铂碳催化剂、碳材料及其制备方法和应用与流程
1.本发明涉及铂碳催化剂、碳材料及其制备方法和应用。
背景技术:
2.燃料电池是一种不经过燃烧,直接将燃料的化学能以电化学反应方式转变为电能的发电装置,其具有能量密度高、启动快、能量转化效率高、污染小等优点,被认为是最有可能大规模替代现有能源技术的新型能源技术。其中,氢燃料电池是目前主流的燃料电池技术,而催化剂和质子交换膜又是氢燃料电池技术的核心。
3.铂碳催化剂是目前最成熟的氢燃料电池催化剂,但对于大规模商业应用来说,铂碳催化剂仍有不足之处。一方面,铂的价格昂贵,其成本约占燃料电池总成本的40%。另一方面,氢燃料电池阴极的铂溶解和团聚导致铂表面积随时间下降明显,影响燃料电池寿命。因此,提高铂碳催化剂的铂利用率和稳定性变得十分迫切。此外,实际应用的氢燃料电池铂碳催化剂的铂载量至少在20wt%以上,其比化工用铂碳催化剂(一般铂载量低于5wt%)的制造难度大很多。
4.影响铂碳催化剂质量比活性、稳定性的因素很多且复杂,一些文献认为,铂碳催化剂的质量比活性、稳定性与铂的粒径、形貌、结构,以及载体的种类、性质和铂载量有关。现有技术主要是通过控制铂的粒径、形貌、结构以及载体的比表面积、孔结构来改进铂碳催化剂的性能;也有文献报道在碳表面连上修饰基团,通过对碳载体改性来提高铂碳催化剂的性能。
5.提高载铂量有利于制造更薄、性能更好的膜电极,但大幅度提高载铂量更容易造成铂金属颗粒间的堆积,导致活性位点利用率急剧下降。如何更有效地利用铂金属颗粒的催化活性位点并增加可接触的三相催化反应界面,从而提高铂利用率以及燃料电池及金属空气电池的综合性能,是本领域亟需解决的关键问题。
6.碳载体的缺陷位越多越有利于提高载铂量,但同时加剧碳腐蚀。石墨程度高的碳载体能有效缓解碳腐蚀,但石墨化程度高也使碳载体表面呈化学惰性,很难将铂均匀分散在碳载体上,在载铂量较高时尤为困难。
7.化学还原法是常用的铂碳催化剂的制造方法,优点是工艺简单,缺点是铂的利用率低,催化活性较低。其原因可能是碳载体孔结构不规则导致铂纳米颗粒分散不均匀。
8.前述背景技术部分所公开的信息仅用于加强对本发明的背景理解,它可以包括不属于本领域普通技术人员已知的信息。
技术实现要素:
9.本发明的目的之一是提供一种铂碳催化剂,该催化剂的质量比活性和其稳定性均优于商业催化剂。本发明的目的之二是,在目的一的基础上,改进铂金属的分散,提供一种性能更佳的高载铂量的铂碳催化剂。本发明的目的之三是改进制造铂碳催化剂的水相化学还原法,提高铂碳催化剂的质量比活性和其稳定性。
10.为了实现前述目的,本发明提供了如下的技术方案。
11.1、一种铂碳催化剂,包括碳载体和负载于其上的铂金属,所述的碳载体为硫氮掺杂导电炭黑。
12.2、按照1所述的铂碳催化剂,其特征在于,在其xps分析的s
2p
谱峰中,在162ev~166ev之间,只有噻吩型硫的特征峰,且在168
±
1ev处有特征峰。
13.3、按照2所述的铂碳催化剂,其特征在于,所述噻吩型硫的特征峰为双峰,分别位于163.4
±
0.5ev和164.6
±
0.5ev。
14.4、按照前述任一的铂碳催化剂,其特征在于,在其xps分析的n
1s
谱峰中,除398.5ev~400.5ev之间有特征峰外,在390ev~410ev之间没有其他的特征峰。
15.5、按照前述任一的铂碳催化剂,其特征在于,以催化剂的质量为基准,铂的质量分数为0.1%~80%,优选为20%~70%,更优选为40%~70%。
16.6、按照前述任一的铂碳催化剂,其特征在于,所述铂碳催化剂的电阻率《10.0ω
·
m,优选《2.0ω
·
m。
17.7、按照前述任一的铂碳催化剂,其特征在于,所述铂碳催化剂的比表面80m2/g~1500m2/g,优选为100m2/g~200m2/g。
18.8、按照前述任一的铂碳催化剂,其特征在于,所述导电炭黑为ec-300j、ec-600jd、ecp600jd、vxc72、black pearls 2000、printex xe2-b、printex l6或hiblaxk 40b2。
19.9、按照前述任一的铂碳催化剂,其特征在于,在其xps分析的s
2p
谱峰中,所述噻吩型硫的特征峰与168
±
1ev处的特征峰的峰面积比为1:5~10:1;或者在其xps分析的s
2p
谱峰中,所述噻吩型硫的特征峰与168
±
1ev处的特征峰的峰面积比大于10。
20.10、一种铂碳催化剂的制备方法,包括:(1)制造硫氮掺杂导电炭黑的步骤;和(2)以步骤(1)中所得硫氮掺杂导电炭黑为载体,负载铂的步骤;
21.其中的步骤(1)包括掺杂硫的操作和掺杂氮的操作;
22.所述掺杂硫的操作包括:将导电炭黑与单质硫混合,在惰性气体中于400℃~1500℃处理(优选恒温处理)0.5h~10h;
23.所述的掺杂氮的操作在所述掺杂硫的操作之前、之后或同时进行。
24.11、按照10所述的制备方法,其特征在于,所述导电炭黑与硫的质量比为20:1~2:1;优选为10:1~4:1,更优选为8:1~4:1。
25.12、按照前述任一的制备方法,其特征在于,所述掺杂硫的操作中,所述温度为1150℃~1450℃。
26.13、按照前述任一的制备方法,其特征在于,所述掺杂硫的操作和/或所述掺杂氮的操作中,所述处理的时间为1h~5h,优选为2h~4h。
27.14、按照前述任一的制备方法,其特征在于,氮源的质量以其所含氮的质量计,所述导电炭黑与氮源的质量比为30:1~1:2;优选为25:1~1:1.5。
28.15、按照前述任一的制备方法,其特征在于,所述导电炭黑为ec-300j、ec-600jd、ecp-600jd、vxc72、black pearls 2000、printex xe2-b、printex l6或hiblaxk 40b2。
29.16、按照前述任一的制备方法,其特征在于,所述导电炭黑的xps分析中,氧质量分数大于2%,可以为2%~15%,优选为2.5%~12%。
30.17、按照前述任一的制备方法,其特征在于,所述导电炭黑的电阻率《10ω
·
m,优
选《5ω
·
m,更优选《2ω
·
m。
31.18、按照前述任一的制备方法,其特征在于,所述导电炭黑的比表面积为10m2/g~2000m2/g,优选为200m2/g~2000m2/g。
32.19、按照前述任一的制备方法,其特征在于,所述负载铂的步骤包括:
33.(a)将(1)中得到的硫氮掺杂导电炭黑与铂前驱体分散在水相中,调节ph为8~12(优选调节ph值为10
±
0.5);
34.(b)加入还原剂进行还原;
35.(c)分离出固体,经后处理得到所述的铂碳催化剂。
36.20、按照前述任一的制备方法,其特征在于,所述铂前驱体为氯铂酸、氯铂酸钾或氯铂酸钠;所述铂前驱体的浓度为0.5mol/l~5mol/l。
37.21、按照前述任一的制备方法,其特征在于,(a)中,用碳酸钠水溶液、碳酸钾水溶液、氢氧化钾水溶液、氢氧化钠水溶液或氨水调节水相的ph值。
38.22、按照前述任一的制备方法,其特征在于,(b)中,所述还原剂为柠檬酸、抗坏血酸、甲醛、甲酸、乙二醇、柠檬酸钠、水合肼、硼氢化钠或丙三醇中的一种或几种;所述还原剂与铂的摩尔比为2~100;还原温度为50℃~150℃,优选为60℃~90℃;还原时间为4h~15h,优选为8h~12h。
39.23、按照前述任一的制备方法,其特征在于,所述的后处理包括:洗涤、过滤和干燥。
40.24、一种铂碳催化剂,其特征在于,由10~23中的任一方法制得。
41.25、一种氢燃料电池,其特征在于,所述氢燃料电池的阳极和/或阴极中,使用了1~9和24中的任一铂碳催化剂。
42.26、一种碳材料,其特征在于,该碳材料为硫氮掺杂的导电炭黑。
43.27、按照26所述的碳材料,其特征在于,在其xps分析的s
2p
谱峰中,在162ev~166ev之间,只有噻吩型硫的特征峰,且在168
±
1ev处有特征峰。
44.28、按照前述任一的碳材料,其特征在于,在其xps分析的n
1s
谱峰中,除398.5ev~400.5ev之间有特征峰外,在390ev~410ev之间没有其他的特征峰。
45.29、按照前述任一的碳材料,其特征在于,在其xps分析的s
2p
谱峰中,所述噻吩型硫的特征峰与168
±
1ev处的特征峰的峰面积比为1:5~10:1;或者在其xps分析的s
2p
谱峰中,所述噻吩型硫的特征峰与168
±
1ev处的特征峰的峰面积比大于10。
46.氮的电负性比碳更大,原子半径与碳接近;硫的电负性与碳相当,但原子半径比碳更大,用硫和氮同时对碳材料掺杂,通过二者的协同作用,可以制造出性质独特的碳载体,提高铂碳催化剂的催化性能和稳定性。
47.与现有技术相比,本发明具有以下有益技术效果。
48.一、本发明的制造方法简单,通过对导电炭黑同时掺硫和氮,制造了掺杂位点性质更均一的碳载体,由其制造的铂碳催化剂具有优于商业催化剂的质量比活性及其稳定性。
49.二、实际应用的氢燃料电池铂碳催化剂的载铂量一般在20wt%以上,本领域希望制造出性能优异的更高载铂量的催化剂。然而在提高载铂量时,铂金属颗粒更容易团聚;而本发明在提高载铂量时,可使铂金属颗粒分散得更均匀。
50.三、化学还原法工艺简单,但是铂的利用率低,催化活性较低。然而,以本发明制造
的硫氮掺杂导电炭黑为载体,采用水相的化学还原法,即可容易的制造出质量比活性和稳定性俱佳的高载铂量催化剂。
51.四、通常认为硫对铂催化剂产生不可逆的毒害作用,然而本发明发现,通过对导电炭黑进行掺硫改性,反而显著提高了铂碳催化剂的催化活性及其稳定性。
52.本发明的其他特征和优点将在具体实施方式部分中详细说明。
附图说明
53.图1为实施例1的硫氮掺杂碳载体的硫的xps谱图。
54.图2为实施例1的硫氮掺杂碳载体的氮的xps谱图。
55.图3为实施例5的铂碳催化剂的硫的xps谱图。
56.图4为实施例5的铂碳催化剂的氮的xps谱图。
57.图5为实施例6的铂碳催化剂的硫的xps谱图。
58.图6为实施例7的铂碳催化剂的硫的xps谱图。
59.图7为实施例7的铂碳催化剂的氮的xps谱图。
60.图8为对比例5铂碳催化剂的硫的xps谱图。
具体实施方式
61.以下结合具体实施方式详述本发明,但需说明的是,本发明的保护范围不受这些具体实施方式和原理性解释的限制,而是由权利要求书来确定。
62.本发明中,除了明确说明的内容之外,未提到的任何事宜或事项均直接适用本领域已知的那些而无需进行任何改变。而且,本文描述的任何实施方式均可以与本文描述的一种或多种其他实施方式自由结合,由此形成的技术方案或技术思想均视为本发明原始公开或原始记载的一部分,而不应被视为是本文未曾披露或预期过的新内容,除非本领域技术人员认为该结合明显不合理。
63.本发明所公开的所有特征可以任意组合,这些组合应被理解为本发明所公开或记载的内容,除非本领域技术人员认为该组合明显不合理,均应被视为被本发明所具体公开和记载。本说明书所公开的数值点,不仅包括实施例中具体公开的数值点,还包括说明书中各数值范围的端点,这些数值点所任意组合的范围都应被视为本发明已公开或记载的范围。
64.本发明中的技术和科学术语,给出定义的以其定义为准,未给出定义的则按本领域的通常含义理解。
65.本发明中的“掺杂元素”是指氮、磷、硼、硫、氟、氯、溴和碘。
66.本发明中,除了根据上下文或自身限定可以唯一确定为“含掺杂元素的碳材料”,其他提及的“碳材料”均指不含掺杂元素的碳材料;碳材料的下位概念也如此。
67.本发明中,“炭黑”与“碳黑”为可相互替换的技术术语。
68.本发明中的“惰性气体”是指,在本发明的制备方法中,对硫、氮掺杂导电炭黑的性能不造成任何可察觉影响的气体。
69.本发明中,除了根据上下文或自身限定可以明确的之外,其他提及的“孔体积”均指p/p0最大时的单点吸附总孔容。
70.本发明提供了一种铂碳催化剂,包括碳载体和负载于其上的铂金属,所述的碳载体为硫氮掺杂导电炭黑。
71.根据本发明的铂碳催化剂,其不含除硫和氮外的其他掺杂元素。
72.根据本发明的铂碳催化剂,其不含除铂外的其他金属元素。
73.根据本发明的铂碳催化剂,所述硫和氮以化学键的方式与导电炭黑结合。
74.根据本发明的铂碳催化剂,在其xps分析的s
2p
谱峰中,在162ev~166ev之间,只有噻吩型硫的特征峰,且在168
±
1ev处有特征峰。除此之外,在其xps分析的s
2p
谱峰中,在160ev~170ev之间没有其他的特征峰。
75.根据本发明的铂碳催化剂,所述噻吩型硫的特征峰为双峰,分别位于163.4
±
0.5ev和164.6
±
0.5ev。
76.根据本发明的铂碳催化剂,一些实施例中,在其xps分析的s
2p
谱峰中,所述噻吩型硫的特征峰与168
±
1ev处的特征峰的峰面积比为1:5~10:1。
77.根据本发明的铂碳催化剂,一些实施例中,在其xps分析的s
2p
谱峰中,所述噻吩型硫的特征峰与168
±
1ev处的特征峰的峰面积比大于5。一些实施例中该峰面积比大于10。
78.根据本发明的铂碳催化剂,在其xps分析的n
1s
谱峰中,除398.5ev~400.5ev之间有特征峰外,在390ev~410ev之间没有其他的特征峰。
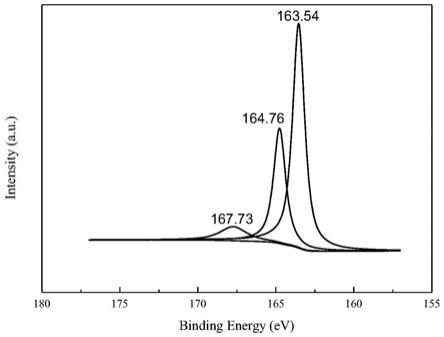
79.根据本发明的铂碳催化剂,在其xps分析的n
1s
谱峰中,398.5ev~400.5ev之间有一个特征峰。
80.根据本发明的铂碳催化剂,在其xps分析的n
1s
谱峰中,在399.1ev
±
0.5ev处有一个特征峰。
81.根据本发明的铂碳催化剂,以催化剂的质量为基准,铂的质量分数为0.1%~80%,优选为20%~70%,更优选为40%~70%。
82.根据本发明的铂碳催化剂,其电阻率《10.0ω
·
m,优选《2.0ω
·
m。
83.根据本发明的铂碳催化剂,所述铂碳催化剂的比表面可以为80m2/g~1500m2/g。
84.根据本发明的铂碳催化剂,所述导电炭黑可为ketjen black(科琴黑)系列超导碳黑、cabot系列导电碳黑和赢创德固赛公司生产的系列导电碳黑中的一种或几种;优选为ketjen black ec-300j、ketjen blackec-600jd、ketjen blackecp-600jd、vxc72、black pearls 2000、printex xe2-b、printex l6或hiblaxk 40b2。
85.一种铂碳催化剂的制备方法,包括:
86.(1)制造硫氮掺杂导电炭黑的步骤;和
87.(2)以步骤(1)中所得硫氮掺杂导电炭黑为载体,负载铂的步骤;
88.其中的步骤(1)包括掺杂硫的操作和掺杂氮的操作;
89.所述掺杂硫的操作包括:将导电炭黑与单质硫混合,在惰性气体中于400℃~1500℃处理(优选恒温处理)0.5h~10h;
90.所述的掺杂氮的操作在所述掺杂硫的操作之前、之后或同时进行。
91.根据本发明铂碳催化剂的制备方法,所述掺杂氮的操作在所述掺杂硫的操作之前或之后进行时,其可以采用任何现有已知的掺杂氮的方法。一种实施方式是,所述掺杂氮的操作在所述掺杂硫的操作之前进行时,将碳材料与氮源混合,在惰性气体中于300℃~1500℃处理(优选恒温处理)0.5h~10h。另一种实施方式是,所述掺杂氮的操作在所述掺杂硫的
操作之后进行时,将硫掺杂碳材料与氮源混合,在惰性气体中于300℃~1500℃处理(优选恒温处理)0.5h~10h。
92.根据本发明铂碳催化剂的制备方法,所述掺杂氮的操作在所述掺杂硫的同时进行时,采用所述掺杂硫的操作条件。一种实施方式是,先将碳材料与氮源和硫源预先混合,然后在所述掺杂硫的操作条件下,同时对碳材料进行掺杂氮和掺杂硫的操作。
93.根据本发明铂碳催化剂的制备方法,所述导电炭黑与硫的质量比为20:1~2:1;优选为10:1~4:1,更优选为8:1~4:1。
94.根据本发明铂碳催化剂的制备方法,(1)中所述硫氮掺杂导电炭黑,在其xps分析的s
2p
谱峰中,在162ev~166ev之间,只有噻吩型硫的特征峰,且在168
±
1ev处有特征峰。
95.根据本发明铂碳催化剂的制备方法,(1)中所述硫氮掺杂导电炭黑,在其xps分析的s
2p
谱峰中,所述噻吩型硫的特征峰为双峰,分别为于163.5
±
0.5ev和164.8
±
0.5ev。
96.根据本发明铂碳催化剂的制备方法,(1)中所述硫氮掺杂导电炭黑,在其xps分析的n
1s
谱峰中,在399.6
±
0.5ev处有一个特征峰。
97.根据本发明铂碳催化剂的制备方法,所述掺杂硫的操作中,温度优选为1100℃~1450℃。
98.根据本发明铂碳催化剂的制备方法,所述掺杂硫的操作和所述掺杂氮的操作中,所述处理(优选恒温处理)的时间各自独立为1h~5h,优选为2h~4h。
99.根据本发明铂碳催化剂的制备方法,如需升温,所述掺杂硫的操作和所述掺杂氮的操作中,升温速率各自独立为3℃/min~15℃/min,优选为6℃/min~15℃/min。
100.根据本发明铂碳催化剂的制备方法,所述导电炭黑可以是普通导电炭黑(conductive blacks)、超导电炭黑(super conductive blacks)或特导电炭黑(extra conductive blacks),比如所述导电炭黑可以为ketjen black(科琴黑)系列超导碳黑、cabot系列导电碳黑和赢创德固赛公司生产的系列导电炭黑中的一种或几种;优选为ec-300j、ec-600jd、ecp-600jd、vxc72、black pearls 2000、printex xe2-b、printex l6或hiblaxk 40b2。
101.根据本发明铂碳催化剂的制备方法,对导电炭黑的制法、来源没有限制。所述导电炭黑可以为乙炔黑、炉法炭黑等。
102.根据本发明铂碳催化剂的制备方法,所述导电炭黑的电阻率可以《10.0ω
·
m,优选为《5.0ω
·
m,更优选《2.0ω
·
m。
103.根据本发明铂碳催化剂的制备方法,所述导电炭黑的id/ig值一般为0.8~5,优选为1~4。在拉曼光谱中,位于1320cm-1
附近的峰为d峰,位于1580cm-1
附近的峰为g峰,id代表d峰的强度,ig代表g峰的强度。
104.根据本发明铂碳催化剂的制备方法,所述导电炭黑的比表面积和孔体积可以在较大范围内变化。比表面积可以为10m2/g~2000m2/g,孔体积可以为0.02ml/g~6ml/g。
105.根据本发明铂碳催化剂的制备方法,一种实施方式中的导电炭黑,其比表面积为200m2/g~2000m2/g。
106.根据本发明铂碳催化剂的制备方法,一种实施方式的掺杂硫的操作中,将导电炭黑与硫粉均匀混合,置于管式炉中,以4℃/min~15℃/min(优选6℃/min~15℃/min)的速率将管式炉升温至400℃~1500℃(优选1150℃~1450℃)处理(优选恒温处理)0.5h~10h。
107.根据本发明铂碳催化剂的制备方法,所述导电炭黑的xps分析中,氧质量分数大于2%,可以为2%~15%,优选为2.5%~12%。
108.根据本发明铂碳催化剂的制备方法,所述负载铂的步骤包括:
109.(a)将(1)中得到的硫氮掺杂导电炭黑与铂前驱体分散在水相中,调节ph为8~12(优选调节ph值为10
±
0.5);
110.(b)加入还原剂进行还原;
111.(c)分离出固体,经后处理得到所述的氢燃料电池铂碳催化剂。
112.根据本发明铂碳催化剂的制备方法,所述铂前驱体为氯铂酸、氯铂酸钾或氯铂酸钠;所述铂前驱体的浓度为0.5mol/l~5mol/l。
113.根据本发明铂碳催化剂的制备方法,(a)中,用碳酸钠水溶液、碳酸钾水溶液、氢氧化钾水溶液、氢氧化钠水溶液或氨水调节水相的ph值。
114.根据本发明铂碳催化剂的制备方法,(b)中,所述还原剂为柠檬酸、抗坏血酸、甲醛、甲酸、乙二醇、柠檬酸钠、水合肼、硼氢化钠或丙三醇中的一种或几种。
115.根据本发明铂碳催化剂的制备方法,(b)中,所述还原剂与铂的摩尔比为2~100。
116.根据本发明铂碳催化剂的制备方法,(b)中,还原温度为50℃~150℃,优选为60℃~90℃;还原时间为4h~15h,优选为8h~12h。
117.根据本发明铂碳催化剂的制备方法,所述的后处理包括:洗涤、过滤和干燥。
118.根据本发明铂碳催化剂的制备方法,(1)中制得的硫氮掺杂导电炭黑可以容易的分散于水相。而对于某些导电炭黑,比如科琴黑,很难直接分散于水相。
119.一种铂碳催化剂,该催化剂由前述任一的方法制得。
120.一种氢燃料电池,该氢燃料电池的阳极和/或阴极使用了前述任一的铂碳催化剂。
121.一种碳材料,该碳材料为硫氮掺杂的导电炭黑。
122.根据本发明的碳材料,其中硫和氮以化学键的方式与导电炭黑结合。
123.根据本发明的碳材料,在其xps分析的s
2p
谱峰中,在162ev~166ev之间,只有噻吩型硫氮的特征峰,且在168
±
1ev处有特征峰。
124.根据本发明的碳材料,所述噻吩型硫的特征峰为双峰,分别位于163.5
±
0.5ev和164.8
±
0.5ev。
125.根据本发明的碳材料,一些实施例中,在其xps分析的s
2p
谱峰中,所述噻吩型硫的特征峰与168
±
1ev处的特征峰的峰面积比为1:5~10:1。
126.根据本发明的碳材料,一些实施例中,在其xps分析的s
2p
谱峰中,所述噻吩型硫的特征峰与168
±
1ev处的特征峰的峰面积比大于10。
127.根据本发明的碳材料,在其xps分析的n
1s
谱峰中,除398.5ev~400.5ev之间有特征峰外,在390ev~410ev之间没有其他的特征峰。
128.根据本发明的碳材料,在其xps分析的n
1s
谱峰中,在399.6ev
±
0.5ev之间有一个特征峰。
129.下面结合具体实施例对本发明进行详细说明。以下实施例将有助于本领域的技术人员进一步理解本发明,但不以任何形式限制本发明。
130.试剂、仪器与测试
131.如无特殊说明,本发明所采用试剂均为分析纯,所用试剂均为市售可得。
132.本发明中的分析测试均采用以下仪器、方法进行。
133.本发明通过x射线光电子能谱分析仪(xps)检测材料表面的元素。所采用x射线光电子能谱分析仪为vg scientifc公司生产配备有avantage v5.926软件的escalab220i-xl型射线电子能谱仪,x射线光电子能谱分析测试条件为:激发源为单色化a1kαx射线,功率为330w,分析测试时基础真空为3
×
10-9
mbar。另外,电子结合能用单质碳的c1s峰(284.3ev)校正,后期分峰处理软件为xpspeak。谱图中噻吩硫的特征峰为分峰后的特征峰。
134.元素分析的仪器和方法、条件:元素分析仪(vario el cube),反应温度1150℃,称取样品5mg,还原温度850℃,载气氦气流速200ml/min,氧气流速30ml/min,通氧时间70s。
135.测试铂碳催化剂中铂质量分数的仪器、方法、条件:取30mg制备好的pt/c催化剂,加入30ml王水,120℃冷凝回流12h,冷却至室温后,取上清液稀释后,用icp-aes测试其中pt含量。
136.本发明所采用高分辨透射电镜(hrtem)的型号为jem-2100(hrtem)(日本电子株式会社),高分辨透射电镜测试条件为:加速电压为200kv。样品中纳米颗粒的粒径通过电镜图片测量得到。
137.bet测试方法:本发明中,样品的孔结构性质由quantachrome as-6b型分析仪测定,催化剂的比表面积和孔体积由brunauer-emmett-taller(bet)方法得到,孔分布曲线根据barrett-joyner-halenda(bjh)方法对脱附曲线进行计算得到。
138.本发明的拉曼检测采用的是日本horiba公司生产的labram hr uv-nir型激光共聚焦拉曼光谱仪,激光波长为532nm。
139.电化学性能测试,仪器型号solartron analytical energylab和princeton applied research(model 636a),方法及测试条件:催化剂的极化曲线lsv在1600rpm的转速下,o2饱和的0.1m hclo4中测试,cv曲线在ar气氛下0.1m hclo4中测试,以此计算电化学活性面积ecsa。稳定性测试时在o2饱和的0.1m hclo4中,0.6v~0.95v范围内扫描5000个循环后,按上述方法测试lsv和ecsa。以上测试时将催化剂配成均匀分散的浆液,涂于直径5mm的玻碳电极上,电极上催化剂的铂含量为3μg~4μg。
140.电阻率测试四探针电阻率测试仪,仪器型号kdy-1,方法及测试条件:施加压力3.9
±
0.03mpa,电流为500
±
0.1ma。
141.vxc72(vulcan xc72,美国卡博特公司生产)购自苏州翼隆晟能源科技有限公司。采用前述的仪器方法测试,结果表明:比表面积258m2/g,孔体积0.388ml/g,氧质量分数8.72%,id/ig为1.02,电阻率为1.22ω
·
m。
142.ketjenblack ecp600jd(科琴黑,日本lion公司生产)购自苏州翼隆晟能源科技有限公司。采用前述的仪器方法测试,结果表明:比表面积1362m2/g,孔体积2.29ml/g,氧质量分数6.9%,id/ig为1.25,电阻率为1.31ω
·
m。
143.商业铂碳催化剂(牌号hispec4000,johnson matthey公司生产)购自alfa aesar。测试结果表明:铂的质量分数为40.2%。
144.实施例1
145.本实施例用于说明本发明硫氮掺杂碳载体的制备。
146.向1g ketjenblack ecp600jd中加入10ml无水乙醇,后加入20ml 20wt%的氨水溶液浸渍24h,于100℃烘箱中烘干后与0.25g单质硫混合均匀,置于管式炉内,以6℃/min的速
率将管式炉升温至1200℃,然后恒温处理3h,自然降温后得到硫氮掺杂导电炭黑,编号为碳载体a。
147.样品表征及测试
148.xps分析的硫质量分数为0.86%;xps分析的氮质量分数为1.32%;比表面积为1395m2/g,电阻率1.38ω
·
m。
149.图1为实施例1的硫氮掺杂碳载体的硫的xps谱图。
150.图1中,噻吩型硫的特征峰与168
±
1ev处的特征峰的峰面积比为11。
151.图2为实施例1的硫氮掺杂碳载体的氮的xps谱图。
152.实施例2
153.本实施例用于说明本发明硫氮掺杂碳载体的制备。
154.将1g vulcan xc72加入15ml 1wt%的尿素溶液浸渍24h,于100℃烘箱中烘干后与0.167g单质硫混合均匀,置于管式炉内,以3℃/min的速率将管式炉升温至1000℃,然后恒温处理3h,自然降温后得到硫氮掺杂导电炭黑,编号为碳载体b。
155.样品表征及测试
156.xps分析的硫质量分数为1.2%;xps分析的氮质量分数为0.6%;xps分析的氧质量分数为5.8%;比表面积为256m2/g;电阻率为1.28ω
·
m。
157.实施例3
158.本实施例用于说明本发明硫氮掺杂碳载体的制备。
159.将1g vulcan xc72加入20ml 10wt%的氨水溶液浸渍24h,于100℃烘箱中烘干后与0.2g单质硫混合均匀,置于管式炉内,以6℃/min的速率将管式炉升温至1300℃,然后恒温处理2h,自然降温后得到硫氮掺杂导电炭黑,编号为碳载体c。
160.样品表征及测试
161.xps分析的硫质量分数为0.89%;氮的质量分数为0.51%;xps分析的氧质量分数为4.9%;比表面积为241m2/g;电阻率为1.26ω
·
m。
162.实施例4
163.本实施例用于说明本发明硫氮掺杂碳载体的制备。
164.将1g vulcan xc72加入20ml 6wt%的氨水溶液浸渍24h,于100℃烘箱中烘干后置于管式炉内,以8℃/min的速率将管式炉升温至1100℃,自然降温后得到氮掺杂导电炭黑,将其与0.143g单质硫混合均匀,置于管式炉内,以8℃/min的速率将管式炉升温至1300℃,然后恒温处理3h,自然降温后得到硫氮掺杂导电炭黑,编号为碳载体d。
165.样品表征及测试
166.xps分析的硫质量分数为1.04%,氮质量分数为0.13%;xps分析的氧质量分数为6.4%;比表面积为220m2/g;电阻率为1.26ω
·
m。
167.实施例5
168.本实施例用于说明本发明铂碳催化剂的制备。
169.按每克碳载体使用250ml水的比例,将碳载体a分散于去离子水中,按每克碳载体加入12mmol氯铂酸,超声分散形成悬浮液,加入1mol/l氢氧化钾水溶液调节体系的ph值为10;将上述悬浮液加热至80℃,搅拌下加入硼氢化钠进行还原反应,还原剂与铂前驱体的摩尔比为5:1,维持反应12h;将反应后的混合物过滤,洗涤至溶液ph为中性,100℃烘干后得到
铂碳催化剂。
170.样品表征及测试
171.铂碳催化剂的铂质量分数为69.7%。
172.图3为实施例5的铂碳催化剂的硫的xps谱图。
173.图3中,噻吩型硫的特征峰与168
±
1ev处的特征峰的峰面积比为10.1。
174.图4为实施例5的铂碳催化剂的氮的xps谱图。
175.铂碳催化剂性能测试结果见表1。
176.实施例6
177.本实施例用于说明本发明铂碳催化剂的制备。
178.按每克碳载体使用250ml水的比例,将碳载体b分散于去离子水中,按每克碳载体加入1.3mmol氯铂酸,超声分散形成悬浮液,加入1mol/l碳酸钠水溶液使体系的ph值为10;将上述悬浮液加热至80℃,搅拌下加入甲酸进行还原反应,甲酸与氯铂酸的摩尔比为50:1,继续维持反应10h;将反应后的混合物过滤,用去离子水洗涤至滤液的ph值为中性,过滤,然后在100℃烘干,得到铂碳催化剂。
179.样品表征及测试
180.铂碳催化剂的铂质量分数为20.2%。
181.图5为实施例6的铂碳催化剂的硫的xps谱图。
182.图5中,噻吩型硫的特征峰与168
±
1ev处的特征峰的峰面积比为5。
183.铂碳催化剂性能测试结果见表1。
184.实施例7
185.本实施例用于说明本发明铂碳催化剂的制备。
186.按每克碳载体使用250ml水的比例,将碳载体c分散于去离子水中,按每克碳载体加入3.4mmol氯铂酸,超声分散形成悬浮液,加入1mol/l碳酸钠水溶液使体系的ph值为10;将上述悬浮液加热至80℃,搅拌下加入甲酸进行还原反应,甲酸与氯铂酸的摩尔比为50:1,继续维持反应10h;将反应后的混合物过滤,用去离子水洗涤至滤液的ph值为中性,过滤,然后在100℃烘干,得到铂碳催化剂。
187.样品表征及测试
188.铂碳催化剂的铂质量分数为40.2%。
189.图6为实施例7的铂碳催化剂的硫的xps谱图。
190.图6中,噻吩型硫的特征峰与168
±
1ev处的特征峰的峰面积比为13。
191.图7为实施例7的铂碳催化剂的氮的xps谱图。
192.铂碳催化剂性能测试结果见表1。
193.实施例8
194.本实施例用于说明本发明铂碳催化剂的制备。
195.按照实施例7的方法制备铂碳催化剂,区别仅在于:使用实施例4制备的碳载体d。
196.样品表征及测试
197.铂碳催化剂的铂质量分数为40.1%。
198.铂碳催化剂性能测试结果见表1。
199.对比例1
200.按每克碳载体用200ml水和50ml乙醇的比例,将ketjenblack ecp600jd分散于去离子水中,按每克碳载体加入12mmol氯铂酸,超声分散形成悬浮液,加入1mol/l氢氧化钾水溶液使体系ph为10;将上述悬浮液加热至80℃,搅拌下加入硼氢化钠进行还原反应,还原剂与铂前驱体的摩尔比为5:1,维持反应12h;将反应后的混合物过滤,洗涤至溶液ph为中性,100℃烘干后得到铂碳催化剂。
201.样品表征及测试
202.铂碳催化剂的铂质量分数为69.7%。
203.铂碳催化剂性能测试结果见表1。
204.对比例2
205.按每克碳载体使用250ml水的比例,将vulcan xc72分散于去离子水中,按每克碳载体加入3.4mmol氯铂酸,超声分散形成悬浮液,加入1mol/l碳酸钠水溶液使体系的ph值为10;将上述悬浮液加热至80℃,搅拌下加入甲酸进行还原反应,甲酸与氯铂酸的摩尔比为50:1,继续维持反应10h;将反应后的混合物过滤,用去离子水洗涤至滤液的ph值为中性,过滤,然后在100℃烘干,得到铂碳催化剂。
206.样品表征及测试
207.铂碳催化剂的铂质量分数为40.1%。
208.铂碳催化剂性能测试结果见表1。
209.对比例3
210.铂碳催化剂为购买的商业催化剂,牌号hispec4000。
211.样品表征及测试
212.铂碳催化剂的铂质量分数为40.2%。
213.铂碳催化剂性能测试结果见表1。
214.对比例4
215.向1g ketjenblack ecp600jd中加入10ml无水乙醇,然后加入25ml 10wt%的氨水溶液浸渍24h;在100℃下于烘箱中烘干;然后放入管式炉内,以8℃/min的速率将管式炉升温至1100℃,恒温处理3h;自然降温后得到氮掺杂碳载体。
216.按每克碳载体使用250ml水的比例,将前述氮掺杂碳载体分散于去离子水中,按每克碳载体加入12mmol氯铂酸,超声分散形成悬浮液,加入1mol/l氢氧化钾水溶液调节体系的ph值为10;将上述悬浮液加热至80℃,搅拌下加入硼氢化钠进行还原反应,还原剂与铂前驱体的摩尔比为5:1,维持反应12h;将反应后的混合物过滤,洗涤至溶液ph为中性,100℃烘干后得到铂碳催化剂。
217.样品表征及测试
218.碳载体的xps分析的氮质量分数为1.48%。
219.铂碳催化剂的铂质量分数为70.0%。
220.铂碳催化剂性能测试结果见表1。
221.对比例5
222.将ketjenblack ecp600jd与单质硫混合均匀,二者的质量比为4:1,置于管式炉内,以5℃/min的速率将管式炉升温至1000℃,然后恒温处理2h,自然降温后得到硫掺杂碳载体。
223.按每克碳载体使用250ml水的比例,将前述硫掺杂碳载体分散于去离子水中,按每克碳载体加入12mmol氯铂酸,超声分散形成悬浮液,加入1mol/l氢氧化钾水溶液调节体系的ph值为10;将上述悬浮液加热至80℃,搅拌下加入硼氢化钠进行还原反应,还原剂与铂前驱体的摩尔比为5:1,维持反应12h;将反应后的混合物过滤,洗涤至溶液ph为中性,100℃烘干后得到铂碳催化剂。
224.样品表征及测试
225.碳载体的xps分析的硫质量分数为0.72%。
226.铂碳催化剂的铂质量分数为70.1%。
227.图8为对比例5铂碳催化剂的硫的xps谱图。
228.图8中,噻吩型硫的特征峰与168
±
1ev处的特征峰的峰面积比为1.49。
229.铂碳催化剂性能测试结果见表1。
230.表1
231.
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1