剂为在XPS表面分析中检出作为金属成分的铁和钴中的至少一种的碳催化剂。含有这样的金属成分的碳催化剂,能够表现优异的催化剂活性。从得到良好催化剂活性的观点来看,铁原子数和钴原子数的合计相对于碳原子数的比优选为0.0001-0.010,更优选为0.0002-0.008。
[碳催化剂的制备方法]
如上述的本发明的碳催化剂的制备方法,没有特别的限定,优选通过使包括含有氮原子的有机物和金属的原料碳化的方法进行制备。例如可举出通过在含有氮原子的树脂的溶液中,加入过渡金属粉末并混合,在干燥和不熔化处理之后,进行焙烧,酸洗净后,再次进行焙烧的碳化方法。更具体地,可以通过后述实施例中记载的方法制备碳催化剂。
被碳化的原料中的所述有机物为能够碳化的有机物,含有氮原子即可,没有特别的限定,可以使用任意一种以上。例如,可以使用选自热固化性树脂和热塑性树脂等的高分子有机化合物,以及低分子量的有机化合物中的中的至少一种以上。另外,可以使用生物质。
作为所述有机物的具体例子,可举出吡咯、咪唑、吡啶、哌啶、三唑、四唑等的环状有机化合物及其衍生物;聚丙烯腈(PAN)、丙烯腈-甲基丙烯酸共聚物(PAN-PMA)、聚酰亚胺、脲醛树脂、聚苯胺、聚吡咯等的高分子化合物等。其中,优选使用丙烯腈-甲基丙烯酸共聚物(PAN-PMA)、聚丙烯腈(PAN)。
作为被碳化的原料中的所述金属,从有效地使碳催化剂的催化剂活性提高的观点来看,优选含有过渡金属,其中,优选为钪、钛、钒、铬、锰、铁、钴、镍、锌或铜,更优选为铁、钴或铜,进一步优选为铁或钴。它们中,可以单独含有一种,也可以含有2种以上。
所述金属可以作为单体添加,也可以作为金属化合物添加。作为金属化合物,例如可举出金属盐、金属氧化物、金属氢氧化物、金属氮化物、金属硫化物、金属碳化物、金属络合物等。
被碳化的原料中的所述金属的含量,在能够体现碳催化剂期望的催化剂活性的范围内即可,没有特别的限定,相对于所述有机物100质量份优选为1-90质量份,更优选为2-80质量份。
在不妨碍碳催化剂的催化剂活性的范围内,所述原料还可以含有其它有机物、其它金属和其它成分。例如,可举出所述有机物的固化剂和交联剂;用于该固化反应或交联反应的催化剂;炭黑和石墨粉末等的导电性碳材料等。
所述原料加热至能够碳化的温度以上,进行碳化。用于碳化的加热温度,根据使用的原料的种类和配合量等适当设定,例如可以为300-3000℃,优选加热至700-2000℃。此外,优选碳化在氮等的惰性气体气氛中进行。
另外,升温速度和在碳化温度下的保持时间,在碳化能够充分进行的范围内即可,没有特别的限定。例如,升温速度可以为0.5-300℃/分钟,保持时间可以为5分钟-24小时。
将所述原料碳化得到的碳催化剂,根据碳催化剂的使用状态等,从使催化剂活性进一步提高的观点来看,可以进一步进行热处理,另外,还可以实施洗净、粉碎等的处理。
所述洗净为用于降低碳催化剂中的金属的量的处理,例如,优选通过酸进行洗净处理。作为洗净处理中使用的酸,能够得到降低金属量的效果的酸即可,没有特别的限定,例如可举出浓盐酸、浓硝酸、浓硫酸等。洗净处理的方法没有特别的限定,例如,可以通过将碳催化剂在含有酸的溶液中浸渍并保持的方法进行。
此外,为了得到同样的效果,也可以进行电解处理来代替洗净处理。
所述洗净处理后,优选进行将碳催化剂在规定的温度下保持的热处理。热处理温度优选为300-3000℃,更优选为700-2000℃。至热处理温度的升温速度,例如可以为0.5-300℃/分钟。在热处理温度下的保持时间,例如可以为5分钟-24时间。热处理优选在氮气等的惰性气体氛围中进行。
所述粉碎可以使用球磨机或珠磨机等公知的粉碎手段进行,由此,可以以粒子状或粉末状获得碳催化剂。
如上述得到的碳催化剂,还可以在35-90℃下用0.01-13mol/L的硫酸水溶液进行表面处理。该表面处理为在硫酸水溶液中浸渍碳催化剂的处理。
对于通过XPS表面分析测定的O/C比小的该碳催化剂,通过这样使用硫酸的表面处理,能够实现增大O/C比,促进耐久性和电极反应。另外,即使进行了所述洗净处理的情况下金属的量也不能充分降低时,能够实现进一步降低碳催化剂中残留的金属成分的效果。
此外,使用硝酸代替硫酸时,虽然所述O/C比增大,但是通过XPS表面分析测定的N/C比也增大,另外,导致耐久性降低。
所述表面处理中,从有效地使所述O/C比增大的观点来看,处理温度更优选为45-80℃,另外,硫酸水溶液的浓度更优选1-4mol/L。
另外,所述表面处理时间,根据处理温度和硫酸水溶液的浓度适当设定,从确保充分获得增大所述O/C比的效果的时间的观点来看,优选为1-28天。
[形态]
本发明的碳催化剂的形态没有特别的限定,优选通过在碳化后的处理工程中进行粉碎,成为粉末状或粒子状的碳催化剂。
另外,通过将原料用公知的纺织方法,加工成纤维状之后,通过进行碳化,能够获得纤维状的碳催化剂。另外,使用该纤维状的碳催化剂制作织布等,能够形成片状的碳催化剂。
[电极]
本发明的氧化还原液流电池用电极包括如上述的本发明的碳催化剂。
通过使用所述碳催化剂,促进电极反应,能够构成显示出良好充放电循环特性的电极。另外,伴随着电极特性的提高,相比以往的电极能够实现薄层化。
适用于本发明的碳催化剂的电极材料没有特别的限定,作为氧化还原液流电池用电极公知的电极材料即可,通常使用碳电极。碳电极中,由于优选表面积大、电极反应场多的碳电极,因此优选使用玻璃碳、碳毡、碳纤维不织布。
包括所述碳催化剂的电极的制作方法,没有特别的限定,优选使用粘结剂在电极材料的表面使碳催化剂固定。例如,可以使用采用涂布机或喷雾机在电极材料的表面将含有碳催化剂和粘结剂的液体进行涂布的方法,或者,使用通过使电极材料在含有碳催化剂和粘结剂的液体中浸渍后,使其干燥,在电极材料的表面使粘结剂黏着的方法。根据这样的方法,能够使碳催化剂均匀地固定在电极材料的整个表面上。
此外,固定在电极材料表面的碳催化剂的量,只要是得到充分的催化剂活性的量即可,没有特别的限定,可根据电极的形态等适当设定。
[氧化还原液流电池]
本发明的氧化还原液流电池包括所述电极。所述电极能够在氧化还原液流电池的正极和负极中的任意一个中使用。
通过使用包括本发明的碳催化剂的所述电极,能够得到促进电极反应、降低电池电阻的氧化还原液流电池。另外,在不使用铂等贵金属形成的高价催化剂的情况下,实现促进电极反应,可以以低成本提供实用性更高的氧化还原液流电池。
本发明的碳催化剂,特别能够适用于钒系氧化还原液流电池。
实施例
以下,通过实施例对本发明进行更详细说明,但是本发明不受其限定。
[碳催化剂的制作]
(实施例1)
在四口烧瓶中加入丙烯腈(和光纯药工业株式会社制)30.93g、甲基丙烯酸(和光纯药工业株式会社制)4.07g和纯水300mL,使用氮气鼓泡15分钟。将该烧瓶用70℃的油浴加热,添加将过硫酸钾(和光纯药工业株式会社制)100mg溶解在纯水50ml中的溶液,在氮气气氛下边搅拌边聚合4小时。放冷后,将得到的乳白色液的溶液浓缩,在60℃下真空干燥,得到聚丙烯腈-聚甲基丙烯酸共聚物(PAN-PMA)约20g。
使得到的PAN-PMA 1.0g溶解在N,N-二甲基甲酰胺15g中,调制溶液A。另外,将2-甲基咪唑1.0g和氯化锌5.78g溶解在N,N-二甲基甲酰胺15g中,调制溶液B。然后,将溶液A和B混合,在其中加入铁粉0.187g并混合。
将得到的混合液在60℃下真空干燥一昼夜。将真空干燥得到的混合物在大气中加热,用30分钟从室温(25℃)升温至150℃,接着用2小时从150℃升温至220℃。然后,在220℃下保持3小时,使所述混合物不熔化,调制碳化原料。
放冷后,将调制的碳化原料用行星球磨机(P-7,Fritsch Japan株式会社制)粉碎。
将粉碎的碳化原料放入石英管中,在氮气气氛中,用聚焦炉加热,从室温(25℃)以50℃/分钟的升温速度升温至1100℃,在1100℃下保持1小时,进行了碳化。
放冷后,将得到的碳化物用与上述同样的行星球磨机和珠磨机(RMB型批次珠磨机,AIMEX株式会社制)粉碎。
通过在粉碎的碳化物1.0g中加入浓盐酸20mL并搅拌30分钟后,使碳化物沉淀,通过除去上清液,进行碳化物的酸洗净。然后,用浓盐酸和蒸馏水1:1的混合溶液洗净并过滤,接下来,进行用蒸馏水的洗净和过滤,用pH试纸确认滤液为中性后,将过滤物真空干燥。
将上述得到的碳化物放入石英管中,在氮气气氛中,用聚焦炉加热,从室温(25℃)以50℃/分钟的升温速度升温至700℃,在700℃下保持1小时,进行热处理。
放冷后,得到的碳化物即为粉末状的碳催化剂。
(实施例2)
在具有冷却管的100mL茄形烧瓶中,加入实施例1中制备的碳催化剂0.5g、4mol/L硫酸水溶液20mL和搅拌棒,边搅拌,边用85℃的油浴加热回流14天。放冷后,过滤,反复用蒸馏水洗净和过滤,用pH试纸确认滤液为中性后,将过滤物用60℃的减压干燥器(5000Pa)干燥一夜。
将干燥的过滤物用研钵粉碎至粒径约1μm以下,得到碳催化剂的表面处理品。
(实施例3-5)
在实施例2中,除了将加热回流的温度和时间变更为下述表1所示条件以外,与实施例2同样地得到碳催化剂的表面处理品。
(实施例6)
在20mL玻璃制样品瓶中,加入实施例1中制备的碳催化剂0.5g、4mol/L硫酸水溶液20mL和搅拌棒,在室温(25℃)下搅拌28天。过滤,其后的操作与实施例2相同,得到碳催化剂的表面处理品。
(比较例1)
将实施例1中制备的碳催化剂0.5g加入石英管中,在氮气气氛中,用聚焦炉加热,从室温(25℃)以50℃/分钟的升温速度升温至1300℃,在1300℃下保持1小时,进行热处理。
放冷后,将得到的热处理品用研钵粉碎至粒径约1μm以下,得到碳催化剂的表面处理品。
(比较例2)
在实施例2中,除了使用16mol/L硝酸水溶液代替4mol/L硫酸水溶液,加热回流1天以外,与实施例2同样地得到碳催化剂的表面处理品。
[测定评价]
针对在上述实施例和比较例中制作的各碳催化剂,进行了以下的各种测定评价。
(O/C比·N/C比)
通过XPS表面分析,求出碳催化剂表面的O/C比和N/C比。XPS表面分析用X射线光电子能谱仪(AXIS NOVA,KRATOS社制),使用AlKα射线(10mA,15kV)作为X射线源进行。对于来自在碳质粒子表面的各原子的核心级的光电子能谱,将C1s能谱峰作为284.5eV进行结合能校正,根据各能谱的峰面积和检测灵敏度系数,算出碳质粒子表面的O/C比和N/C比。此外,定量下限为0.0001。
(耐久性)
通过在4mol/L硫酸水溶液20mL中加入碳催化剂500mg,在45℃下搅拌3小时的耐酸试验,进行耐久性评价。
将该试验后的悬浊液过滤回收碳催化剂,用蒸馏水反复洗净和过滤,用pH试纸确认滤液为中性后,用60℃的减压干燥器(5000Pa)干燥一夜。然后,测定干燥后的回收碳催化剂的质量,相对于试验前的碳催化剂的质量的质量变化率为5%以下时评价为“A”,超过5%时评价为“B”。
(氧化还原电位测定)
首先,使用上述实施例和比较例中制作的各碳催化剂,通过下述示出的方法制作电极样品。然后,使用制作的各电极样品,通过用下述测定条件的循环伏安法进行氧化还原电位测定,求得氧化还原电位差、氧化电流密度和还原电流密度。
<电极样品的制作>
在碳催化剂5.0mg中加入水-异丙醇(质量比8:2)混合液500μL和5质量%Nafion(注册商标)水溶液50μL,用超音波浴进行10分钟超声波分散处理后,用均质器(Sonifier(注册商标)MODEL S-150D,BRANSON社製)搅拌混合2分钟,调制催化剂浆料。
将1.382μL该催化剂浆料用微型注射器滴到玻璃碳旋转电极(直径4mm,长度2.6cm)上,用微型注射器前端铺展在电极整个表面上之后,用烘干机热风干燥,使碳催化剂以0.1mg/cm2黏着在电极表面。
向该黏着有碳催化剂的电极滴加0.5mol/L硫酸水溶液,并进行脱气处理,用0.5mol/L硫酸水溶液置换电极中的空气,作为电极样品。
<测定条件>
工作电极:用上述制作的电极样品的旋转电极
对电极:铂电极
参比电极:可逆氢电极(0.5mol/L硫酸水溶液)
电解液:含有钒离子(V4+)0.1mol/L和硫酸根离子(SO42-)2mol/L的硫酸钒的水溶液(60mL)
温度:室温(25℃)
气氛:氮气0.35L/分钟×10分钟脱气后,流动
扫描电位:1.5-0.3V(对比基准电极)
扫描速度:0.05V/s
扫描循环:5个循环
由所述循环伏安法求得的氧化还原电位差越小,氧化还原的可逆反应性越高。另外,氧化电流密度和还原电流密度的电流值的绝对值越大,催化剂活性越高。
上述的各种测定评价的结果合并示于下述表1中。
此外,关于比较例2,在氧化还原电位测定的循环伏安法中,难以测定氧化还原峰,不能求得氧化还原电位差、氧化电流密度和还原电流密度。
[表1]
表1
由表1所示结果可知,O/C比为0.05-0.20时(实施例1-6),与O/C比不足0.05时(比较例1)相比,氧化还原电位差小,氧化还原的可逆反应性高。另外,实施例1-6的氧化电流密度和还原电流密度的绝对值大,催化剂活性更高。
另外,通过比较实施例1和实施例2-6可知,通过使用硫酸进行规定的表面处理,能够进一步提高氧化还原的可逆反应性和催化剂活性。
附图标记说明
1 电池
1a 正极室
1b 负极室
2 正极液罐
3 负极液罐
4 隔膜
5 正极
6 负极
7 正极液泵
8 负极液泵
- 还没有人留言评论。精彩留言会获得点赞!