使用α-羟基羰基化合物作为还原剂的制作方法
技术领域
本发明涉及某些化合物作为还原剂的应用。特别是,本发明涉及该化合物在还原还原活化的(reduction-activated)前药中的应用,所述前药生成用于抑制疾病的活性物质。所述活性物质特别是DNA交联剂,其可用于抑制不良细胞的生长或增生。
背景技术:
在本说明书中已发表文献的清单和讨论不应当将其确认为本技术领域状况的一部分或共知常识。
α-羟基羰基化合物(尽管其分子能够被氧化)在低碱性(pH<11)环境下并非有效的还原剂。
在US5,831,097和EP0364752中说明的某些α-羟基羰基化合物(偶姻)在染料工业中是有效的还原剂。然而,EP0364752中说明的条件要求pH最小为13以实现还原效果。同样地,US4,950,306公开某些化合物(包括α-羟基羰基化合物)能够在染料工艺中作为有效的还原剂,但条件是需要向反应介质加入足量的碱使pH至少为11。
而且,尽管其它文件(例如US3,208,999和Textil-Praxis20(11),916-20(1965))提及某些α-羟基羰基化合物(例如单羟基丙酮和二羟基丙酮)有效作为某些化合物(氰乙化淀粉(cyanoethylated starches)和瓮染料(vat dye))的还原剂的使用,这些文件提及反应条件是高碱性的(即需要使用大量的氢氧化铵或氢氧化钠的浓溶液,从而使反应的pH值超过13)。
另外,上述文件没有公开(涉及使用α-羟基羰基化合物的还原作用)含有超过10wt%有机溶剂的溶剂体系的使用。
技术实现要素:
本发明人意外地发现某些α-羟基羰基化合物在低pH值下具有有效的还原能力,因此能够在温和环境(和/或在大量非水性(有机)溶剂的存在下)下使用,以还原各种结构(包括多种有机化合物)。
因此,本发明的第一方面是提供式I的化合物作为用于还原有机化合物中可还原基团的试剂的应用,所述有机化合物含有一个或多个可还原基团,其中式I的化合物具有如下结构
其中
R1表示H、芳基、Het或C1-12烷基,所述C1-12烷基任选被一个或多个取代基取代,所述取代基选自OH、卤素和C1-3烷氧基,
R2表示H或C1-6烷基,所述C1-6烷基任选被一个或多个OH基取代,
芳基表示C6-10碳环芳基,所述C6-10碳环芳基可被一个或多个选自卤素、C1-6烷基和C1-6烷氧基的取代基取代,
Het表示4元至14元杂环基,其含有一个或多个选自氧、氮和/或硫的杂原子,所述杂环基可含有一个、两个或三个环,其可以被一个或多个选自卤素、C1-6烷基和C1-6烷氧基的取代基取代,
所述式I的化合物的特征在于其能够形成下述式Ia的环二聚体:
其中R1和R2如上述所定义。
除非另有说明,本文定义的烷基和烷氧基可以是直链,或当具有足够数目(即至少为3)的碳原子,可以是支链和/或环状的。而且,当具有足够数目(即至少为4)的碳原子,该烷基和烷氧基也可以是部分环化的/无环的(part cyclic/acyclic)。所述烷基和烷氧基还可以是饱和的或不饱和的和/或当具有足够数目(即至少为2)的碳原子时,其中间插入一个或多个氧和/或硫原子。除非另有说明,烷基和烷氧基还可以被一个或多个卤素尤其是氟原子取代。
芳基的实例包括苯基和萘基等。
杂环(Het)可以是完全饱和的,部分不饱和的,完全芳香的或部分芳香的。杂环(Het)基的实例包括1-氮杂双环[2.2.2]辛基、苯并咪唑基、苯并[c]异噁唑烷基、苯并异噁唑基、苯并二噁烷基、苯并二氧杂环庚烷基、苯并二氧杂环戊烷基、苯并呋喃基、苯并呋咱基、苯并吗啉基、2,1,3-苯并噁唑二唑基、苯并噁唑烷基、苯并噁唑基、苯并吡唑基、苯并[e]嘧啶、2,1,3-苯并噻二唑基、苯并噻唑基、苯并噻吩基、苯并三唑基、苯并二氢吡喃基、苯并吡喃基、噌啉基、2,3-二氢苯并咪唑基、2,3-二氢苯并[b]呋喃基、1,3-二氢苯并-[c]呋喃基、1,3-二氢-2,1-苯并异噁唑基、2,3-二氢吡咯[2,3-b]吡啶基、二噁基、呋喃基、六氢嘧啶基、乙内酰脲基、咪唑基、咪唑[1,2-a]吡啶基、咪唑[2,3-b]噻唑基、吲哚基、异喹啉基、异噁唑啉基、异噁唑基、马来酰亚胺基、吗啉基、萘并[1,2-b]呋喃基、噁二唑基、1,2-噁嗪基或1,3-噁嗪基、噁唑基、酞嗪基、哌嗪基、哌啶基、嘌呤基、吡喃基、吡嗪基、吡唑基、吡啶基、嘧啶基、吡咯烷酮基、吡咯烷基、吡咯啉基、吡咯并[2,3-b]吡啶基、吡咯并[5,1-b]吡啶基、吡咯并[2,3-c]吡啶基、吡咯基、喹唑啉基、喹啉基、环丁砜基、3-环丁烯砜基、4,5,6,7-四氢苯并咪唑基、4,5,6,7-四氢苯并吡唑基、5,6,7,8-四氢苯并[e]嘧啶、四氢呋喃、四氢吡喃基、3,4,5,6-四氢吡啶基、1,2,3,4-四氢嘧啶基、3,4,5,6-四氢嘧啶基、噻二唑基、噻唑烷基、噻唑基、噻吩基、噻吩并[5,1-c]吡啶基、硫代苯并二氢吡喃基、三唑基、1,3,4-三唑并[2,3-b]嘧啶基和吨基(xanthenyl)等。
杂环(Het)基上的取代基可以是(视乎情况)位于包括杂原子的环系统中的任何原子上。杂环(Het)基的连接点可以是包括(视乎情况)杂原子的环系统中的任何原子或作为环系统一部分的任何稠碳环上的原子。
本文所用的术语“卤素”包括氟、氯、溴和碘。
式I的化合物可以(对于本发明的所有方面)包括下述化合物,其中:
(1)R1不是H;
(2)R1表示H或特别是被一个或多个OH基取代的C1-2烷基(例如CH2OH);
(3)R2表示被一个或多个OH基取代的C1-4烷基(例如CH2OH或CH(OH)CH2OH)或特别是H。
而且,具体的式I的化合物(对于本发明的所有方面)包括二羟基丙酮(DHA,另称作1,3-二羟基-2-丙酮)、乙醇醛、甘油醛、赤藓糖、木酮糖和它们的二聚体。其它具体的式I的化合物包括赤藓酮糖和3-羟基-2-丁酮。在本发明的一个具体实施方式中(其所有方面),式I的化合物是DHA或其二聚体。
因为式I的化合物可以二聚体形式或单体形式存在,为了免除疑惑,本文参照的式I化合物包括上述两种形式(除非本文另有所指)。而且,式I化合物以式Ia的环二聚体形式存在的能力是这些化合物的结构内在的性质。因此,本文中一般或具体定义的式I化合物(全部为能够形成式Ia的环状二聚体的化合物)包括(一般或具体定义的)上述化合物本身(即其没有上述涉及形成环二聚体的特征表现)。式I的化合物是否能够形成Ia的环二聚体例如可以通过研究分离的化合物形成所述二聚体的固体形式或溶液形式(例如水性溶液或有机溶液)的倾向而决定。
本发明的另一方面提供上述定义的式I的化合物作为还原可还原无机化合物的试剂的应用。
在本发明这方面的一个实施方式中,无机化合物不是碘或不含有铜(II)离子或铁氰化物。
无机化合物,在具体的实施方式中,可包含镧系元素(如铈)或特别是过渡金属(即族IIIA到族IIB的金属)。无机化合物可包括所述镧系元素或过渡金属的配位络合物。具体的过渡金属包括在钴和镍族中的过渡金属(即在中性状态下具有d9或d10电子构型的过渡金属),例如钴或铂。金属(配位络合物等中)可以在高于该金属稳定的氧化态的氧化态。例如,对于钴,III氧化态可以被还原成II氧化态;对于铂,IV氧化态可以被还原成II氧化态。在本发明这方面的某些实施方式中,过渡金属是铁或铜以外的过渡金属。
在本发明的第一方面的一个具体实施方式中,式I的化合物作为用于还原有机基团中的可还原基团的试剂的应用包括该化合物作为酶以外的化合物的还原剂使用。
在本发明的第一方面的另一具体实施方式中,可还原化合物在非水性的溶剂体系中进行还原。应用于本文时,术语“非水性的溶剂体系”包括氯化烃(如二氯甲烷)、烃(如己烷)、芳香烃(如甲苯或二甲苯)、偶极非质子溶剂(如N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲基亚砜或N-甲基吡咯烷酮)、乙腈、酯(如乙酸乙酯)或特别是低级(C1-4)烷基醇(如异丙醇、乙醇或甲醇)。术语“非水性的溶剂体系”还包括这些溶剂的混合物。
发现添加碱可促使式I的化合物的还原。因此,本发明的第一方面的一个具体实施方式涉及上述定义的式I的化合物和碱用于还原有机化合物中的可还原基团的应用,所述有机化合物包括一个或多个可还原基团。
在该实施方式中,碱可以是有机的或无机的。例如,碱可以是胺(伯胺、仲胺、或特别是叔胺,如三乙胺、三甲胺或二乙基异丙胺)、氮基杂环(如N-甲基吗啉或吡啶)、醇盐(如碱金属醇盐,如乙醇钠)、氢氧化物盐(如氢氧化铵或碱金属氢氧化物,如氢氧化钠或氢氧化钾)或特别是碳酸盐或碳酸氢盐(如碱土金属碳酸盐或特别是碱土金属碳酸氢盐)。
碱的使用量可根据以下因素变化,例如,具体选用的式I化合物、待还原的化合物的性质、所需的反应速度等。然而,在本发明的第一方面的某些具体实施方式中,碱的使用量例如可以是式I化合物的四个当量或以下(如三个、二个或一个),例如催化量(如0.1当量或以下)的碱。N,N-二甲基甲酰胺。另选地,碱可以为反应混合物提供一定的pH。因此,在本发明的第一方面的其它实施方式中,用作还原剂的式I的化合物在pH7~11(如pH7.1(如7.2、7.3、7.4或7.5)~10.9(如10.8、10.7、10.6、10.5、10.4、10.3、10.2、10.1或10.0))(如水性溶液)下引发还原。
本文使用的术语“水性溶液”意指物质的溶液,其中溶剂体系包含水,和进一步任选包含一种或多种溶剂,如与水混溶的有机溶剂(如低级(如C1-4)烷基醇,如乙醇、异丙醇或特别是甲醇)。而且,本文所指的pH意指例如在室温(如25°C)下通过本领域技术人员已知的方法(如通过电位测量使用工作電極和参比电极)确定的pH值。
式I的化合物也可以在还原可还原化合物的方法中使用。因此,根据本发明的第二方面,提供了还原有机化合物中的可还原基团的方法,所述方法包括将所述化合物与上述定义的式I的化合物接触。
在本发明的第二方面的一个具体实施方式中,所述方法用于还原酶以外的化合物。
在本发明的第二方面的另一实施方式中,所述方法包括在上述定义的非水性的溶剂体系的存在下将可还原化合物与式I的化合物接触。在一个具体实施方式中,还原在所述非水性的溶剂体系中进行。
在本发明的第二方面的另一具体实施方式中,所述方法在碱(如在本发明的第一方正中定义的碱)的存在下进行。碱的使用量例如可以是式I的化合物的一个当量或以下,例如催化量(如0.1当量或以下)的碱。
在本发明的第二方面的另一具体实施方式中,所述方法包括在溶液(如水性溶液)或悬浮液的存在下将可还原化合物与上述定义的式I的化合物接触,所述溶液(如水性溶液)或悬浮液的pH为7~11(如7.1(如7.2、7.3、7.4或7.5)~10.9(如10.8、10.7、10.6、10.5、10.4、10.3、10.2、10.1或10.0))。另选地,所述方法包括在溶液(如水性溶液)或悬浮液(在一定体积的基本为非水性的溶剂体系中)的存在下将可还原化合物与上述定义的式I的化合物接触,所述溶液(如水性溶液)或悬浮液含有一定量的碱,该碱溶解或悬浮在当量体积的水中,使生成7~11(如7.1(如7.2、7.3、7.4或7.5)~10.9(如10.8、10.7、10.6、10.5、10.4、10.3、10.2、10.1或10.0))的pH值。
在本发明的第一和第二方面中,被还原的基团例如可以是硝基、氧代(如酮,诸如醌、羰基)、亚氨基、偶氮基、N-氧化物或吡啶盐基团(pyridinium group)。特别是,例如硝基或吡啶盐基团。
如上所述,意外地发现可以在足量的(或全部为)非水性的(有机)溶剂的存在下使用α-羟基羰基化合物进行还原。
因此,本发明的另一方面涉及使用上述定义的式I的化合物作为还原剂在基本为非水性的溶剂体系中的应用。
本文使用的术语“基本为非水性的溶剂体系”包括含有至多为80wt%(如70wt%、60wt%、50wt%、40wt%、30wt%、20wt%、10wt%、5wt%、1wt%或0.1wt%)的水的溶剂体系。在这些溶剂体系中,其余的溶剂(即至少为20wt%)是有机溶剂。有机溶剂可以是包括上述与术语“非水性的溶剂体系”有关的溶剂。术语“基本为非水性的溶剂体系”还包括完全的非水性的溶剂体系(如全部为有机溶剂的溶剂体系)。
同上,本发明这一方面的一个具体实施方式涉及上述定义的式I的化合物和碱作为还原剂在基本为非水性的溶剂体系中的应用。
在本发明的这方面中,式I的化合物可用于还原有机化合物或无机化合物(如下述的有机化合物)。
本发明进一步提供相应的还原方法,所述方法使用式I的化合物和基本为非水性的溶剂体系。因此,本发明进一步涉及还原可还原化合物的方法,所述方法包括将所述的可还原化合物与上述定义的式I的化合物和基本为非水性的溶剂体系接触。
在本发明的这方面的一个具体的实施方式中,所述方法包括将所述的可还原化合物与上述定义的式I的化合物和碱接触。
而且,在本发明的这方面中,所述的可还原化合物可以是有机化合物或无机化合物(如下述的有机化合物)。
在本发明的这方面中,当可还原的化合物为无机化合物时,无机化合物在具体的实施方式中可以:
(i)不是碘或不含有铜(II)离子或铁氰化物;
(ii)包含镧系元素(如铈)或特别是过渡金属(即IIIA族到IIB族的金属);和/或
(iii)包含所述的镧系元素(如铈)或过渡金属的配位络合物。
具体的过渡金属可以包括在钴和镍族中的过渡金属(即在中性状态下具有d9或d10电子构型的过渡金属),例如钴或铂。金属(配位络合物等中)可以在高于该金属稳定的氧化态的氧化态。例如,对于钴,III氧化态可以被还原成II氧化态;对于铂,IV氧化态可以被还原成II氧化态。在本发明这方面的某些实施方式中,过渡金属是铁或铜以外的过渡金属。
还原的产物可以是生物学活性物质。因此,本发明的第三和第四方面分别提供:
(i)上述定义的式I的化合物作为活化剂用于将还原活化前药转化成相应的活性物质;和
(i)还原活化前药的还原方法,所述方法包括将还原活化药物前与上述定义的式I的化合物接触。
本发明的第三方面的一个具体的实施方式涉及作为活化剂用于将还原活化的前药转化成相应的上述定义的式I的化合物的活性物质和碱的应用。所使用的碱的性质和量以及式I的化合物诱发前药的活化(通过还原)的pH可以如本发明的第一方面所述的应用中所定义的。
相对地,在本发明的第四方面的一个具体的实施方式中,所述方法在碱(如本发明的第一方面中定义的碱)的存在下进行。所用的碱的性质和量可以是如本发明的第一方面所述的应用中定义的碱。而且,在本发明的第四方面的另一具体实施方式中,所述方法包括在溶液(如水性溶液)或悬浮液的存在下将还原活化前药与上述定义的式I的化合物接触,所述溶液(如水性溶液)或悬浮液的pH为7~11(如7.1(如7.2、7.3、7.4或7.5)~10.9(如10.8、10.7、10.6、10.5、10.4、10.3、10.2、10.1或10.0))。另选地,所述方法包括在溶液(如水性溶液)或悬浮液(在一定体积上述定义的基本为非水性的溶剂体系中)的存在下将还原活化前药与上述定义的式I的化合物接触,所述溶液(如水性溶液)或悬浮液含有一定量的碱,该碱溶解或悬浮在当量体积的水中,使生成的7~11(如7.1(如7.2、7.3、7.4或7.5)~10.9(如10.8、10.7、10.6、10.5、10.4、10.3、10.2、10.1或10.0))的pH值。
术语“前药”是本领域技术人员熟知的。然而,为了免除疑惑,本文所用的术语“还原活化前药”包括具有药理学活性的化合物或不具有药理学活性但能够通过涉及还原步骤的工艺转化成具有药理学活性(即“相应的活性物质”),或至少稍微大于“前药”部分的药理学活性的物质的化合物。
本文使用的术语“活化剂”包括通过还原工序将前药转化成相应的生物学活性物质或引发其转化的式I的化合物
这方面(以及本发明所有相关的方面)的还原活化前药包括:
(a)甲硝唑(2-甲基-5-硝基-1H-咪唑-1-乙醇);
(b)氯霉素(2,2-二氯-N-[(αR,βR)-β-羟基-α-羟甲基-4-硝基苯乙基]乙酰胺);
(c)呋喃西林(2-[(5-硝基-2-呋喃基)亚甲基]肼甲酰胺);甲硝唑、氯霉素和呋喃西林在活化后对哺乳动物细胞是细胞毒性的(Bailey等人(1996));
(d)E09(3-[5-氮丙啶基-4,7-二氧代-3-羟甲基-1-甲基-1H-吲哚-2-基]-丙-β-烯-α-醇);
(e)SR-4233(“替拉扎明(tirapazamine)”,3-氨基-1,2,4-苯并三嗪-1,4-二氧化物);
(f)RSU-1069(1-(1-氮丙啶基)-3-(2-硝基-1-咪唑基)-2-丙醇);
(g)RB-6145(1-[3-(2-溴乙基氨基)-2-羟丙基]-2-硝基咪唑);
(h)AQ4N(1,4-二([2-(二甲基氨基-N-氧化物)乙基]氨基)5,8-二羟基-蒽-9,10-二酮);
(i)RB90003X
(j)丝裂霉素C;
(k)Mitosene;
(l)Cyclopropamitosene;
(m)Dynemycin A;
(n)下式化合物
其中
每个RA独立地表示氯、溴、碘或-OS(O)2RC,
RC表示C1-8烷基(任选被一个或多个氟原子取代)或苯基(任选被一个或多个选自卤素、硝基、C1-4烷基和C1-4烷氧基的取代基取代),
RB1~RB4独立地表示H、CN、C(O)N(RD)RE、C(S)N(RD)RE、C(O)OH、S(O)2NHRF,
或RB1还可以表示NO2,
RD和RE独立地表示H或C1-4烷基(其任选被一个或多个选自OH、N(H)-C1-2烷基、N(C1-2烷基)2、4-吗啉基和C(O)OH的取代基取代),
或RD和RE与连接的N-原子一起表示4-吗啉基,和
RF表示H或S(O)2CH3,
当RB1不是H,RB2是H,
例如在1992和1995年Anlezark等人公开的上式化合物中的任何一个SN23163,SN23849,SN23777,SN23428,SN23759,SN24927,SN24928,SN24926,SN25402,SN25079,SN24939,SN24935,SN25923,SN25313,SN23856,SN25066,SN23816,SN25015,SN24971,SN25260,SN25261,SN25263,SN25084,SN25188,SN25507或特别是SN23862(5-{N,N-二[2-氯乙基]胺}-2,4-二硝基苯甲酰胺);
(o)下式化合物
其中
RA如上述所定义(如Cl),和
X1表示NH2,X2和X3两者均表示H,或
-X1-X2-表示-NH-CH2CH2-,X3表示H或
-X1-X3-表示-NH-,X2表示H;
(p)下式化合物
其中Y表示
1-氮丙啶基(任选在2-位被甲基取代),
甲氧基(由此形成米索硝唑化合物)或
N(H)CH2CH2Br(由此形成RB6145化合物);
(q)下式的自杀性前药
其中
R表示-O-R’或-NH-R’,
R’表示
或
其中波浪线表示为片段连接的位置,
RA如上述所定义(如Cl),和
R’’表示以下的肽内酯
其中波浪线表示为片段连接的位置,
例如,在上式的化合物中,R表示:
-NH-R’,或其中R表示-O-R’和R’表示
其中RA如上述所定义(如Cl);
Hu等人(2003);Li等人(2003)和Manger等人(1994)说明的自杀性前药。
(r)下式的硝基二氢吲哚化合物
(s)吖啶-CB1954
(t)采他子卡(tretazicar,5-(氮丙啶-1-基)-2,4-二硝基苯甲酰胺);
(u)抗癌化学疗法或疾病治疗中使用的苯醌、萘醌或蒽醌,其效用依赖于醌基的还原,例如;
下式的25-二(1-氮丙啶基)-14-苯醌
其中每个R独立地表示H或NR’C(O)OR’’,其中R’表示H或C1-4烷基和R’’表示C1-4烷基(如每个R表示NHC(O)OC2H5,由此形成亚胺醌(“AZQ”),或每个R表示H,由此形成2,5-二(1-氮丙啶基)-1,4-苯醌(“DZQ”)),
苯醌芥(benzoquinone mustard)(2-(N,N-二[2-氯乙基]氨基)-1,4-苯醌),
阿霉素或
丝裂霉素;
(v)含有醌型残基的共轭前药(conjugate prodrugs)在还原活化(reductive activation)(如在WO99/61409中说明的,在此通过引用的方式将该文件的全部内容并入本文)中释放细胞毒性剂(cytotoxic agent),例如下式的化合物,
其中Ra1表示甲基、N(C1-2烷基)2或与Ra2一起表示吡咯稠环或呋喃稠环(该环任选被一个或多个选自甲基和羟甲基的取代基取代),
Ra2表示甲基或与Ra1一起表示吡咯稠环或呋喃稠环(该环任选被一个或多个选自甲基和羟甲基的取代基取代),
Ra3表示下次的结构片段
其中波浪线表示为片段连接的位置,
Rb1表示H或甲基,
Rb2表示N(CH2CH2Cl)2(如在3-或4-位)和
Rb3表示甲基(如在4-位)或ORc1(如在3-位,其中Rc1表示C1-6烷基(如n-丁基)或C3-6环烷基甲基(如环丙基甲基或环丁基甲基);
(w)可还原的苯醌、萘醌、蒽醌或吲哚醌作为共轭前药的非细胞毒性平台,其中所述醌用作释放药物的引发组分(trigger component)(如WO97/23456或WO98/35701中公开的,在此通过引用的方式并入上述文件),诸如下式的化合物
其中Rd1表示C1-4烷氧基、氮丙啶-1-基(任选被一个或两个甲基取代),-N(H)CH2C(CH3)2OH或下式的结构片段
其中波浪线表示为片段连接的位置,
Rd5表示H或甲基和RD表示药物部分(drug moiety),
Rd2表示甲基、-C(O)O(C1-4烷基)或-OCH2ORe1,
Re1表示药物部分、H、Re2-C(O)ORe3或-C(O)NH2,
Re2和Re3独立地表示苯基、苄基或环己基,所述的三个基团任选被一个或多个选自卤素、硝基、酰氧基和-SH的取代基取代,
Rd3表示C1-3烷基,其任选被OH(如甲基、异丙基或2-羟基乙基)或C3-6环烷基(如环丙基或环己基)取代,
Rd4表示H、C1-4烷基、N(Re4)Re5或N(Re6)C(O)ORe7,
Re4~Re6独立地表示H或C1-4烷基和
Re7表示C1-4烷基,
或下式的化合物
其中Rf1和Rf2独立地表示H、C1-4烷基或与Rf1和Rf2一起表示苯稠环(该环任选被一个或多个选自甲基和甲氧基的取代基取代),和
Rd1和Rd4如上述定义;和
(w)硝基芳香化合物或硝基杂环化合物,通过药物释放体系中还原活化后的“自烷基化”作为前药引发平台(如在WO00/10611中说明的,在此通过引用的方式并入上述文件的公开内容),如下式的化合物
其中Ar表示芳香环(如苯基、萘基或蒽基)或杂芳环(如吡咯基、咪唑基、(苯并)呋喃基、(苯并)噻吩基、(苯并)噁唑基、(苯并)噻唑基、吡啶基、嘧啶基、吡嗪基、吲哚基或(异)喹啉基),所述的环任选被一个或多个选自C1-4烷基、C1-4烷氧基、OH、卤素、N(Re4)Re5和C(O)ORe6(其中Re4~Re6如上述定义)的取代基取代,
Rg表示下式的结构片段
或
其中波浪线表示为片段连接的位置,且每个Rd5和RD如上定义且各自独立出现,
或者是药学可接受的盐和/或其溶剂化物(如上述(a)~(t)中定义的化合物)。
在本发明的这方面中,实施方式可以包括通过硝基的还原作用将还原活化前药转化成相应的活性物质。
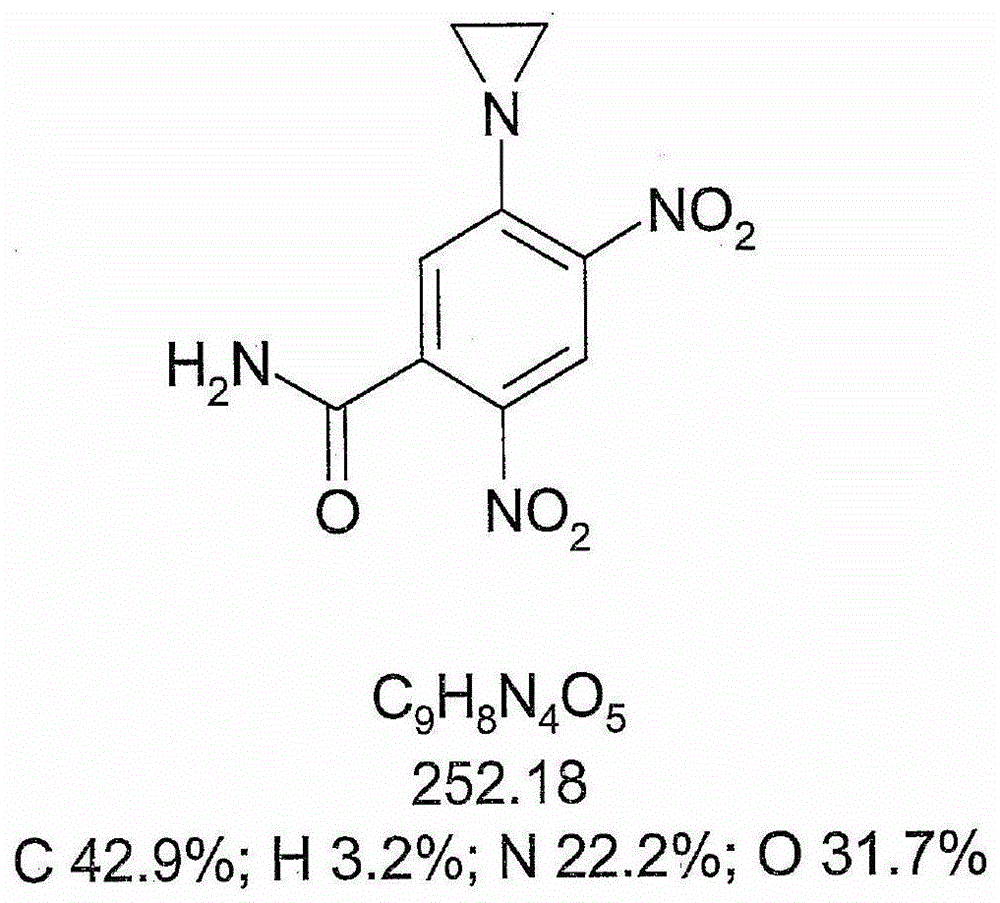
事实上,在一个具体的实施方式中,式I化合物用于活化前药采他子卡(5-(氮丙啶-1-基)-2,4-二硝基苯甲酰胺;也称作CB1954;图1见其结构)。
采他子卡是能够抑制增生疾病的试剂的一个实例。有很多疾病是可以预防的或减少当中的细胞生长或增生。其中的一些疾病如癌症会对生命造成威胁等,即便不对生命造成威胁也会使变得虚弱(如牛皮癣)或起刺激作用和令人感到不舒适(如疣)。抑制这些疾病(尤其是癌症)的一个策略是利用化学试剂,其能够诱发DNA的交联,以及预防或减少细胞生长或增生。采他子卡只有在被还原(于硝基)成相应的羟基胺(其然后进一步如下所述被活化)后实现所述交联。
采他子卡受大众关注超过35年。采他子卡在1960s末首度在一系列有潜力的抗癌化合物的合成中作为当中的一部分被合成,这些抗癌化合物自1950s早期开始一直被研究。合成和测试后,采他子卡似乎代表了癌症化学疗法的焦点,其为一种毒性副作用低的能够治疗肿瘤的细小的低分子化合物。作为抗癌药物,其代表真正表现抗肿瘤选择性的化合物的少数实例中的一种。不幸的是,对于人类癌症的治疗,此抗肿瘤选择性只出现在某些大鼠肿瘤中。采他子卡的抗肿瘤选择性的原理在于其为前药,其在酶促活化后生成的双功能试剂,能够形成DNA-DNA链间交联。大鼠细胞中的采他子卡的生物活化涉及通过酶NQO1(DT-硫辛酰胺脱氢酶)从4-硝基转化成4-羟基胺的需氧还原作用(图2)。人类形式的NQO1代谢采他子卡的效率比大鼠的NQO1低。因此,人类细胞和肿瘤对采他子卡不敏感。
其它内生的人类肿瘤细胞中的采他子卡还原酶及其活性远大于NQO1所导致的(Knox等人,2000)(Wu等人,1997)。然而,此活性是潜在的且只有在二氢烟碱核糖甙(NRH)而不是在NADH或NADPH的存在下是可检测的(Knox等人,2000)。与此活性相关的酶是人类NAD(P)H醌氧化还原酶2(NQO2)(Knox等人,2000)(Wu等人,1997)。在NRH的存在下,NQO2能够催化醌的二电子还原和采他子卡的四原子硝基还原(Wu等人,1997)。NQO2可视作为人类的NRH-依赖型硝基还原酶。
5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺的细胞毒性高,即便对耐受采他子卡的细胞亦然,且能够在它们的DNA中形成链间交联。细胞活化采他子卡时细胞的选择性归因于化合物的形成。与生物活化采他子卡的能力无关,所有细胞类型对于还原的4-羟基氨基衍生物的选择性几乎相同(Boland等人,1991)。尽管5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺能够在细胞中产生DNA-DNA链间交联,其不能在裸DNA中造成损害(Knox等人,1991a)。还有从5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺转化成最接近的DNA交联的细胞毒物质的活化步骤。提出了类似于通过4-硝基喹啉-N-氧化物和N-乙酰氨基芴的代谢作用所形成的羟基胺的酶促酯化和活化(Knox等人,1991a)。事实上,5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺能够通过乙酰基-辅酶A和其它硫酯直接的化学反应非酶促地活化成能够与裸DNA反应生成链间交联的形式,(Knox等人,1991a)(图2)。最终的DNA-反应性采他子卡衍生物可能是4-(N-乙酸基)-5-(氮丙啶-1-基)-2-硝基苯甲酰胺。
采他子卡的生物活化导致其细胞毒性激增,且最终的剂量改变(dose modification)可高达100,000倍。甚至比从单官能试剂转化成双官能试剂预测的还要大。在提供双官能试剂的单官能同源物的情况中(例如具有半芥子和单官能铂化合物),当量毒性(equitoxicity)的剂量改变似乎仅为大约50-200倍(Knox等人,1991b;Knox等人,1987)。然而,观察到的DNA链间交联的形成及特性解释了为什么采他子卡细胞毒性在其活化后会有如此大的增加。
(i)采他子卡诱发的链间交联以高频率形成,且构成了总损害(total lesions)中的70%(Friedlos等人,1992)。此频率远远高于其它试剂所报导的。例如,链间交联占顺铂(Cisplatin)或卡铂(Carboplatin)的总DNA反应的2%或以下(Knox等人,1986)。在摩尔效用方面,与单链双加成物和单官能损害相比,链间交联是本质上毒性更大的损害。预料生成交联比例更高的试剂比仅由低频率生成的试剂具有更大的毒性。
(ii)所述交联难以被修补,这样使它们与通过双官能试剂诱发的试剂相比,本质上具有更大的细胞毒性(Friedlos等人,1992)。
(iii)由于采他子卡的生物活化,与不能还原采他子卡的细胞相比,Walker细胞中DNA-结合药物的量增加了10-倍(Friedlos等人,1992)。
采他子卡诱发的链间交联这种不正常的特性显示其与由其它试剂形成的链间交联不一样。未仍能全面识别由采他子卡诱发的链间交联损害。然而,4-羟基胺(如上所述在活化后)主要与脱氧鸟苷的8-位进行反应。在DNA中,这样会使氮丙啶基团随时准备在反链上反应并形成观察到的交联。分子模拟研究显示此第二臂反应(second arm reaction)将优选在DNA的反链上的脱氧鸟苷的O6位置进行反应(Knox等人,2003;Knox等人,1991a)。该C8-O6DNA链间交联将会是唯一的,因为其不会由其它烷化型或铂酸盐(platination)型试剂生成,并可说明采他子卡的独特的特性。
此特性与生物活化步骤中的选择性一起解释了何故采他子卡在大鼠中作为抗肿瘤试剂异常地有效,并提供了其在G(V)DEPT中作用的基本原理,以及在人类中通过NQO2的活化(Knox等人,2003)(Burke&Knox,1998)。
采他子卡还能够在转基因动物中用于选择性细胞消融。在活性转基因动物中特异的细胞群体的条件性目标消融是用于确定活体内的细胞功能的一个非常有效的方法。目标消融是通过以下工序进行的:并入细菌性硝基还原酶(其能够需氧地活化采他子卡(NTR))(Anlezark等人,1992;Knox等人,1992)和合适的组织特异性启动子构成的转基因,以及将其注入研究中的动物(一般为小鼠)的受精卵中。出生后,确定转基因的染色体整合,以及首建转基因鼠品种(founder mice bred)以建立转基因系。在动物发展过程中的不同阶段用采他子卡进行处理,以评估研究中的细胞群体的特异消融的效果。细胞消融进行得非常快,最早于施用前药后7小时开始,并似乎不依赖官能团p53(Cui等人,1999)。此体系的应用的实例包括转基因小鼠的乳腺的内管腔细胞。对NTR表达动物的处理,结果为快速地和选择性地杀灭这些细胞群体,然而与其密相关的肌上皮细胞则不受影响。在脂肪细胞(Felmer等人,2002)、星形胶质细胞(Cui等人,2001)和神经细胞(Isles等人,2001;Ma等人,2002)中也观察到使用此体系的选择性消融的其它实例。
明显没有对邻近的细胞群体造成影响。已知羟基能够在短距离移位,因此预计其会对邻近的细胞群体造成影响(Bridgewater等人,1997;Friedlos等人,1998)。在以上实验中,基本分别在于目标细胞群体是分裂的,然而,邻近的组织大概并非如此的。采他子卡的活化已知有效对抗非分裂(非周期)细胞(Bridgewater等人,1995)。然而,此影响只能通过使细胞进入分裂而测量。这样,出现的似乎是由采他子卡诱发的DNA损害(即DNA链间交联)在分裂和非分裂细胞群体中都诱发,当细胞试图在其DNA受损的情况下进行分裂,DNA受损的结果只导致对细胞的细胞毒性。相反,S-期特异性试剂,如抗叶酸只对试剂存在下进行分裂的细胞造成影响。
在消融实验中,待消融的组织进行分裂,然而邻接的组织则大概不会,即结果很清楚。DNA损害不是永久性的,且其会慢慢修复(Friedlos等人,1992)。结果是一个窗口(window),其中非分裂组能够修复损害,但进入分裂的任何细胞将会死亡。此窗口的长度将依赖于诱发的初始DNA损害量。在细胞系中,采他子卡诱发的交联用约55小时的半衰期得到修复(Friedlos等人,1992)。该半衰期远长于其它DNA交联剂的半衰期(Friedlos等人,1992),且将其延伸到体内的情况中,给出约3周的窗口(10x半衰期)。
在癌症治疗中,存在一种类似的情况,非增生组织应该对活化的采他子卡有耐受性,除非其在窗口内进入细胞分裂。
类似的论点适于其它双官能烷化剂和铂酸盐化剂。然而,它们是全身施用的,结果对正常的快速分裂的宿主组织(如骨髓、肠粘膜和淋巴系统)具有毒性。由于采他子卡是前药,其可避开活性酶(NQO2)的良好分布或病毒(G(V)DEPT)的施用途径。活性4-羟基胺衍生物具有非常短的半衰期,且位移不能离活性位点很远,因此不会出现储库效应。而且,归因于采他子卡诱发的交联的修复较差,多数人类实体癌症没有高比例的在任何时间下增生的细胞和较宽的杀灭窗口可能是一个优点。
较早前,已经完成了对采他子卡的I期和药代动力学进行的研究。(Chung-Faye等人,2001),其中其得到的结论是能够对人类安全地施用高达24mg/m2的采他子卡,也不具有任何毒性(MTD为37.5mg/m2)。
人类中施用采他子卡受G(V)DEPT技术的实用性或NQO2的分布限制。然而,如果采他子卡能够在近距离被活化,其对治疗其它癌症(或其它不良的细胞生长或增生)是有药学益处的。因此,活化例如子宫颈、膀胱、脑、胸腔或局部位置中的采他子卡的体系会将体循环和活化体系分隔,并使其发生。理论上,能够通过使用酶和辅酶实现,尽管会遇到困难。然而,也有另选的方法还原硝基。电解还原不能在体内实现,但能够化学还原硝基。
报导了在丙酮中使用锌粉和碳酸铵将采他子卡化学还原成5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺),产率为13%(Knox等人,1988),或使用四氢呋喃中的肼水合物/Pd-C,产率为28%(Knox等人,1993)。但这些合成途径都不能在体内应用。
考虑了其它还原活化的前药。
本发明人意外地发现一种新颖的化学还原体系,其使还原活化的前药能够例如在水溶液和乳液中还原成相应的活性物质。此发明使获得新的治疗,其中优选直接向患者施用活性剂(例如用于治疗某些癌症、癌前疾病(如恶性小痣或特别是光化性角化病)和皮肤病如牛皮癣(其基本为过度生殖的皮肤细胞)。
如实施例中详细说明的,本发明人发现式I化合物如DHA能够用于将还原活化的前药(如采他子卡)转化成活性物质(如采他子卡的情况,5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺)。
因此,本发明的第五方面是提供一种组合物,其包括:
(a)如上定义的还原活化的前药;和
(b)如上定义的式I化合物。
当前药是采他子卡时,例如所述组合物在下述合适的条件下会随时间生成5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺,其能够诱发上述的细胞中的DNA的交联。
理应明白,上述包括还原活化的前药和式I化合物的组合物常规地通过混合包括还原活化前药的组合物和包括式I化合物的组合物而制得。因此,本发明的第六方面是提供试剂盒(kit of parts),其包括含有如上定义的还原活化的前药的第一部分,和含有如上定义的式I化合物的第二部分。
一般而言,试剂盒的两部分是相互兼容的组合物(如它们均具有相同的物理形式如乳液、水性溶液和凝胶),且混合时在合适的条件下通过还原活化的前药和式I化合物(如DHA)的反应能够生成相应的活性试剂(如采他子卡的情况,为5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺)。
上述试剂盒也可是一种治疗体系,其能够如下所述用于抑制不良的细胞生长或增生。
便利地,试剂盒或治疗体系包含还原活化前药和式I化合物的使用说明,特别是,它们包含混合含有还原活化前药和式I化合物的部分的说明,包括将所得的含有还原活化前药和式I化合物的组合物用于治疗(如用于抑制不良的细胞生长和增生)前将含有它们的部分混合的时间点。
一般而言,本发明的第五方面包含还原活化的前药和式I化合物的组合物,以及本发明的第六方面试剂盒中的各部分的组合物是药物组合物,其中还原活化的前药和式I化合物的组合,以及试剂盒中单独的还原活化的前药和式I化合物都与药学可接受载体混合。
同样地,本发明还包括含有上述定义的式I化合物和药学可接受载体的药物组合物,以及含有5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺和药学可接受载体的药物组合物。
药物组合物的特性取决于其用于治疗患者的形式,特别是取决于向患者给药的途径。
一般而言,为了治疗皮肤的或指状突可及膜(membranes accessible by digitations)(如口腔膜、阴道膜、子宫颈膜、肛门膜和直肠膜)的疾病,本发明的第五方面的组合物或药物组合物,或本发明的第六方面的试剂盒或治疗体系适于外用给药(topical administration)。该药物组合物包括乳液、药膏、洗液、喷雾和凝胶。
一般而言,治疗体内(特别是体腔中)疾病时,所述药物组合物是无菌的,无热原水溶液或悬浮液。一般而言,式I化合物可以是溶液形式。一般而言,还原活化前药可制成悬浮液。
制造如乳液、药膏、洗液、喷雾和凝胶等药物组合物以及无菌的,无热原水性溶液或悬浮液的方法是本领域已知的。例如,可通过将组合物的组分,试剂盒或治疗体系悬浮或溶解在例如含有以下的一种或多种组分的混合物中制得药膏:矿物油、液体石油、白色凡士林、丙二醇、聚氧乙烯尿氧丙烯化合物、乳化蜡和水。而且,可通过将组合物的组分,试剂盒或治疗体系悬浮或溶解在例如含有以下的一种或多种组分的混合物中制得乳液或洗液:矿物油、山梨醇酐单硬脂酸酯、聚乙二醇、羊毛脂、液体石蜡、白色软石蜡、聚山梨醇酯60、鲸腊酯蜡、十六硬脂酸酯、2-辛基十二醇、苄醇和水。
在本发明中,优选本发明的第五方面的组合物或药物组合物是弱碱性的。对于包含采他子卡和式I的化合物的组合的组合物,特别优选组合物是弱碱性的,这是由于弱碱性有利于从采他子卡和式I的化合物生成5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺,且采他子卡和式I的化合物的组合在中性或酸性pH下是非反应性的。
对于表示本发明的第六方面的试剂盒或治疗体系的组分的组合物或药物组合物(即单独含有还原活化前药和式I的化合物的组合物),特别优选当混合它们时,所得组合物是弱碱性的。
优选式I的化合物的单独的组分是略酸性的。优选还原活化前药组分是弱碱性的(特别是如采他子卡的化合物,其在酸性pH下是不稳定的)。优选组合产品是弱碱性的。
“弱碱性”意指组合物的pH为8~10.5,更优选9~10。
理应明白,当药物组合物是水性溶液时,其碱性(pH)可例如通过使用pH值试纸或溶液,或使用pH电极直接进行测量。对于对其它组合物如乳液的碱性(pH)的评估,可将组合物与水混合或用水萃取,可测量通过混合或萃取所得的水性溶液的pH。
例如,对于乳液,通过将0.2mg乳液与1mL水旋涡10秒钟可容易地测得乳液的pH。通过离心分离,悬浮液变清,测得水的pH。
组合物的碱性可通过使用本领域已知的缓冲液得到控制。生理学可接受以及允许在药物中使用的缓冲液也是本领域已知的。
优选的缓冲液体系是碳酸氢钠(NaHCO3)和碳酸钠(Na2CO3)的组合。此缓冲液特别适用于乳液。
本发明人发现通过改变初始缓冲液浓度,还原活化前药(如采他子卡)和式I的化合物(如DHA)之间的反应时间和程度可得到控制。pH对特定时间内的反应率以及反应程度有影响。缓冲液的强度可控制反应的时间质子似乎是在反应中形成的,其会消耗掉缓冲液。当缓冲液被消耗掉时,pH会下降且反应会停止。通过改变缓冲液的强度(浓度),可改变其发生的时间。优选选择缓冲液体系,从而使混合还原活化前药和式I的化合物时,于60分钟内完成反应的50%。
优选本发明的第五方面中包含还原活化前药和式I的化合物的组合物所含有的式I的化合物与还原活化的前药相比摩尔数过量。同样地,优选在试剂盒或治疗体系中各部分组合时,式I的化合物与还原活化前药相比摩尔数过量。优选地,式I的化合物相对于还原活化前药的摩尔数过量>4:1,如>5:1(式I的化合物:还原活化前药,如DHA:采他子卡),更优选>10:1。
在本发明的第五方面的组合物或药物组合物,或本发明的第六方面的试剂盒或治疗体系中,优选:
(a)还原活化前药(如采他子卡)在含有所述组分的组合物中的浓度为0.5~5%w/w(如0.5~1%w/w)组合物(如每克组合物5~10mg);且
(b)式I的化合物(如DHA)在含有所述组分的组合物中的浓度为2.5~10%w/w(如5~10%w/w)组合物(如每克组合物50~100)。
在本发明发明人发现之前,没有人提出在外用给药(例如乳液或洗液或药膏)中使用采他子卡。因此,本发明的进一步的方面是提供外用给药用的含有采他子卡的组合物;优选为药物组合物。组合物可以是乳液或洗液或药膏形式。
药物治疗(微生物感染的治疗或杀灭癌症细胞)中一些式I的化合物(DHA和甘油醛)的使用在WO2006/003492,Naturwissenschaften51,217-218(1964)and Cancer Chemother.Rep.(Part1)52(7),687-696(1968)有说明。然而,在本发明发现前,没有人提出DHA和甘油醛以外的式I的化合物可在药物中使用。因此,本发明的进一步的目的是提供上述定义的式I的化合物在药物中的应用,条件是该化合物不是DHA或甘油醛。式I的化合物被封装并用于药物中。
本发明还包括含有上述定义的式I的化合物和药学可接受的载体的药物组合物,条件是该化合物不是DHA或甘油醛。在一个实施方式中,所述药物组合物不是乳液或洗液或药膏或喷雾。优选含有上述定义的式I的化合物的药物组合物是注射用的无菌的,无热原水性溶液。
尽管5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺已知是采他子卡的硝基还原产品,在本发明人发现之前,没有人提出组合物(如上述的药物组合物)可在含有该化合物的药物中的使用。因此,本发明的进一步的方面是提供5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺在药物中的应用。
如上所述,本发明的第四方面涉及还原活化前药的还原方法,所述方法包括将还原活化前药与上述定义的式I的化合物接触。一般而言,还原活化前药和式I的化合物在合适的水性溶液中接触。优选如上所述溶液是弱碱性的。
本发明人发现当使用采他子卡和DHA进行此方法时,在pH9下进行反应的反应速度比pH10下进行反应快,且主要得到高产率的优选的4-羟基胺产品(与2-羟基胺产品相比)。而且,此方法优选式I的化合物的相对于还原活化前药摩尔过量,且摩尔过量的范围和优选的摩尔过量如上所述。当式I的化合物应用于采他子卡的反应时,所述方法还可包含进一步的步骤:例如通过使用任何合适的分离方法如HPLC从反应混合物中纯化5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺。
明显地,化合物(如采他子卡的化合物)中的硝基仅还原至羟基胺基团(且没有还原至其它基团如亚硝基或胺基)。
因此,本发明的进一步方面是提供上述定义的式I的化合物将有机硝基化合物转化成相应的羟基胺的选择性还原中的应用。在本发明的这方面的一个实施方式中,所述应用是式I的化合物和碱的应用。所使用的碱的性质和量以及式I的化合物诱发硝基化合物的还原的pH可以如本发明的第一方面所述的应用中定义的。
同样地,本发明还提供将有机硝基化合物选择性还原至相应的羟基胺的方法,所述方法包括将所述有机硝基化合物与上述定义的式I的化合物接触。在一个特别的实施方式中,所述方法包括将有机硝基化合物与式I的化合物和碱接触。所使用的碱的性质和量可以如本发明的第一方面所述的应用中定义的。而且,在另一特别的实施方式中,所述方法包括在pH7~11(如pH7.1(如7.2、7.3、7.4或7.5)~10.9(如10.8、10.7、10.6、10.5、10.4、10.3、10.2、10.1或10.0))的溶液(如水性溶液)或悬浮液中将有机硝基化合物与上述定义的式I的化合物接触。
有机硝基化合物可以例如是任何上述的硝基化合物。然而,特别是有机硝基化合物是芳香族硝基化合物或杂芳族硝基化合物。
本文使用的“芳香族硝基化合物”包括包含C6-14碳环芳香族基团(如苯基、萘基、蒽基或菲基)的化合物,芳香族基团具有一个硝基取代基。除了具有一个硝基取代基,芳香族基团任选进一步被一个或多个选自以下基团的取代基取代:C1-6烷基、C1-6烷氧基(上述两个基团任选被一个或多个选自卤素、OH、C1-4烷氧基、Het和芳基的取代基取代)、OH、卤素(如氟、氯、溴或碘)、氰基、硝基、CO2Ra、C(O)NRbRc、S(O)1-2Rd、S(O)2NRbRc、N(Rb)Rc、Het、氮丙啶基和芳基,其中Ra~Rd独立地表示C1-6烷基(任选被一个或多个选自卤素、OH、C1-4烷氧基、Het和芳基的取代基取代)、Het或芳基,或Ra~Rc可另选地(和独立地)表示H,其中Het和芳基如上述定义。
在一个具体实施方式中,碳环芳香基还具有至少一个吸电子取代基,如一个或多个(如一个或两个)选自卤素(如氟、氯、溴或碘)、氰基、硝基、CO2Ra、C(O)NRbRc、S(O)1-2Rd和S(O)2NRbRc的取代基,其中Ra~Rd如上述定义。如本领域技术人员理解的,碳环芳香基的取代模式会影响与该基团连接的硝基的还原电位。一般,向该基团加入吸电子取代基会增加(即使其负电性降低)还原电位(E2或E0,如通过极谱法测得的)。在一个更具体的实施方式中,碳环芳香基还具有至少一个硝基取代基。
本文所用的术语“杂芳香族硝基化合物”包括含有5元~14元杂芳香族基团的化合物,所述杂芳香族基团含有一个或多个选自氧、氮和/或硫的杂原子,其中杂芳香族基团可包含一个、两个或三个环,且杂芳香族基团具有硝基取代基。杂芳香族基团任选具有一个或多个选自C1-6烷基、C1-6烷氧基(上述两种基团任选被一个或多个选自卤素、OH、C1-4烷氧基、Het和芳基)、OH、氧代、卤素(如氟、氯、溴或碘)、氰基、硝基、CO2Ra、C(O)NRbRc、S(O)1-2Rd和S(O)2NRbRc、N(Rb)Rc、Het、氮丙啶基和芳基的取代基取代,其中Ra~Rd如上述定义。为了免除疑惑,术语“杂芳香族硝基化合物”包括部分-芳香族杂环基,其包含两个或三个环,其中至少一个环(但非所有环)是芳香族的。在这些环体系中,优选硝基与芳香环连接。
“杂芳香族硝基化合物”的具体的杂芳香环体系包括苯并咪唑基、苯并[c]异噁唑烷基、苯并异噁唑基、苯并呋喃基、苯并呋咱基、2,1,3-苯并噁唑二唑基、苯并噁唑基、苯并吡唑基、苯并[e]嘧啶、2,1,3-苯并噻二唑基、苯并噻唑基、苯并噻吩基、苯并三唑基、噌啉基、呋喃基、咪唑基、咪唑并[1,2-a]吡啶基、咪唑并[2,3-b]噻唑基、吲哚基、异喹啉基、异噁唑基、萘并[1,2-b]呋喃基、噁二唑基、噁唑基、酞嗪基、嘌呤基、吡嗪基、吡唑基、吡啶基、嘧啶基、吡咯并[2,3-b]吡啶基、吡咯并[5,1-b]吡啶基、吡咯并[2,3-c]吡啶基、吡咯基、喹唑啉基、喹啉基、噻唑烷基、噻唑基、噻吩基、噻吩并[5,1-c]吡啶基、三唑基、1,3,4-三唑并[2,3-b]嘧啶基、吨基、苯并二噁烷基、苯并二氧杂环庚烷基、苯并二氧杂环戊烷基、苯并吗啉基、苯并噁唑烷基、苯并二氢吡喃基、苯并吡喃基、2,3-二氢苯并咪唑基、2,3-二氢苯并[b]呋喃基、1,3-二氢苯并-[c]呋喃基、1,3-二氢-2,1-苯并异噁唑基、2,3-二氢吡咯[2,3-b]吡啶基、4,5,6,7-四氢苯并咪唑基、4,5,6,7-四氢苯并吡唑基、5,6,7,8-四氢苯并[e]嘧啶和硫代苯并二氢吡喃基等。
本发明人意外地发现通过向可还原化合物(例如可还原的有机化合物,如有机硝基化合物或还原活化的前药)和碱的预形成混合物(例如溶液或悬浮液)加入式I的化合物,可利用上述定义的式I的化合物有效地对可还原化合物进行还原。
因此,本发明的进一步方面提供了可还原化合物的还原方法,所述方法包括向所述可还原的化合物和碱的混合物加入上述定义的式I的化合物,其中所述碱如本发明第一方面中所定义的。
可还原的化合物可以是可还原的有机化合物,如有机硝基化合物或如上述定义的还原活化前药(如采化子卡)。在本发明的这方面中,式I的化合物可以特别是DHA。
可还原化合物和碱的混合物可以例如是水性或特别是有机溶剂体系的溶液或悬浮液。具体的溶剂体系包括低级(C1-4)烷基醇(如异丙醇、乙醇或甲醇)、氯化烃(如二氯甲烷)、水及混合物(单相或二相混合物)。具体的碱包括碱金属(如钠或钾)碳酸氢盐或特别是碱金属碳酸盐(例如碳酸钾,如无水碳酸钾)。
例如可在室温或更高温度进行还原。在本发明具体的实施方式中,反应在高温(如25°C以上),如30~100°C(如40~70°C,如约60°C)下进行。还原也可以在基本不含氧化剂(如大气氧气)下进行。因此,在本发明的具体实施方式中,反应在惰性气氛(如氮气气氛或氩气气氛)下和/或通过使用去氧(或去气)溶剂和/或试剂进行。
在本发明的一个更具体的实施方式中,可还原化合物维持在化学计量过量(相对于计算所得实现还原所需的式I的化合物的摩尔数)。在这方面中(不受理论限制),认为式I的化合物在每摩尔式Ia的二聚体中提供1摩尔氢化物当量(即每2摩尔式I的化合物提供1摩尔氢化物当量)。因此,例如将硝基还原至羟基胺(二电子还原需要提供两个氢化物当量),实现所需还原的式I化合物的摩尔数是每1摩尔硝基化合物需要4摩尔的式I化合物。
式I的化合物可以任何速度加入到可还原化合物和碱的混合物中。然而,在本发明的一些实施方式中,式I的化合物慢慢地加入到混合物中,如每分钟至多2摩尔当量(相对于可还原化合物)(如每分钟至多1.75、1.5、1.25、1.0、0.9、0.8、0.7、0.6、0.5、0.4、0.3或特别是每分钟0.2或0.15摩尔当量)。
如上所述,可还原化合物可以是采他子卡。因此,本发明的这方面的一个具体的实施方式涉及制备5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺(或5-(氮丙啶-1-基)-2-羟基氨基-4-硝基苯甲酰胺的方法,所述方法包括:
(a)提供采他子卡和碱的混合物;和
(b)加入至多四摩尔当量式I的化合物(或另选地,加入至多两摩尔当量的式I的化合物的二聚体(即式Ia的化合物)。
式I的化合物在具体的实施方式中可以是DHA。
此反应可以在上述涉及可还化合物的还原方法中所述的条件(以及使用碱和/或溶剂)下进行。本发明这方面的进一步的实施方式包括方法中进一步包括从副产品5-(氮丙啶-1-基)-2-羟基氨基-4-硝基苯甲酰胺(如果有的话)分离产品5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺的步骤。可通过本领域技术人员已知的技术(如分步结晶、重结晶、层析法、溶剂萃取、真空升华等)实现分离。
本发明的进一方面是提供抑制个体某部位(site in the individual)细胞的不良生长或增生的方法,所述方法包括向个体中的该部位提供抗增生试剂的还原活化前药(如采他子卡)和上述定义的式I的化合物。
本文中使用的术语“抗增生试剂的还原活化前药”包括特别是上述的还原活化前药,如丝裂霉素C、E09、RSU-1069、RB-6145和尤其是采他子卡。
还原活化前药和式I化合物可以在向个体给药之前进行组合或它们可以依次进行给药。
例如,对于外用给药,含有还原活化前药的组合物和含有式I的化合物的组合物(两者中的组合物是适于外用给药的)在向个体给药前组合是便利的。在下面的实施例2和3(外用给药的组合物为乳液)中将详细说明(参考采他子卡和DHA的组合)此实施方式。
同样地,对于体腔内给药,含有还原活化前药的组合物和含有式I的化合物的组合物(两者中的组合物是适于体腔内给药的)在给药前混合是便利的。然而,也可以在体腔内进行混合(例如,单独的无菌的,无热原的还原活化前药和式I的化合物的溶液或悬浮液可以在体腔内施药,还原活化前药和式I的化合物的初次接触可以在体腔内进行。在一个实施方式中,还原活化前药和式I的化合物例如可以在中性条件下共同施用,而施用的另一试剂(如缓冲液)使体腔内环境弱碱性。一般而言,然而,还原活化前药(或其与式I的化合物的组合)可以在碱性介质中存在。
理应明白,当还原活化前药和式I的化合物在向个体施药前在合适的条件下进行混合,会生成相应的活性物质(如采他子卡的情况,为5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺),其然后施用于个体。因此,本发明的进一步的方面提供抑制细胞在个体中位点处的不良生长或增生的方法,所述方法包括向个体中的位点施用或提供含有5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺的组合物。
本发明的进一步方面提供:
(i)上述定义的抗增生试剂的还原活化前药和上述定义的式I的化合物的组合在制备用于抑制个体细胞不良生长或增生中的使用;以及
(ii)5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺在制备用于抑制个体细胞不良生长或增生中的使用。
涉及治疗细胞不良生长或增生的方法的相应的方面,所述方法包括向需要该治疗的患者施用:
(i)含有抗增生试剂的还原活化前药或其药学可接受盐和/或溶剂化物,和式I的化合物的组合产品;或
(ii)有效量的5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺。
组合产品可以是试剂盒或组合制剂。因此,本发明的这方面包括向患者施用:
(a)组合物,其包括
(I)抗增生试剂的还原活化前药或其药学可接受盐和/或溶剂化物,
(II)上述定义的式I的化合物,以及,任选
(III)药学可接受的佐剂、稀释剂或载体;或
(b)试剂盒,其包括
(I)第一部分,其含有抗增生试剂的还原活化前药或其药学可接受盐和/或溶剂化物,和
(II)第二部分,其含有上述定义的式I的化合物。
本发明的另一方面涉及组合产品(即组合物或试剂盒)其本身。在本发明的这些方面中,抗增生试剂的还原活化前药如上述定义。
一般而言,试剂盒的两个部分是相互兼容的组合物(如它们具有相同的物理形式,如乳液、水性溶液和凝胶),且混合时能够在合适的条件下通过还原活化前药与式I的化合物(如DHA)的反应生成相应的活性试剂(例如,在采他子卡的情况下,为5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺)。
所述试剂盒可以是治疗体系,其可用于抑制不良的细胞生长或增生。
便利地,试剂盒或治疗体系包含抗增生试剂的还原活化前药和式I的化合物的使用说明,特别是,它们包含混合含有抗增生试剂的还原活化前药和式I的化合物的部分的说明,包括将所得的含有还原活化前药和式I的化合物的组合物用于治疗(如用于抑制不良的细胞生长和增生)前将所述部分混合的时间点。
一般而言,包含抗增生试剂的还原活化前药和式I的化合物的组合物,以及试剂盒中的各部分的组合物是药物组合物,其中抗增生试剂的还原活化前药和式I的化合物的组合,以及试剂盒的部分中的单独的抗增生试剂的还原活化前药和式I的化合物与药学可接受载体混合。
在治疗不良的细胞生长或增生中,组合产品(组合物的组合或试剂盒的组合)或5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺可以作为单独的治疗体系或试剂施用,或可另选地与一种或多种另外的活性试剂(如已知用于治疗某些讨论中的疾病的活性试剂)一起施用(同时或依次地)。
待抑制的不良的细胞生长或增生可以是任何不良的生长或增生,特别是对于容易出现DNA交联的细胞。
不良的细胞生长或增生可以是良性的,如一般的疣或牛皮癣或其它涉及不良的细胞生长或增生的皮肤疾病,或其可以是赘生物(neoplastic),如肿瘤(tumor)或其转移。
因此,本发明的方法和药物治疗(即包括抗增生试剂的还原活化前药、5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺,或其两者之一的应用)可用于治疗疣和其它其皮肤疾病,如牛皮癣。其它良性生长、癌前疾病和癌症,包括子宫颈癌、膀胱癌、脑癌、胸腔癌和子宫癌。
所述方法和药物治疗特别优选以外用的方式(topically)抑制疾病。
所述方法和药物治疗还特别优选通过向体腔内(膀胱或腹膜或胸腔)施药抑制疾病。对于采他子卡,其活性4-羟基胺衍生物施用或递送后的半衰期非常短,且位移不能离活性位点很远,因此不会出现储库效应,且活性药物不会进入循环产生全身效应(systemic effects)。
本发明的具体的实施方式可包括:
(a)外用组合物(如乳液、洗液或药膏),其包括抗增生试剂的还原活化前药(如RSU-1069、丝裂霉素C或特别是采他子卡或E09)、式I的化合物(如DHA)和外用可接受的佐剂、稀释剂或载体(如洗液、乳液或药膏基质(ointment base));
(b)小丸药或类似的固体递送媒介(delivery vehicle),其包括抗增生试剂的还原活化前药(如RSU-1069、丝裂霉素C或特别是采他子卡或E09)、式I的化合物(如DHA),和药学可接受佐剂、稀释剂或载体;和
(c)含有抗增生试剂的还原活化前药(如RSU-1069、丝裂霉素C或特别是采他子卡E09)、式I的化合物(如DHA)和药学可接受佐剂、稀释剂或载体(如无菌的溶剂体系,如无菌的水性溶剂体系)的溶液或悬浮液。
对于上述(a)和(b),具体的组合物还可以包括碱(如上述本发明的第一方面所定义的碱)和/或pH缓冲液体系,所述pH缓冲液体系于施用组合物时在施药位点提供局部pH7~11)(例如:对于含有采化子卡的制剂,提供pH7.5、8.0、8.5或9.0和10.5或特别是10;或形成pH7.1~8.0(如约pH7.5)的含有E09的制剂)。
可以使用上述(a)所述的外用组合物(或乳液、洗液或药膏)例如治疗皮肤癌、皮肤上的非黑素瘤、前列腺癌、癌前疾病(如恶性小痣或特别是光化性角化病)、疣或牛皮癣。这样的组合物还可作为直肠药膏用于治疗诸如肠癌等疾病。
当其包含E09时,上述(b)的固体传递媒介可特别适用于前列腺癌的治疗。这是由于前列腺已知对高pH特别敏感,且本发明人发现在较低pH(如约pH7.5)下与诸如采他子卡的化合物相比,E09的还原更迅速。
可通过注射、导管插入法(transcatheterisation)或其它灌输方法将上述(c)的溶液或悬浮液,导入体腔内或出现不良的细胞生长或增生的空间。例如,可将溶液引入膀胱以治疗诸如膀胱癌等疾病、使与子宫颈接触以治疗子宫颈癌、注射至腹膜以治疗例如卵巢癌,或注射至脑部以治疗脑癌。
在一个特别的实施方式中,含有式I的化合物(如DHA)和抗增生试剂的还原活化前药(如采化子卡或E09)可通过膀胱内导尿管灌输(intravesical urinary catheter infusion)进行给药以治疗膀胱癌。
因此,本发明的进一步方面涉及治疗膀胱癌的方法,其包括通过膀胱内导尿管灌输施用有效量的含有式I的化合物(如DHA)和抗增生试剂(如采他子卡或E09)的还原活化前药的溶液或悬浮液。
上述包含5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺的组合物也可以是特别的药物形式,如:
(i)外用组合物(如乳液、洗液或药膏),其包括5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺和外用可接受的佐剂、稀释剂或载体(如洗液、乳液或药膏基质);
(ii)含有5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺和药学可接受佐剂、稀释剂或载体(如无菌的溶剂体系,如无菌的水性溶剂体系)的溶液或悬浮液。
上述组合物(i)和(ii)可分别具有与上述组合物(a)和(c)相同的最终应用。
另选地,上述(ii)的溶液或悬浮液(以及上述(c)的溶液或悬浮液)可以通过喷雾对口、鼻腔、喉咙(throat)、咽(pharynx)、喉(larynx)、气管或肺施药以治疗上述位置中不良的细胞增生(如癌症)。因此,本发明的进一步方面涉及可喷射溶液或悬浮液,其包括:
(A)抗增生的还原活化前药、上述定义的式I的化合物和药学可接受佐剂、稀释剂或载体;或
(B)5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺和药学可接受佐剂、稀释剂或载体。
通过喷雾直接对口、鼻腔、喉咙、咽、喉、气管或肺施用5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺(或提供该化合物的混合物),包括5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺通过血清蛋白的失活。而且,相同的失活机理将确保5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺不具有全身效应。
因此,本发明的进一步方面涉及治疗口癌、鼻腔癌、喉咙癌、咽癌、喉癌、气管癌或肺癌的方法,所述方法包括通过喷雾、溶液或悬浮液进行给药:
(A)抗增生的还原活化前药、上述定义的式I的化合物和药学可接受佐剂、稀释剂或载体;或
(B)5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺和药学可接受佐剂、稀释剂或载体。
适于传递喷雾形式的溶液或悬浮液的装置是本领域技术人员已知的。合适的装置(包括:机械式喷雾器,其从储存器抽吸溶液或悬浮液;和雾化装置(aerosol devices),其利用推进剂气体(propellant gases)通过喷嘴生成喷雾)包括在例如WO2006/005845中说明的。
因此,本发明的另一方面涉及:
(a)机械式喷雾器,其具有负载溶液或悬浮液的贮器,所述溶液或悬浮液包括以下物质:
抗增生的还原活化前药、上述定义的式I的化合物和药学可接受佐剂、稀释剂或载体;或
5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺和药学可接受佐剂、稀释剂或载体;和
(b)雾化装置,其包括含有一种或多种推进剂气体的溶液或悬浮液,和
抗增生的还原活化前药、上述定义的式I的化合物和药学可接受佐剂、稀释剂或载体;或
5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺和药学可接受佐剂、稀释剂或载体。
上述(b)的装置在一个实施方式中可以是计量式(metered dose)吸入装置。
作为液体喷雾的另一选择,干粉可以被雾化,从而使药物能够传递至位点(如肺)。因此,根据本发明的进一步方面,提供了干粉气溶胶组合物(aerosol composition),其包含5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺。
术语“干粉气溶胶组合物”包括干粉制剂,其能够通过吸入装置传递至口、鼻腔、喉咙、咽、喉、气管或肺。同样地,术语包括平均粒径为100μm(如50或10μm)或以下的干粉。在此方面中,粒径可以通过本领域技术人已知的方法(如通过激光散射技术,例如使用粒径分析仪(如MastersizerTM)的技术)测定。
递送干粉气溶胶(如干粉未吸入器)是本领域技术人员已知的,在例如WO2004/110536中有说明。因此,本发明的进一步方面提供一种治疗体系,其包括:
(i)干粉吸入装置,其任选包含一种推进剂气体源;和
(ii)所述干粉气溶胶组合物的一个或多个单独剂量(discrete doses),所述干粉气溶胶组合物含有5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺。
所述治疗体系可包括作为物理单独个体(physically separate entities)(即有效作为试剂盒)的装置和组合物剂型。另选地,吸入装置可以预先上载有一个或多个组合物剂型。
在治疗不良的细胞生长或增生中,上述组合物(包括外用组合物、固体传递介质、溶液或悬浮液、干粉未气溶胶等)可单独用于治疗某一疾病(不良的细胞生长或增生,如膀胱癌、子宫颈癌、腹膜癌、或脑癌、或口癌、鼻腔癌、喉咙癌、咽癌、喉癌、气管癌或肺癌),即作为主要的治疗体系或试剂给药。另选地,然而,组合物可以与一种或多种另一活性试剂(如已知用于治疗某些讨论中的疾病的活性试剂)一起施用(即同时或依次地施用)。
在这方面,已知的用于治疗膀胱癌的活性试剂包括顺铂、阿霉素和丝裂霉素C。
合适的人类使用的还原活化前药(如采他子卡)包括10~30mg/m2,一般为15~25mg/m2,例如24mg/m2。
本发明的方法和药物治疗能够用于治疗动物(如非人类哺乳动物,特别是马、牛和羊等,猫和狗)和人类。优选他们用于治疗人类。
本发明的进一步方面提供在细胞中交联DNA的方法,其包括向细胞施用还原活化前药和DNA交联剂(如采他子卡)的组合和上述定义的式I的化合物(如DHA),或提供的方法包括向细胞施用包括5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺的组合物。一般而言,细胞是培养细胞,方法是体内方法。
本文使用的术语“DNA交联剂的还原活化前药”包括特别是以下化合物,或其药学可接受的盐和/或溶剂化物:
(1)E09(3-[5-氮丙啶基-4,7-二氧代-3-羟甲基-1-甲基-1H-吲哚-2-基]-丙-β-烯-α-醇);
(2)RSU-1069(1-(1-氮丙啶基)-3-(2-硝基-1-咪唑基)-2-丙醇);
(3)RB-6145(1-[3-(2-溴乙基氨基)-2-羟丙基]-2-硝基咪唑);
(4)丝裂霉素C;
(5)下式化合物
其中
每个RA独立地表示氯、溴、碘或-OS(O)2RC,
RC表示C1-8烷基(任选被一个或多个氟原子取代)或苯基(任选被一个或多个选自卤素、硝基、C1-4烷基和C1-4烷氧基的取代基取代),
RB1~RB4独立地表示H、CN、C(O)N(RD)RE、C(S)N(RD)RE、C(O)OH、S(O)2NHRF,
或RB1还可以表示NO2,
RD和RE独立地表示H或C1-4烷基(其任选被一个或多个选自OH、N(H)-C1-2烷基、N(C1-2烷基)2、4-吗啉基和C(O)OH的取代基取代),
或RD和RE与连接的N-原子一起表示4-吗啉基,和
RF表示H或S(O)2CH3,
条件是:当RB1不是H时,RB2是H,
例如在1992和1995年Anlezark等人公开的上式化合物中的任何一个SN23163,SN23849,SN23777,SN23428,SN23759,SN24927,SN24928,SN24926,SN25402,SN25079,SN24939,SN24935,SN25923,SN25313,SN23856,SN25066,SN23816,SN25015,SN24971,SN25260,SN25261,SN25263,SN25084,SN25188,SN25507或特别是SN23862(5-{N,N-二[2-氯乙基]胺}-2,4-二硝基苯甲酰胺);
(6)下式化合物
其中
RA如上述所定义(如Cl),和
X1表示NH2,X2和X3两者均表示H,或
-X1-X2-表示-NH-CH2CH2-,X3表示H或
-X1-X3-表示-NH-,X2表示H;
(7)下式化合物
其中Y表示
1-氮丙啶基(任选在2-位被甲基取代),
甲氧基或
N(H)CH2CH2Br;
(8)下式的自杀性前药
其中
R表示-O-R’或-NH-R’,
R’表示
其中波浪线表示为片段连接的位置,
RA如上述所定义(如Cl),和
(9)吖啶-CB1954
(10)采他子卡(5-(氮丙啶-1-基)-2,4-二硝基苯甲酰胺)。
事实上,术语“DNA交联剂的还原活化前药”包括特别是诸如E09、RSU-1069、RB-6145、丝裂霉素C和特别是采他子卡的化合物。
附图说明
以下将参照附图和实施例详细地说明本发明,其中
图1示出采他子卡(5-(氮丙啶-1-基)-2,4-二硝基苯甲酰胺)的结构。
图2示出采他子卡的生物活化。
图3示出二羟基丙酮(DHA;CAS No:62147-49-3,Beil.8,266,Merck Index13,3166)的结构。正常的形式是二聚体,但在溶液中迅速转化成单体。
图4示出采他子卡通过DHA进行的还原。
图5示出采他子卡在E45基乳液中通过DHA进行的还原(如下面实施例2中说明的)。在图5的A、B和C的每一个中,两条线中上面的一条是在325nm下测量的,而下面的一条是在260nm下测量的。
图6示出图5中保留时间为5分钟所见的光谱匹配,其来自合成标准物5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺。连续线是标准物,虚线是样品。光谱260nm下的吸收率被标准化。
图7示出在pH7.5和温度37°C的含有140mM NaCl和以下物质的10mM磷酸盐缓冲液中2小时后中国仓鼠V79细胞的存活百分比:
A:对照(即没有其它物质);
B:10mM DHA;
C:10μM E09;
B+C:10mM DHA和10μM E09的组合;
D:100μM E09;和
B+D:10mM DHA和100μM E09的组合。
使用下述方法测定V79细胞的存活:
在37°C下培养含有140mM NaCl的10mM磷酸盐缓冲液(pH7.5)中的V79细胞(体积为1mL,2x105细胞/mL),然后加入试剂(对照实验除外)(如上所述,在规定的浓度下)。培养2小时后,通过离心分离收获细胞,进行连续稀释(4x10倍),将细胞置于生长介质中,在湿度5%为的CO2气氛为生长1星期后,评价它们形成的群居的能加。
图7的图表显示,E09的细胞毒性效果通过加入DHA而大大提高,其有助于形成E09的活性形式。
对于下面的实施例,采他子卡是市售的,其从购自Morvus Technology Limited和Sigma化学公司作为研究之用。
实施例1:采他子卡的化学活化
从采他子卡制造活性4-羟基氨基衍生物报导的化学方法在有机溶剂中使用苛刻的还原条件获得低于30%的产率(Knox等人,1993;Knox等人,1988)。发明人发现二羟基丙酮(DHA)能够在弱碱性条件下的水溶液中将采他子卡还原至所需的羟基胺。在pH9下,产率>85%,且采他子卡还原所测得的唯一的其它产物是5-(氮丙啶-1-基)-2-羟基氨基-4-硝基苯甲酰胺。
二羟基丙酮(DHA;1,3-二羟基-2-丙酮;CAS No:62147-49-3,Beil.8,266,Merck Index13,3166;图3)是防晒或美黑乳(self-tanning lution)中的活性成分,并获得FDA的准许。DHA是无色的糖,其通过染色(staining)加深皮肤的颜色。其与表皮中死去的表面细胞作用产生颜色的转变。一般在施用后5~7天内,随着死去的皮肤细胞自然脱落,颜色逐渐变淡。防晒美黑产品含有的二羟基丙酮浓度至多5%。浓度越高,晒后颜色越黑。作为美黑剂,其在pH4~6下稳定。太碱或太酸都会形成棕色化合物,减少溶液作为美黑剂的效用。赤藓酮糖(1,3,4-三羟基-2-丁酮)的作用类似于DHA,但其不能很快地产生深黑色。DHA通过甘油的发酵产生,其为简单的三碳糖,天然无毒的白色粉末。正常形式为二聚体(C6H12O6),但其在溶液中迅速转化成单体。DHA获FDA准许可外用,并建议用者应该采取保护措施以避免接触眼睛、鼻子和粘膜。报导的唯一副作用是过敏性接触性皮炎。但这种副作用很少出现,且美黑乳中导致过敏性大多数归因于其它成分如制剂中的防腐剂。见美国联邦法规,第376页:
题目21--食品和药品(TITLE21--FOOD AND DRUGS)
第I章--食品和药品管理,卫生与社会服务部(CHAPTER I--FOOD AND DRUG ADMINISTRATION,DEPARTMENT OF HEALTH AND HUMAN SERVICES)
第73部分--免证书的着色添加剂列表--目录(PART73--LISTING OF COLOR ADDITIVES EXEMPT FROM CERTIFICATION--Table of Contents)
亚部分C--化妆品(Subpart C--Cosmetics)
第73.2150节二羟基丙酮(Sec.73.2150Dihydroxyacetone)。
从未曾报导DHA作为还原剂。还原糖是已知的,但其为醛醣,在其中的一个末端具有醛。醛为还原剂,且在温和的氧化剂(如Cu2+或Fe3+)的存在下,醛会氧化成羧酸。作为酮醇,与DHA的当量反应是不可能的。然而,显示二羟基丙酮在铁(III)螯合物的存在下促进羟基团的形成(Malisza&Hasinoff,1995)。这种反应可能涉及其反还原能力及其碱不稳定性,使导致形成棕色化合物。
在碱性条件下,使用过量的DHA,采他子卡随时间以线性速度减少(图4)。
向采他子卡(100μM)和0.1M碳酸氢钠缓冲液(pH9或pH10)的混合物加入100μL的100mM DHA水溶液获得最终体积1ml开始进行分析。在37°C下培养所述混合物,每6分钟取试样(10μL)一次,随即通过HPLC[Partisil10SCX(4.2×150mm)(Whatman,Maidstone,Kent,U.K.]进行分析,按1.5mL/min的速度以0.13M磷酸钠(pH5)等度洗脱。通过对应的外标物的峰面积(通过325nm下的吸收率定量)测定每个样品中的采他子卡浓度。通过曲线拟合计算初始率(FigP,Biosoft,Cambridge,U.K.)。通过相对于可信的标准物的保留时间鉴别还原产品。
只观察到采他子卡还原的两个产物,2-羟基氨基衍生物和4-羟基氨基衍生物。采他子卡的还原率和羟基胺产品的比例依赖于pH。pH10下的还原(0.69nmol/min)比pH9下的还原(0.92nmol/min)慢。30min后,pH10下4-羟基胺对2-羟基胺的比例为2.7:1,pH9下则为9.7:1。因此,pH9下的还原较快,并且主要得到高产率的优选的4-羟基胺产品(图4)。
通过上述的HPLC方法没有测得来自反应的DHA或DHA-相关的产品。
实施例2:外用给药的乳液中的采他子卡的活化
制得指定为A和B的二种乳液。使用时,将它们等量混合。乳液A由E45基质(白色软石蜡BP14.5%w/w、轻液体石蜡Ph Eur12.6%w/w、低过敏性无水羊毛脂(Medilan)1.0%w/w、Crookes Healthcare Ltd,诺丁汉,UK)组成),每克含有10mg采他子卡、10mg NaHCO3和90mg Na2CO3。乳液B含有E45,每克含有100mg DHA二聚体。混合组分A和B,制得淡黄色乳液。数小时后转为棕色,24小时内持续加深。200μg乳液在1mL水中的悬浮液在猛烈搅拌下生成如pH试纸显示的约pH10的溶液。含有10%上述量的缓冲盐的乳液的初步实验给出初始pH相同的溶液。然而,4小时后,如上制备的溶液变中性,且乳液不再加深。这暗示通过改变初始缓冲液浓度,反应的时间和程度能够得到控制。
混合后,在不同时间,提取200μg乳液至1mL的DMSO中。然后用50mM碳酸氢铵缓冲液(pH10)将提取物稀释至1/100,用反相HPLC进行分析。开始时,325nm下提取物的分析显示只有一个主要峰,其为采他子卡(与可信的标准物具有相同的保留时间和UV吸收光谱的光谱匹配)。4小时后,260nm和325nm线下提取物分析观察到更多的峰(图5)。
将每克含有10mg采他子卡、10mg NaHCO3和90mg Na2CO3的乳液A(由E45基质(白色软石蜡BP14.5%w/w、轻液体石蜡Ph Eur12.6%w/w、低过敏性无水羊毛脂(Medilan)1.0%w/w、Crookes Healthcare Ltd,诺丁汉,UK组成)与乳液B(含有E45,每克含有100mg DHA二聚体)混合,在不同时间,用1mL的DMSO提取200μg乳液,并猛烈搅拌。通过离心分离将提取物变澄清,然后用50mM碳酸氢铵缓冲液(pH10)将提取物稀释至1/100,用HPLC进行分析。将样品(10μL)注入Waters Symmetry Shield RP18柱(150x3.9mm)中,用乙腈的10mM甲酸铵(pH4.5)溶液线性梯度洗脱(1-40%于30分钟内)。利用TSP UV3000扫描检测器持续监测洗脱液在230~400nm的UV吸收率。A.)混合乳液后立即制备的提取物。B.)混合后4小时来自乳液的提取物。C.)5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺的合成标准物。蓝线(在每对线中下面的线)是在260nm下获得的线,红线(在每对线中上面的线)是在325nm下获得的线。约13分钟获得的峰是CB1954,约19分钟获得的大峰(260nm)来自E45。无法测量提取方法的效率。
仍然可以检测到采他子卡,在保留时间约5.0分钟下的峰鉴别为采他子卡的4-羟基胺(与可信的标准物具有相同的保留时间且与其UV吸收光谱相匹配)(图6)。其它峰不对应已知的采他子卡的还原产物,其可能来自DHA的氧化,或还原的采他子卡产物与乳液或DHA的反应。
采他子卡(5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺)的活性形式在E45基乳液中形成。
实施例3:采他子卡乳液的外用
混合上述制得的乳液,将约0.1g涂覆在健康的人类志愿者手指上的疣(成长中,坡面高1.5mm),用膏药(plaster)覆盖。记录涂覆乳液的初始温度(warmth)。约4小时后,移去膏药,观察到疣已脱落,留下~1mm深的坑。周围的组织变黄。数天后逐渐转白,6星期后没有观察到疣的再生。也没有明显的副作用的报告。
将每克含有10mg采他子卡、10mg NaHCO3和90mg Na2CO3的乳液A(由E45基质(白色软石蜡BP14.5%w/w、轻液体石蜡Ph Eur12.6%w/w、低过敏性无水羊毛脂(Medilan)1.0%w/w、Crookes Healthcare Ltd,诺丁汉,UK组成)与乳液B(含有E45,每克含有100mg DHA二聚体)混合,并涂覆(100μg)至健康的人类志愿者手指上的疣(成长中,坡面高1.5mm),用膏药(plaster)覆盖。约4小时后,移去膏药,观察到疣已脱落,留下~1mm深的坑。周围的组织变黄。数天后逐渐转白,6星期后没有观察到疣的再生。也没有明显的副作用的报告。1星期后拍摄照片。
外用中,采他子卡的活化对疣有明显的影响,但对周围的正常组织只有最小的影响。
实施例4:膀胱癌的治疗
膀胱灌注化疗或膀胱内化疗是通过对患有浅表膀胱癌的患者的膀胱填充药物以对抗癌细胞。尽管浅表膀胱癌是癌症的早期形式,许多情况下早期移除可将其治愈。然而,直接将药物与膀胱壁接触的治疗,可有效预防复发或延缓复发。膀胱内化疗是一个简易程序。导管经过尿道插入膀胱。含采他子卡的碳酸氢盐缓冲液(pH9)慢慢地灌输2~3分钟,随后进行2~3分钟DHA水溶液灌输。移去导管,2小时后通过排尿排去药物。
实施例5:接近中性pH下前药的还原
分析:HPLC
分析混合物含有在最终反应体积为1mL的磷酸钠缓冲液(所需pH)中的测试化合物(100μM)和DHA(10mM)。加入DHA开始反应,混合物在37°C下培养。每20min抽取试样(10μL),随即通过HPLC使用Partisphere5C18(4.2x150mm)柱(Whatman,Maidstone,Kent,U.K.]进行分析,以每分钟2.0mL乙腈水溶液进行梯度洗脱(1-95%,10分钟内)。使用光电二极管阵列UV-VIS检测器持续监测洗脱液的吸收率。通过外标物的相应的峰面积以及PDA扫描确定的合适的波长下的吸收率的定量测定每个样品中的药物浓度。通过曲线拟合计算初始率(FigP软件)。
注
a使用1mM的DHA(代替10mM的DHA)
实施例6:不同的pH值下硝基化合物和前药的DHA还原
样品如实施例5所述进行分析。
注
a使用1mM的DHA(代替10mM的DHA)
实施例7:NADP+通过α-羟基羰基化合物的还原
将10μL测试化合物的试样(水中浓度为100mM)加入到200μMNADP+水溶液(约990μL)中,缓冲至pH10(1mM NaHCO3缓冲液)。分析溶液中的测试化合物的最终浓度为1mM。然后,通过光谱光度计测量350nM下吸收率的增加(在2分钟内)监测NADP+的还原。随着每分钟A350的变化记录初始率。
注
1.应该在溶液中形成单体。
2.使用工业级(>90%)。350nm下观察到吸收率的任何增加都滞后约60s。
3.计算的单体浓度。
4.立体异构体的混合。向反应混合物加入1mM EDTA后没有观察到对率造成的影响。
5.仅~50%纯。350nm下观察到吸收率的任何增加都滞后约90s。
不受理论限制,相信在上述分析中具有还原活性的化合物为α-羟基羰基化合物,其能够形成式Ia所示类型的环二聚体。
相反,上述分析中率测得低于0.001(即没有检测到还原活性)的化合物包括:
丙三醇;
乙二醛;
D-葡萄糖;
二甘醇酸酐;
(±)-四氢糠基醇;
1,4-二噁烷-2,3-二醇;和
2-(羟甲基)四氢吡喃。
实施例8:采他子卡通过α-羟基羰基化合物的还原
向采他子卡(100μM)和0.1M碳酸氢钠缓冲液(pH9或pH10)的混合物加入100μL的100mM测试化合物的水溶液获得最终体积1ml,开始进行分析。在37°C下温育所述混合物,每6分钟取试样(10μL)一次,随即通过HPLC[Partisil10SCX(4.2×150mm)(Whatman,Maidstone,Kent,U.K.]进行分析,按1.5mL/min的速度以0.13M磷酸钠(pH5)等度洗脱。通过对应的外标物的峰面积(通过325nm下的吸收率定量)测定每个样品中的采他子卡浓度。通过曲线拟合计算初始率(FigP,Biosoft,Cambridge,U.K.)。通过相对于可信标准物的保留时间鉴别还原产品。
相反,上述分析中测得的初始率低于0.001(即没有检测到还原活性)的化合物包括:
丙三醇;
乙二醛;
D-葡萄糖;
二甘醇酸酐;
(±)-四氢糠基醇;
2-(羟甲基)四氢吡喃;
4-羟基-2-丁酮;
二氯丙酮;
1,4-二噁烷-2,3-二醇;和
二氯乙酰氯。
实施例9:采他子卡大规模还原至相应的羟基胺
5-(氮丙啶-1-基)-2,4-二硝基苯甲酰胺(“CB1954”,1.00g,3.97mmol)的甲醇(‘AnalaR’-级别,40mL)溶液用过量的无水K2CO3粉末(10.0g,72mmol;aprox.18equiv.)处理,搅拌下将混合物加热至60°C。60°C下N2流气氛中搅拌悬浮液。然后,在快速搅拌下于15min内将1,3-二羟基丙酮(1.50g作为DHA二聚体,8.33mmol;2.1mol equiv)的甲醇(‘AnalaR’-级别,40mL)溶液(预先充入N2气除气)加入到反应混合物中,期间保持60°C的温度。观察到快反应,颜色从淡黄橙色转为棕色。薄层层析法(TLC;铝板上Merck silica gel60GF254,9:1v/v CH2Cl2:MeOH)显示定量除去CB1954原料(Rf0.73),形成两个极性产品(分别在Rf0.53和Rf0.47)。该两个产品分别表现出与可信的样品5-(氮丙啶-1-基)-2-羟基氨基-4-硝基苯甲酰胺和5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺相同的TLC行为,所述可信的样品由公开的方法[Knox,R.J.,Friedlos,F.,Jarman,M.,Roberts,J.J.Biochemical Pharmacology37,4661–4669(1988);Knox,R.J.,Friedlos,F.,Biggs,P.J.,Flitter,W.D.Gaskell,M.,Goddard,P.,Davies,L.,Jarman,M.Biochemical Pharmacology46,797–803(1993)]制得。
将反应混合物热过滤,用低温甲醇(10mL)洗涤不溶物。冷却合并的滤液,旋转蒸发(30°C,高真空)获得粘稠的黄棕色油(1.02g,>100%)。TLC检查确定两个主要产品的存在。在碱性条件下进行O2氧化所得的2-亚硝基氧化产品和4-亚硝基氧化产品所对应的较小的点(总计<1–2%,Rf0.62、0.66),在操作过程中和处理时都明显可见。(注意:应尽量向所有装置充N2气以减小与空气的接触,避免羟胺氧化至亚硝基产品和无谓的带色的副产品)。判断2-羟基胺(Rf0.53)和4-羟基胺(Rf0.47)反应产物形成比例为~40:60。由于异构体洗脱特性相当接近,使用目前的溶剂体系难以对它们进行层析分离。然而,粗混合物样品(100mg)的快速色谱分离(Merck硅胶,60-200目,9:1v/v CH2Cl2–MeOH)在除去馏分中的溶剂后得到5-(氮丙啶-1-基)-2-羟基氨基-4-硝基苯甲酰胺(31mg)和5-(氮丙啶-1-基)-4-羟基氨基-2-硝基苯甲酰胺(49mg)。两个产品具有与报导的羟基胺的同质异构体的特性一致的NMR图谱,且TLC行为与可信的化合物无法区分[Knox,R.J.,Friedlos,F.,Jarman,M.,Roberts,J.J.Biochemical Pharmacology37,4661–4669(1988)]。
注:
(1)优选的反应溶剂为甲醇。1,3-二羟基丙酮(DHA)二聚体在许多普通溶剂(包括丙酮和高级醇)中的溶解性有限度。.
(2)60°C下反应几乎是即时的,但在较低温度下较慢。使用较高温度体系对于相对产率有不良的影响。
在碱的存在下,产物羟基胺对空气氧化表现较敏感,因此建议尽快从反应混合物除去K2CO3试剂。含有2-羟基胺产物和4-羟基胺产物的混合物的层析分离要求在处理过程中尽可能减少溶解的物质与O2(空气)接触。
参考文献
Anlezark,G.M.,Melton,R.G.,Sherwood,R.F.,Coles,B.,Friedlos,F.&Knox,R.J.(1992).The bioactivation of5-(aziridin-1-yl)-2,4-dinitrobenzamide(CB1954)-I.Purification and properties of a nitroreductase enzyme from Escherichia coli-a potential enzyme for antibody-directed enzyme prodrug therapy(ADEPT).Biochem Pharmacol,44,2289-95.
Anlezark,G.M.,Melton,R.G.,Sherwood,R.F.,Wilson,W.R.,Denny,W.A.,Palmer,B.D.,Knox,R.J.,Friedlos,F.&Williams,A.(1995).Bioactivation of dinitrobenzamide mustards by an E.coli B nitroreductase.Biochem Pharmacol,50,609-18.
Bailey,S.M.,Knox,R.J.,Hobbs,S.M.,Jenkins,T.C.,Mauger,A.B.,Melton,R.G.,Burke,P.J.,Connors,T.A.&Hart,I.R.(1996).Investigation of alternative prodrugs for use with E.coli nitroreductase in'suicide gene'approaches to cancer therapy.Gene Ther,3,1143-50.
Boland,M.P.,Knox,R.J.&Roberts,J.J.(1991).The differences in kinetics of rat and human DT diaphorase result in a differential sensitivity of derived cell lines to CB1954(5-(aziridin-1-yl)-2,4-dinitrobenzamide).Biochem Pharmacol,41,867-75.
Bridgewater,J.A.,Knox,R.J.,Pitts,J.D.,Collins,M.K.&Springer,C.J.(1997).The bystander effect of the nitroreductase/CB1954enzyme/prodrug system is due to a cell-permeable metabolite.Hum Gene Ther,8,709-17.
Bridgewater,J.A.,Springer,C.J.,Knox,R.J.,Minton,N.P.,Michael,N.P.&Collins,M.K.(1995).Expression of the bacterial nitroreductase enzyme in mammalian cells renders them selectively sensitive to killing by the prodrug CB1954.Eur J Cancer,31a,2362-70.
Burke,P.J.&Knox,R.J.(1998).Therapeutic systems PCT/GB98/01731);WO98/57662.
Chung-Faye,G.,Palmer,D.,Anderson,D.,Clark,J.,Downes,M.,Baddeley,J.,Hussain,S.,Murray,P.I.,Searle,P.,Seymour,L.,Harris,P.A.,Ferry,D.&Kerr,D.J.(2001).Virus-directed,enzyme prodrug therapy with nitroimidazole reductase:a phase I and pharmacokinetic study of its prodrug,CB1954.Clin Cancer Res,7,2662-8.
Cobb,L.M.(1970).Toxicity of the selective antitumor agent 5-aziridino-2,4-dinitrobenzamide in the rat.Toxicol Appl Pharmacol,17,231-238.
Connors,T.A.&Melzack,D.H.(1971).Studies on the mechanism of action of5-aziridinyl-2,4-dinitrobenzamide(CB1954),a selective inhibitor of the Walker tumour.Int J Cancer,7,86-92.
Cui,W.,Allen,N.D.,Skynner,M.,Gusterson,B.&Clark,A.J.(2001).Inducible ablation of astrocytes shows that these cells are required for neuronal survival in the adult brain.Glia,34,272-82.
Cui,W.,Gusterson,B.&Clark,A.J.(1999).Nitroreductase-mediated cell ablation is very rapid and mediated by a p53-independent apoptotic pathway.Gene Ther,6,764-70.
Felmer,R.,Cui,W.&Clark,A.J.(2002).Inducible ablation of adipocytes in adult transgenic mice expressing the e.Coli nitroreductase gene.J Endocrinol,175,487-98.
Friedlos,F.,Court,S.,Ford,M.,Denny,W.A.&Springer,C.(1998).Gene-directed enzyme prodrug therapy-quantitative bystander cytotoxicity and DNA damage induced by CB1954in cells expressing bacterial nitroreductase.Gene Therapy,5,105-112.
Friedlos,F.,Quinn,J.,Knox,R.J.&Roberts,J.J.(1992).The properties of total adducts and interstrand crosslinks in the DNA of cells treated with CB1954.Exceptional frequency and stability of the crosslink.Biochem Pharmacol,43,1249-54.
Hu,L.,Yu,C.,Jiang,Y.,Han,J.,Li,Z.,Browne,P.,Race,P.R.,Knox,R.J.,Searle,P.F.&Hyde,E.I.(2003).Nitroaryl phosphoramides as novel prodrugs for E.coli nitroreductase activation in enzyme prodrug therapy.J Med Chem,46,4818-21.
Isles,A.R.,Ma,D.,Milsom,C.,Skynner,M.J.,Cui,W.,Clark,J.,Keverne,E.B.&Allen,N.D.(2001).Conditional ablation of neurones in transgenic mice.J Neurobiol,47,183-93.
Knox,R.J.,Burke,P.J.,Chen,S.&Kerr,D.J.(2003).CB1954:from the Walker tumor to NQO2and VDEPT.Curr Pharm Des,9,2091-104.
Knox,R.J.,Friedlos,F.,Biggs,P.J.,Flitter,W.D.,Gaskell,M.,Goddard,P.,Davies,L.&Jarman,M.(1993).Identification,synthesis and properties of5-(aziridin-1-yl)-2-nitro-4-nitrosobenzamide,a novel DNA crosslinking agent derived from CB1954.Biochem Pharmacol,46,797-803.
Knox,R.J.,Friedlos,F.,Jarman,M.&Roberts,J.J.(1988).A new cytotoxic,DNA interstrand crosslinking agent,5-(aziridin-1-yl)-4-hydroxylamino-2-nitrobenzamide,is formed from5-(aziridin-1-yl)-2,4-dinitrobenzamide(CB1954)by a nitroreductase enzyme in Walker carcinoma cells.Biochem Pharmacol,37,4661-9.
Knox,R.J.,Friedlos,F.,Lydall,D.A.&Roberts,J.J.(1986).Mechanism of cytotoxicity of anticancer platinum drugs:evidence that cis-diamminedichloroplatinum(II)and cis-diammine-(1,1-cyclobutanedicarboxylato)platinum(II)differ only in the kinetics of their interaction with DNA.Cancer Res,46,1972-9.
Knox,R.J.,Friedlos,F.,Marchbank,T.&Roberts,J.J.(1991a).Bioactivation of CB1954:reaction of the active4-hydroxylamino derivative with thioesters to form the ultimate DNA-DNA interstrand crosslinking species.Biochem Pharmacol,42,1691-7.
Knox,R.J.,Friedlos,F.,Sherwood,R.F.,Melton,R.G.&Anlezark,G.M.(1992).The bioactivation of5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB1954)-II.A comparison of an Escherichia coli nitroreductase and Walker DT diaphorase.Biochem Pharmacol,44,2297-301.
Knox,R.J.,Jenkins,T.C.,Hobbs,S.M.,Chen,S.,Melton,R.G.&Burke,P.J.(2000).Bioactivation of 5-(aziridin-1-yl)-2,4-dinitrobenzamide(CB1954)by human NAD(P)H quinone oxidoreductase2:a novel co-substrate-mediated antitumor prodrug therapy.Cancer Res,60,4179-86.
Knox,R.J.,Lydall,D.A.,Friedlos,F.,Basham,C.,Rawlings,C.J.&Roberts,J.J.(1991b).The Walker256carcinoma:a cell type inherently sensitive only to those difunctional agents that can form DNA interstrand crosslinks.Mutat Res,255,227-40.
Knox,R.J.,Lydall,D.A.,Friedlos,F.,Basham,C.&Roberts,J.J.(1987).The effect of monofunctional or difunctional platinum adducts and of various other associated DNA damage on the expression of transfected DNA in mammalian cell lines sensitive or resistant to difunctional agents.Biochim Biophys Acta,908,214-23.
Li,Z.,Han,J.,Jiang,Y.,Browne,P.,Knox,R.J.&Hu,L.(2003).Nitrobenzocyclophosphamides as potential prodrugs for bioreductive activation:synthesis,stability,enzymatic reduction,and antiproliferative activity in cell culture.Bioorg Med Chem,11,4171-8.
Ma,D.,Allen,N.D.,Van Bergen,Y.C.,Jones,C.M.,Baum,M.J.,Keverne,E.B.&Brennan,P.A.(2002).Selective ablation of olfactory receptor neurons without functional impairment of vomeronasal receptor neurons in OMP-ntr transgenic mice.Eur J Neurosci,16,2317-23.
Malisza,K.L.&Hasinoff,B.B.(1995).Doxorubicin reduces the iron(III)complexes of the hydrolysis products of the antioxidant cardioprotective agent dexrazoxane(ICRF-187)and produces hydroxyl radicals.Arch Biochem Biophys,316,680-8.
Mauger,A.B.,Burke,P.J.,Somani,H.H.,Friedlos,F.&Knox,R.J.(1994).self-immolative prodrugs:candidates for antibody-directed enzyme prodrug therapy in conjunction with a nitroreductase enzyme.J Med Chem,37,3452-8.
Sheard,C.E.,Double,J.A.&Berenbaum,M.C.(1971).The sensitivity to chemopherapeutic agents of a rat tumour grown in immunosuppressed mice.Br J Cancer,25,838-844.
Workman,P.,Morgan,J.E.,Talbot,K.,Wright,K.A.,Donaldson,J.&Twentyman,P.R.(1986a).CB1954revisited.II.Toxicity and antitumour activity.Cancer Chemother Pharmacol,16,9-14.
Workman,P.,White,R.A.&Talbot,K.(1986b).CB1954revisited.I.Disposition kinetics and metabolism.Cancer Chemother Pharmacol,16,1-8.
Wu,K.,Knox,R.,Sun,X.Z.,Joseph,P.,Jaiswal,A.K.,Zhang,D.,Deng,P.S.&Chen,S.(1997).Catalytic properties of NAD(P)H:quinone oxidoreductase-2(NQO2),a dihydronicotinamide riboside dependent oxidoreductase.Arch Biochem Biophys,347,221-8.
通过参考的方式并入本文中提及的所有文献的全部内容。
- 还没有人留言评论。精彩留言会获得点赞!