用于EUV光刻的底层的制作方法
用于euv光刻的底层
1.发明背景
2.相关申请
3.本技术要求2019年8月21日提交的发明名称为“用于euv光刻的底层”的美国临时专利申请系列号62/889,964的优先权,其通过引用全文纳入本文。
发明领域
4.本发明总体上涉及使用euv(极紫外)光刻技术来制造微电子结构的方法。
5.相关领域描述
6.随着半导体工业继续遵循摩尔定律,不断减小特征尺寸的需求要求使用更薄的膜来防止图案塌陷(pattern collapse)。更薄的膜会需要使用硬掩膜将图案转印到基板上。极紫外(“euv”)曝光有望成为单次曝光光刻实现7nm及以上节点所需的临界尺寸(“cd”)目标的首选方法。然而,euv光刻受到许多问题的阻碍,这些问题包括低通量、随机效应和粘附问题。
7.由含碳层、含硅层和光刻胶组成的传统三层堆叠物往往会出现光刻胶和硅底层之间粘附性差、或硅硬掩膜(“si-hm”)层由于为改善粘附所进行的改变而蚀刻速率低的问题。旋涂硅硬掩膜能够向光刻胶提供更好的粘附、并且具有高蚀刻速率,这将提供一种对改进光刻效果和加工时间极为友好的解决方案。
8.发明概述
9.本发明广义上涉及硅硬掩膜组合物以及在euv工艺中使用这些组合物的方法。
10.在一个实施方式中,本发明提供了一种结构的形成方法。该方法包括:提供基板,该基板任选地包括其上的一个或多个中间层。在基板上或在一个或多个中间层(如果存在)上施涂组合物,以形成硅硬掩膜层。该组合物包含聚硅氧烷,该聚硅氧烷包含:
11.粘附促进单体,其具有选自以下一种或两种的结构:
[0012][0013]
其中:
[0014]
每个r独立地选自c1~约c6烷基和氢,
[0015]
n为1~约6,
[0016]
每个x独立地选自环氧丙氧基、环氧、环氧环烷基、琥珀酸酐、乙酰胺和异氰脲酸酯部分;以及
[0017]
一种或两种以下单体:
[0018]
表面改性单体,其具有选自以下一种或两种的结构:
[0019][0020]
其中:
[0021]
每个r1独立地选自c1~约c6烷基和c6~约c
20
芳基,
[0022]
每个r2独立地选自c1~约c6烷基和氢,
[0023]
每个r3独立地选自c1~约c6烷基和氢,
[0024]
m为1~约6,
[0025]
每个y独立地选自乙酰氧基、酯和芳基部分,以及
[0026]
致密化单体,其具有选自以下一种、两种或三种的结构:
[0027][0028]
其中每个r4独立地选自c1~约c6烷基和氢。
[0029]
任选地在硅硬掩膜层上形成六甲基二硅氮烷底漆层。在六甲基二硅氮烷底漆层(如果存在)上形成光刻胶层,或者如果不存在六甲基二硅氮烷底漆层则在硅硬掩膜层上形成光刻胶层,以及对光刻胶层的至少一部分进行euv辐射。
[0030]
在另一个实施方式中,该方法包括:提供基板,该基板任选地包括其上的一个或多个中间层。在基板上或在一个或多个中间层(如果存在)上施涂组合物,以形成硅硬掩膜层。该组合物包含聚硅氧烷,该聚硅氧烷包含:
[0031]
粘附促进单体,其包含环氧官能化三烷氧基硅烷、酸酐官能化三烷氧基硅烷、乙酰胺基官能化三烷氧基硅烷、三烷氧基硅烷基异氰脲酸酯及其混合物,以及一种或两种以下单体:
[0032]
四烷氧基硅烷;和
[0033]
选自二烷氧基硅烷、三烷氧基硅烷及其组合的表面改性单体;
[0034]
任选地在硅硬掩膜层上形成六甲基二硅氮烷底漆层。在六甲基二硅氮烷底漆层(如果存在)上形成光刻胶层,或者如果不存在六甲基二硅氮烷底漆层则在硅硬掩膜层上形成光刻胶层。对光刻胶层的至少一部分进行euv辐射。
附图说明
[0035]
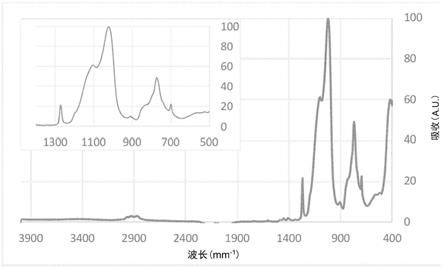
图(fig.)1是硅硬掩膜1(实施例1)的ftir光谱;
[0036]
图2是示出在硅硬掩膜5(实施例5)上测出的交联温度的曲线图;以及
[0037]
图3是示出实施例9中所测试的样品的euv光刻胶聚焦曝光量矩阵(“fem”)测量值的表。
[0038]
发明详述
[0039]
更详细而言,本发明提供了硅硬掩膜组合物以及使用这些组合物利用euv(即,小于约20nm且通常约13.5nm的波长)光刻工艺来形成微电子结构的方法。
[0040]
硅硬掩膜组合物
[0041]
1.用于组合物中的聚合物
[0042]
优选的聚合物为聚硅氧烷,其包括一种或多种的粘附促进单体和一种或两种以下单体:(1)一种或多种的表面改性单体;和/或(2)一种或多种的致密化单体。
[0043]
优选的粘附促进单体产生包含具有选自一种或两种以下结构的重复单元的聚合物:
[0044][0045]
其中:
[0046]
每个r独立地选自c1~约c6烷基(优选c1~约c3烷基)和氢;
[0047]
n为1~约6,更优选1~约3;以及
[0048]
每个x独立地选自环氧丙氧基、环氧、环氧环烷基(优选c3~约c
10
环烷基,并优选c1~约c6环烷基)、琥珀酸酐、乙酰胺和异氰脲酸酯部分。
[0049]
优选的表面改性单体产生包含具有选自一种或两种以下结构的重复单元的聚合物:
[0050][0051]
其中:
[0052]
每个r1独立地选自c1~约c6烷基(优选c1~约c3烷基)和c6~约c
20
芳基(优选c6~约c
14
芳基);
[0053]
每个r2独立地选自c1~约c6烷基(优选c1~约c3烷基)和氢;
[0054]
每个r3独立地选自c1~约c6烷基(优选c1~约c3烷基)和氢;
[0055]
m为1~约6,更优选1~约3;以及
[0056]
每个y独立地选自乙酰氧基、酯和芳基部分。对于y优选的芳基部分为c6~约c
20
、更
优选c6~约c
14
、最优选c6。
[0057]
优选的致密化单体产生包含具有选自一种、两种或三种以下结构的重复单元的聚合物:
[0058][0059]
其中每个r4独立地选自c1~约c6烷基(优选c1~约c3烷基)和氢。
[0060]
应当了解,可调整单体的比率和载量以提供最终组合物的适宜性质。可以调整特性以适应各种光刻胶类型、特征尺寸和特征类型(线/空间、接触孔等)。
[0061]
在一个实施方式中,聚硅氧烷中粘附促进单体或重复单元的摩尔百分比优选为约2%~约50%,更优选为约5%~约35%,进一步优选为约10%~约20%。表面改性单体的摩尔百分比优选约5%~约90%,更优选约10%~约80%,进一步优选约40%~约70%。致密化单体的摩尔百分比优选约0%~约80%,更优选约10%~约70%,进一步优选约15%~约50%。
[0062]
在另一个实施方式中,聚硅氧烷中粘附促进单体或重复单元的摩尔百分比优选为约2%~约50%,更优选为约5%~约35%,进一步优选为约10%~约20%。表面改性单体的摩尔百分比优选约0%~约70%,更优选约10%~约70%,进一步优选约15%~约70%。致密化单体的摩尔百分比优选约2%~约90%,更优选约10%~约80%,进一步优选约15%~约70%。
[0063]
在又一个实施方式中,聚硅氧烷中粘附促进单体或重复单元的摩尔百分比为约0%~约30%,优选约0.01%~约30%,更优选约0.01%~约20%,进一步优选约0.1%~约15%。表面改性单体的摩尔百分比优选约0%~约70%,更优选约5%~约60%,进一步优选约15%~约50%。致密化单体的摩尔百分比优选约30%~约95%,更优选约40%~约80%,进一步优选约50%~约70%。
[0064]
在一个实施方式中,聚硅氧烷基本上或甚至由一种或多种类型的粘附促进单体和一种或两种以下单体构成:一种或多种类型的表面改性单体和/或一种或多种类型的致密化单体。在另一个实施方式中,聚硅氧烷基本上或甚至由一种或多种类型的粘附促进单体、一种或多种类型的表面改性单体和一种或多种类型的致密化单体构成。
[0065]
2.聚合材料和方法
[0066]
用于硅硬掩膜组合物的聚合物优选由可水解硅烷单体来合成,特别优选的合成方法为溶胶-凝胶法。可水解硅烷单体的示例包括选自以下的那些:四乙氧基硅烷(“teos”)、原硅酸四甲酯(“tmos”)、甲基三甲氧基硅烷(“mtms”)、甲基三乙氧基硅烷(“mteos”)、二甲基二甲氧基硅烷(“dmdms”)、二甲基二乙氧基硅烷(“dmdeos”)、苯基三甲氧基硅烷(“ptms”)、苯乙基三甲氧基硅烷(“petms”),2-(甲氧羰基)乙基三甲氧基硅烷(“cmetms”)、乙酰氧基乙基三甲氧基硅烷、乙基三甲氧基硅烷(“etms”)、正丁基三甲氧基硅烷(“butms”)、(3-环氧丙氧基丙基)三甲氧基硅烷(“glytms”)、5,6-环氧己基三乙氧基硅烷
(“epoteos”)、2-(3,4-环氧环己基)乙基三甲氧基硅烷(“echtms”)、[3-(三乙氧基硅基)丙基]琥珀酸酐(“suctos”),(3-乙酰胺基丙基)三甲氧基硅烷(“amitms”),(1,3-二-2-丙烯-1-基)-5-(([3-三乙氧基硅基丙基])-(1,3,5-三嗪-2,4,6(1h,3h,5h)-三酮)(“daica-teos”)及其组合。
[0067]
如前所述,这些组合物中所用的聚合物包括至少一种或多种类型的粘附促进单体,优选为其与一种或两种以下单体的共聚物:(1)一种或多种类型的表面改性单体;以及(2)一种或多种类型的致密化单体。
[0068]
用作会形成聚硅氧烷的前述结构(i)和(ii)的粘附促进单体的优选起始化合物包括三烷氧基硅烷(优选c1~约c6烷氧基,更优选c1~约c3烷氧基),其包含高粘附官能团如环氧、酸酐、乙酰胺基和/或异氰脲酸酯。上述起始化合物的特别优选的示例选自(3-环氧丙氧基丙基)三甲氧基硅烷、5,6-环氧己基-三乙氧基硅烷、2-(3,4-环氧环己基)-乙基三甲氧基硅烷、[3-(三乙氧基硅基)丙基]-琥珀酸酐、(3-乙酰胺基丙基)-三甲氧基硅烷、(1,3-二-2-丙烯-1-基)-5-(([3-三乙氧基硅基丙基]-(1,3,5-三嗪-2,4,6(1h,3h,5h)-三酮)及其混合物。
[0069]
在包含表面改性单体的实施方式中,该单体发挥调节由组合物形成的硬掩膜层的表面能的功能,从而在某些实施方式中提供硬掩膜层和euv光刻胶之间的相容性。用作会产生聚硅氧烷的结构(iii)和(iv)的表面改性单体的优选起始化合物包括二烷氧基硅烷、三烷氧基硅烷及其组合。在任一情况下,烷氧基优选c1~约c6烷氧基,更优选c1~约c3烷氧基。进一步优选所选的表面改性单体的si原子与两个烷基、芳基和/或烷基芳基部分(在二烷氧基硅烷的情况下)连接,或与一个烷基、芳基或烷基芳基部分连接(在三烷氧基硅烷的情况下)。在任一情况下,优选烷基为c1~约c6,优选c1~约c3;优选芳基为c6~约c
20
,更优选c6至~约c
14
,最优选c6;优选烷基芳基为烷基部分c1~约c6(更优选c1~约c3)且芳基部分c6~约c
20
(更优选c6至约c
14
,最优选c6)。在另一个实施方式中,表面改性单体缺少环氧、酸酐、乙酰胺基和/或异氰脲酸酯官能团中的一个或全部。
[0070]
特别优选的表面改性单体选自甲基三甲氧基硅烷、甲基三乙氧基硅烷、二甲基二甲氧基硅烷、二甲基二乙氧基硅烷、苯基三甲氧基硅烷、苯乙基三甲氧基硅烷、2-(甲氧羰基)乙基三甲氧基硅烷、乙酰氧基乙基三甲氧基硅烷、乙基三甲氧基硅烷、正丁基三甲氧基硅烷及其混合物。
[0071]
这些聚合物中所用的致密化单体提供亲水性硅烷醇基团,并且通过在固化的硬掩膜层中为最终交联聚合物的结构提供交联位点以在施涂后烘烤过程中协助提供热驱动交联。用作会形成聚硅氧烷的前述结构(v)、(vi)和(vii)的致密化单体的优选起始化合物包括四烷氧基硅烷(优选c1~约c6烷氧基,更优选c1~约c3烷氧基),其中特别优选四乙氧基硅烷和/或原硅酸四甲酯。
[0072]
聚合物通过将上述所需单体优选以达到前述摩尔比的量溶解于合适的聚合溶剂中来合成。聚合溶剂可包括但不限于丙二醇单甲醚乙酸酯(“pgmea”)、丙二醇甲醚(“pgme”)、丙二醇乙醚(“pgee”)、环己酮、乳酸乙酯、丙醇、丁醇及其混合物。向反应混合物中添加水以使硅烷单体水解,同时添加用于溶胶-凝胶缩合的催化剂。h2o/单体的摩尔比范围为约1:1~约10:1,更优选约2:1~约8:1。合适的催化剂包括但不限于矿物酸(如盐酸或硝酸)、乙酸、马来酸及其组合。优选使水解在室温下进行约1小时~约48小时、更优选约4小
时~约24小时、进一步优选约16小时。然后,水解后的单体在约40℃~约120℃、优选约60℃~约100℃、更优选约90℃的温度下共聚约0.5小时~约72小时、优选约1小时~约48小时、更优选约5小时~约16小时。
[0073]
通过采用聚苯乙烯标准品的气相渗透色谱法(gpc)测定所得聚合物的数均分子量(mn)优选约500g/mol~约3000g/mol,更优选约800g/mol~约2000g/mol。通过gpc测定的聚合物的重均分子量(mw)范围优选约600g/mol~约10000g/mol,更优选约1000g/mol~约8000g/mol,进一步优选约1500g/mol~约5000g/mol。
[0074]
3.组合物制备
[0075]
然后,将所形成的聚合物分散或溶解于溶剂体系中以形成硅硬掩膜组合物。优选的溶剂体系包括选自pgmea、pgme、pgee、丙二醇正丙醚(“pnp”)、乳酸乙酯、环己酮、γ-丁内酯(“gbl”)、甲基异丁基甲醇及其混合物的溶剂。溶剂体系优选以基于组合物总重量100重量%计为约80~约99.9重量%、更优选约90~约99.9重量%、进一步优选约99.0~约99.9重量%的量使用。用于形成硅硬掩膜层的组合物将优选包含基于组合物总重量100重量%计为约0.1~约20重量%、更优选约0.1~约10%重量、进一步优选约0.1~约1.0重量%的固含量。
[0076]
将上述成分在溶剂体系中混合在一起以形成硅硬掩膜层组合物。此外,任何可选成分(例如表面活性剂)也同时分散在溶剂体系中。
[0077]
在一些实施方式中,使用添加剂。优选地,将添加剂简单混入硅硬掩膜层组合物中。优选的添加剂包括催化剂如苄基三乙基氯化铵(“bteac”)、叔丁基溴化膦(“tbpb”)、乙基三苯基溴化膦(“etppb”)和三乙氧基-3-(2-咪唑啉-1-基)丙基硅烷。可在硬掩膜组合物中使用的另一种添加剂是光酸发生剂(“pag”)如三(羟苯基)甲磺酸铵、三(羟苯基)三氟甲磺酸铵及其组合。当存在时,添加剂(累积或单独)应以基于组合物总重量100重量%计为约0.01~约2.0重量%、优选约0.1~约1.0重量%的量存在于组合物中。
[0078]
在一个实施方式中,硬掩膜组合物基本上或甚至由聚硅氧烷、溶剂体系、催化剂和光酸发生剂组成。在一个实施方式中,硬掩膜组合物基本上或甚至由聚硅氧烷、溶剂体系和催化剂组成。在另一个实施方式中,硬掩膜组合物基本上或甚至由聚硅氧烷、溶剂体系和光酸发生剂组成。在又一个实施方式中,硬掩膜组合物基本上或甚至由聚硅氧烷和溶剂体系组成。
[0079]
硅硬掩膜组合物的使用方法
[0080]
在本发明方法中,上述硬掩膜组合物在基板表面上或在基板表面上的中间层(如下所述)上形成为层。可采用任何微电子基板,但基板优选为半导体基板,例如硅、sige、sio2、si3n4、sion、sico:h(例如商品名black diamond(黑钻))、铝、钨、硅化钨、砷化镓、锗、钽、氮化钽、ti3n4、铪、hfo2、钌、磷化铟、玻璃或前述材料的混合物。基板可具有平坦表面,也可以包括形貌特征(通孔、沟槽、接触孔、凸起特征、线条等)。本文所用的“形貌(topography)”是指基板表面内或基板表面上的结构高度或深度。
[0081]
可在基板或任何中间层上形成富碳层。富碳层可通过任何已知的施涂方法形成,其中一种优选方法是以约1000~约5000rpm、优选约1250~约1750rpm的速度旋涂约30~约120秒、优选约45~约75秒的时间。术语“富碳”是指由基于组合物总固量100重量%计包含大于约50重量%碳、优选大于约70重量%碳、更优选约75~约80重量%碳的组合物形成的
层。合适的富碳层选自旋涂碳层(soc)、无定形碳层和碳平面化层。
[0082]
示例性的富碳层通常会削弱溶解或分散于溶剂体系中的聚合物和一种或多种以下可选成分:酸和/或碱淬火剂、催化剂、交联剂和表面改性添加剂。优选的组合物会适于形成厚的层,并且基于所述组合物总重量100重量%计优选含有约0.1~约70重量%、更优选约5~约40重量%、进一步优选约10~约30重量%的固含量。在施涂了富碳组合物后,优选将其加热至约100℃~约400℃、更优选约160℃~约350℃的温度约30秒~约120秒、优选约45秒~约60秒的时间,以蒸发溶剂。烘烤后富碳层的厚度(通过椭偏仪测得的五处测量值的平均值)优选约10nm~约120nm,更优选约20nm~约100nm,进一步优选约50nm~约60nm。富碳层可以通过其他已知的施涂方法如化学气相沉积(“cvd”)、等离子体增强化学气相沉积(“pecvd”)、原子层沉积(“ald”)或等离子体增强原子层沉积(“peald”)来形成。
[0083]
本发明的硅硬掩膜层可施涂在富碳材料附近或基板或任何中间层附近。硅硬掩膜层优选通过以约1000rpm~约3000rpm的速度、优选约1500rpm~约2000rpm的速度旋涂约30秒~约120秒、优选约30秒~约60秒的时间。在施涂了硅硬掩膜后,优选将其加热至约150℃~约300℃、更优选约200℃~约250℃的温度约30秒~约120秒、优选约30秒~约60秒的时间,以蒸发溶剂。在该烘烤过程中,会发生溶胶-凝胶反应,从而使材料交联。烘烤后硬掩膜层的厚度(通过椭偏仪测得的五处测量值的平均值)优选约2nm~约50nm,更优选约5nm~约30nm,进一步更优选约10nm~约25nm。硬掩膜层的蚀刻速率应至少为富氟等离子体气氛中光刻胶(例如,化学放大型、金属氧化物或断链型光刻胶)的1.5倍。此外,富碳层的蚀刻速率应至少为富氧等离子体蚀刻气氛中硅硬掩膜层的1.5倍。
[0084]
在硅硬掩膜层烘烤之后,可将euv光刻胶(即成像层)施涂到硅硬掩膜层以形成光刻胶层。光刻胶层可通过任何常规方法形成,其中一种优选方法是以约350rpm~约4000rpm(优选约1000rpm~约2500rpm)的速度旋涂光刻胶组合物约10秒~约60秒(优选约10秒~约30秒)的时间。然后,光刻胶层任选地在至少约70℃、优选约80℃~约150℃、更优选约100℃~至约150℃的温度下进行施涂后烘焙(“pab”)约30秒~约120秒的时间。烘烤后光刻胶层的厚度(通过椭偏仪测得的五处测量值的平均值)通常为约5nm~约120nm,优选约10nm~约50nm,更优选约20nm~约40nm。
[0085]
在涂覆光刻胶之前,可实施六甲基二硅氮烷(“hmds”)底漆工艺。在该工艺中,晶片在150℃加热下90秒的同时曝光于密封室中的hmds蒸汽中。
[0086]
随后,光刻胶层通过以约5mj/cm2~约100mj/cm2、优选约10mj/cm2~约80mj/cm2、更优选约20mj/cm2~约60mj/cm2的剂量曝光于euv辐射来进行图案化。更具体而言,光刻胶层通过使用位于光刻胶层表面上方的掩膜来曝光。该掩膜具有设计成使euv辐射从掩膜反射并接触到光刻胶层表面的区域。掩膜的其余部分被设计成吸光以防止辐射在某些区域接触到光刻胶层的表面。本领域技术人员将容易地理解,反射和吸收部分的布置是根据光刻胶层中和最终基板或任何中间层中要形成的所需图案来设计的。
[0087]
euv曝光后,光刻胶层优选以低于约180℃、优选约60℃~约140℃、更优选约80℃至约130℃的温度经受约30秒~约120秒(优选约30秒至约90秒)的时间来进行曝光后烘焙(“peb”)。
[0088]
然后,使光刻胶层与显影剂接触来形成图案。根据所用的光刻胶是正性显影还是负性显影,显影剂将移除光刻胶层的曝光部分或移除光刻胶层的未曝光部分以形成图案。
之后,将图案转印至硅硬掩膜层、转印至任何现有的中间层、以及最后转印至基板。该图案转印可通过等离子体蚀刻(例如,cf4蚀刻剂、o2蚀刻剂)或湿法蚀刻或显影工艺来进行。在图案将通过蚀刻从光刻胶层转印至基板的实施方式中,优选硅硬掩膜层对典型euv光刻胶的蚀刻速率至少为约1x,优选为约1.5x~约2x。
[0089]
无论是通过蚀刻还是通过显影来实施图案转印,所得特征都具有高分辨率。例如,采用本发明的方法可以实现小于约40nm半节距、优选小于约30nm半节距的分辨率。硅硬掩膜层会提高最终特征的外边距折叠(collapse margin)。外边距折叠通过从剂量到尺寸的差值来量化,该差值来自于结构仍能经受的最大剂量、或在负色调显影光刻胶或负色调成像光刻胶的情况下的最小剂量。
[0090]
通过浏览本文的公开内容和下述的工作实施例,本发明的各实施方式的额外优点对于本领域技术人员来说将是显而易见的。应当理解,除非本文另有指示,否则本文中所述的各实施方式不必相互排斥。例如,在一个实施方式中描述或描绘的特征也可以包括在其他实施方式中,但非必须包括在其他实施方式中。因此,本发明包括本文中所述的特定实施方式的各种组合和/或集成。
[0091]
本文中,当用于两项或更多项的列举时,短语“和/或”是指可以单独使用所列出的项中的任何一项,或者可以使用所列出的项中的两项或更多项的任何组合。例如,如果组合物被描述成含有或不含组分a、b和/或c,则该组合物可以含有或不含:仅a;仅b;仅c;a和b的组合;a和c的组合;b和c的组合;或a、b和c的组合。
[0092]
本说明书还使用了数值范围来量化与本发明的各实施方式有关的某些参数。应当理解,当提供数值范围时,这种范围应被解释为向仅列出范围下限值的权利要求限定以及仅列出范围上限值的权利要求限定提供书面支持。例如,所公开的约10~约100的数值范围为列出“约大于10”(无上限)的权利要求和列出“约小于100”(无下限)的权利要求提供书面支持。
实施例
[0093]
以下实施例对本发明的方法进行说明。但应理解,这些实施例是以说明性的方式提供的,不应将其中任何内容视为本发明总体范围的限制。
[0094]
实施例1
[0095]
硅硬掩膜1的合成与制剂
[0096]
在本实施例中,将3.95克teos(gelest,美国宾夕法尼亚州莫里斯维尔市)、7.62克mtms(gelest,美国宾夕法尼亚州莫里斯维尔市)、2.62克petm(gelest,美国宾夕法尼亚州莫里斯维尔市)和3.54克glytms(gelest,美国宾夕法尼亚州莫里斯维尔市)溶解于36克pgme(kmg电子化学品公司(kmg electronic chemicals),美国德克萨斯州沃思堡市)中。接着,将8.78克3n乙酸(vwr,巴达维亚,il)滴加到溶液中以使硅氧烷单体水解,并将溶液在室温下保持约16小时。反应保持在n2覆盖物下,并加热至90℃20小时以产生母液1。所得聚合物的分子量约2000,使用tosoh ecosec hlc-8320凝胶渗透色谱以thf为流动相而测得的多分散指数(“pdi”)为1.6。
[0097]
然后,将5.89克母液1溶解于110.9克pgme和12.88克pgmea(kmg电子化学品公司,德克萨斯州沃思堡)(90:10)中,向其添加0.29克tbpb溶液(pgme中0.2重量%)。制得的制剂
在搅拌轮上整晚搅拌,所得制剂为约0.9重量%固体。
[0098]
实施例2
[0099]
硅硬掩膜2的合成与制剂
[0100]
在该过程中,将3.95克teos、4.90克mtms、6.78克petm和3.54克glytms溶解于44克pgme中。接着,将8.78克3n乙酸滴加到溶液中以使硅氧烷单体水解,并将溶液在室温下保持约16小时。反应保持在n2覆盖物下,并加热至90℃20小时以产生母液2。所得聚合物的分子量约2000,使用tosoh ecosec hlc-8320凝胶渗透色谱以thf为流动相而测得的多分散指数(pdi)为1.6。
[0101]
然后,将5.9克母液2溶解于110.1克pgme和12.8克pgmea(90:10)中,向其添加0.29克tbpb溶液(pgme中0.2重量%)。制得的制剂在搅拌轮上整晚搅拌。所得制剂为约0.9重量%固体。
[0102]
实施例3
[0103]
硅硬掩膜3的合成与制剂
[0104]
在该实施例中,将3.95克teos、7.62克mtms、2.26克petm和3.93克epotms(gelest,美国宾夕法尼亚州莫里斯维尔市)溶解于35克pgme中。接着,将8.78克3n乙酸滴加到溶液中以使硅氧烷单体水解,并将溶液在室温下保持约16小时。反应保持在n2覆盖物下,并加热至90℃17.5小时以产生母液3。所得聚合物的分子量约2300,使用tosoh ecosec hlc-8320凝胶渗透色谱以thf为流动相而测得的多分散指数(pdi)为1.7。
[0105]
然后,将7.0克母液3溶解于136.8克pgme和15.9克pgmea(90:10)中,向其添加0.35克tbpb溶液(pgme中0.2重量%)。制得的制剂在搅拌轮上整晚搅拌。所得制剂为约0.9重量%固体。
[0106]
实施例4
[0107]
硅硬掩膜4的合成与制剂
[0108]
在该过程中,将3.95克teos、8.98克mtms和3.54克glytms溶解于33克pgme中。接着,将8.78克3n乙酸滴加到溶液中以使硅氧烷单体水解,并将溶液在室温下保持约16小时。反应保持在n2覆盖物下,并加热至90℃17.5小时以产生母液4。所得聚合物的分子量约1500,使用tosoh ecosec hlc-8320凝胶渗透色谱以thf为流动相而测得的多分散指数(pdi)为1.5。
[0109]
然后,将4.98克母液4溶解于84.9克pgme和9.9克pgmea(90:10)中,向其添加0.26克tbpb溶液(pgme中0.2重量%)。制得的制剂在搅拌轮上整晚搅拌。所得制剂为约1.0重量%固体。
[0110]
实施例5
[0111]
硅硬掩膜5的合成与制剂
[0112]
在该过程中,将11.34克teos、4.76克mtms和2.36克glytms溶解于29克pgme中。接着,将8.78克3n乙酸滴加到溶液中以使硅氧烷单体水解,并将溶液在室温下保持约16小时。反应保持在n2覆盖物下,并加热至90℃4小时以产生母液5。所得聚合物的分子量约3000,使用tosoh ecosec hlc-8320凝胶渗透色谱以thf为流动相而测得的多分散指数(pdi)为2.2。
[0113]
然后,将7.0克母液5溶解于173.17克pgme和19.90克pgmea(90:10)中,在搅拌轮上整晚搅拌。所得制剂为约0.7重量%固体。
[0114]
实施例6
[0115]
硅硬掩膜6的合成与制剂
[0116]
在该过程中,将11.65克teos和7.83克mteos(gelest,美国宾夕法尼亚州莫里斯维尔市)溶解于26克pgme中。接着,将8.78克3n乙酸滴加到溶液中以使硅氧烷单体水解,并将溶液在室温下保持约16小时。反应保持在n2覆盖物下,并加热至90℃4小时以产生母液6。所得聚合物的分子量约2200,使用tosoh ecosec hlc-8320凝胶渗透色谱以thf为流动相而测得的多分散指数(pdi)为1.0。
[0117]
然后,将9.42克母液5溶解于98.77克pgme和11.81克pgmea(90:10)中,在搅拌轮上整晚搅拌。所得制剂为约1.57重量%固体。
[0118]
实施例7
[0119]
硅硬掩膜1的ftir分析
[0120]
使用thermal scientific nicolet is50 ftir光谱仪获取由实施例1得到的硅硬掩膜的ftir光谱。为制备样品,重新生成由实施例1制得的制剂,但添加更少的溶剂以达到约10wt.%的固含量,然后以1500rpm的转速在硅晶片(nestec,测试级)上旋涂60秒。旋涂后,将该层在205℃热板上烘烤60秒,以形成交联膜。该层的总厚度约250nm。使用m-2000椭偏仪(j.a.woollam,内布拉斯加州林肯市)测定膜厚。然后,将膜从晶片上刮下成粉末状。粉末样品以atr-ftir模式进行表征。图1示出ftir光谱。
[0121]
实施例8
[0122]
硅硬掩膜5的表征
[0123]
交联温度通过采用去胶(strip)试验以pgme为去胶溶剂来测定。将硅硬掩膜5(实施例5)以1500rpm的转速旋涂到基板上,并以图2所示的温度烘烤。厚度如实施例7中所述那样测定。然后,将pgme汇集在晶片表面,并以1500rpm的转速旋转-干燥60秒。再次测定厚度,以观察是否有膜损失。这些结果示于图2,负的厚度损失表示薄膜膨胀。
[0124]
硅硬掩膜5的表面接触角使用ast optima(b5rm5208-143)接触角测量工具来测定。实施例5的材料的接触角在不同点测量5次并取平均值。使用水和二碘甲烷作为滴液溶剂。材料的最终表面接触角为61.8(h2o)和51.9(ch2i2)
[0125]
实施例9
[0126]
硅硬掩模1的光刻结果
[0127]
将一层材料(brewer science,密苏里州罗拉市)在硅晶片上以1486rpm的转速旋涂30秒,并在220℃下烘烤60秒以形成60nm膜。硅硬掩膜1(实施例1)在一层材料上以1406rpm旋涂30秒,并在205℃下烘烤60秒以形成25nm膜。euv光刻胶jsr4267(购自imec)通过以1040rpm旋涂25秒来涂敷在硬掩膜上,然后在130℃下烘焙60秒以形成35nm涂层。然后,光刻胶采用表1所示的参数来曝光。成像步骤使用nxe3300 euv扫描仪,晶片加工使用tel pro z轨道。图3示出使用光刻胶下方的实施例1硅硬掩模层的光刻质量。
[0128]
表1
[0129]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1