一种卡格列净的口服固体制剂及其制备方法与流程
本发明属于药物制剂领域,具体涉及一种卡格列净口服固体药物组合物及其制备方法。
技术背景
卡格列净,通用名为canagliflozin,化学名为:1-(β-D-吡喃葡糖基)-4-甲基-3-[5-(4-氟苯基)-2-噻吩基甲基]苯,其结构式如式I所示。
卡格列净属于选择性2型钠葡萄糖协同转运蛋白(SGLT2)抑制剂,目前已获得美国和欧盟批准用于2型糖尿病的治疗。
正常情况下,肾脏在血糖的调节中起重要作用。葡萄糖经过肾小管时全部被重吸收回血液,因此终尿中几乎不含葡萄糖,同时保持了血糖的稳定。葡萄糖是不带电荷的有机物,它的重吸收是逆浓度差进行的主动重吸收过程,主要依赖钠-葡萄糖协同转运蛋白(SGLT)。SGLT分为SGLT1和SGLT2两种类型。SGLT2 是一种低亲和力、高容量的转运载体,对葡萄糖和钠进行1:1的转运,主要分布于肾近曲小管S1节段,对进入肾曲小管高浓度的葡萄糖进行重吸收,SGLT2重吸收葡萄糖占全部重吸收葡萄糖的90%。SGLT1则是一种高亲和力、低负载的葡萄糖载体,转运1分子的葡萄糖同时转运2分子的Na+,分布于肾近曲小管S2~3节段,SGLT1重吸收其余10%的葡萄糖。
卡格列净选择性抑制SGLT2可抑制大部分葡萄糖在体内的重吸收,促使葡糖糖大量从尿中排出达到控制血糖水平的目的。由于卡格列净的作用机制与胰岛素无关,因此和其他任何的糖尿病治疗方案(包括胰岛素)联合使用都能提供额外的降糖作用。既可以与其它口服降糖药联合用于现有治疗方案下血糖控制不佳或胰岛素抵抗问题患者,又可与胰岛素联合用于β细胞功能非常低而现有口服药物无法作用的患者。
目前,美国和欧盟批准的卡格列净的药用形式为卡格列净半水合物,其结构式如图2所示。该半水合物已在CN101573368中公开。
技术实现要素:
本发明的目的在于提供一种卡格列净口服固体药物组合物,其中,卡格列净以无定型形态存在。本发明人发现,无定型形态的卡格列净其溶解性虽优于卡格列净半水合物,但仍属于难溶化合物,且存在可压性差,不易制备成固体制剂,如果采用大量溶剂制粒则容易转化成卡格列净半水合物等技术难题。通过大量研究意外发现通过控制原料药的粒径,可有效提高卡格列净的可压性及溶出度且无需使用造粒溶剂,有效解决了无定型形态的卡格列净在制剂制备过程中转晶和可压性差的重要技术难题。
本发明人通过将无定型形态的卡格列净原料进行微粉化处理,收集了不同体积平均粒径的样品,采用不同的辅料,通过干法制粒工艺制备不同平均粒径原料药的片剂,考察压片时的可压性、片剂外观性状及片剂溶出度。结果发现,当卡格列净原料的体积平均粒径为1µm时,片剂的前期溶出度显著下降,无法满足产品质量要求,而且当卡格列净原料药颗粒的平均粒径为1µm和50µm时,其片剂的压片压力与片剂硬度相关性差,而将卡格列净原料药的平均粒径控制在2.5~30µm范围内,可以有效确保制剂的制备过程中物料的可压性及成品片剂的溶出度和不转晶,从而完成了本发明。
为实现本发明的卡格列净口服固体药物组合物,提供如下实施方案。
在一实施方案中,本发明的一种卡格列净口服固体药物组合物,包含卡格列净和药用辅料,其中,卡格列净为无定型形态,其颗粒平均粒径为2.5~30µm,优选2.5~20µm,更优选为2.5~10µm。
在上述实施方案中,本发明的卡格列净口服固体药物组合物,卡格列净的重量为组合物重量的10%~60%;所述药用辅料包括填充剂、崩解剂或润滑剂,所述填充剂为微晶纤维素、乳糖、预胶化淀粉、甘露醇、淀粉、山梨醇或者它们的任意混合物,优选为微晶纤维素、乳糖或它们的任意混合物,其重量为组合物重量的30%~75%;所述崩解剂为交联羧甲基纤维素钠、低取代羟丙基纤维素、羧甲基淀粉钠、交联聚维酮或者它们的任意混合物,优选为交联羧甲基纤维素钠,其重量为组合物重量的3%~10%;所述润滑剂为硬脂酸镁、硬脂酸钙、硬脂酸、硬脂富马酸钠或它们的任意混合物,优选硬脂酸镁,润滑剂重量为组合物重量的0.5%~5%。
在上述实施方案中,本发明的卡格列净口服固体药物组合物,卡格列净的含量为
50~300mg,优选100~300 mg。
在上述实施方案中,本发明的卡格列净口服固体药物组合物,所述药用辅料还进一
步包含表面活性剂,所述表面活性剂选自十二烷基硫酸钠、聚山梨酯、脂肪酸山梨坦、伯洛沙姆、聚氧乙烯蓖麻油和它们的任意混合物。
在上述实施方案中,本发明的卡格列净口服固体药物组合物,所述组合物为片剂。
在一具体实施方案中,本发明的卡格列净口服固体药物组合物,包含卡格列净和含有填充剂、崩解剂和润滑剂的药用辅料,其中,卡格列净为无定型形态,其颗粒平均粒径为2.5~30µm,优选2.5~20µm,更优选为2.5~10µm,卡格列净重量为组合物重量的10%~60%,填充剂为重量为组合物重量的30%~75%,崩解剂重量为组合物重量的3%~10%,润滑剂重量为组合物重量的0.5%~5%。
在上述具体实施方案中,本发明的卡格列净口服固体药物组合物,所述填充剂为微晶纤维素、乳糖、预胶化淀粉、甘露醇、淀粉、山梨醇或者它们的任意混合物,优选为微晶纤维素、乳糖或它们的任意混合物;所述崩解剂为交联羧甲基纤维素钠、低取代羟丙基纤维素、羧甲基淀粉钠、交联聚维酮或者它们的任意混合物,优选为交联羧甲基纤维素钠;所述润滑剂为硬脂酸镁、硬脂酸钙、硬脂酸、硬脂富马酸钠或它们的任意混合物,优选硬脂酸镁。
在上述具体实施方案中,本发明的卡格列净口服固体药物组合物,卡格列净的含量为50~300mg,优选100~300mg。
在上述具体实施方案中,本发明的卡格列净口服固体药物组合物,还进一步包含表面活性剂,所述表面活性剂选自十二烷基硫酸钠、聚山梨酯、脂肪酸山梨坦、伯洛沙姆、聚氧乙烯蓖麻油和它们的任意混合物。
在一优选实施方案中,本发明的卡格列净口服固体药物组合物,包含卡格列净10%~60%、填充剂 30%~75%、崩解剂3%~10% 和润滑剂0.5%~5%,其中,所述填充剂为微晶纤维素、乳糖或它们的任意混合物,所述崩解剂为交联羧甲基纤维素钠,所述润滑剂为硬脂酸镁,所述卡格列净为无定型形态,其颗粒的平均粒径为2.5~30µm,优选2.5~20µm,更优选为2.5~10µm。
在上述优选实施方案中,本发明的卡格列净口服固体药物组合物,卡格列净的含量为50~300mg,优选为100~300 mg。
在上述优选实施方案中,本发明的卡格列净口服固体药物组合物,根据需要还可进一步包含表面活性剂,所述表面活性剂选自十二烷基硫酸钠、聚山梨酯、脂肪酸山梨坦、伯洛沙姆、聚氧乙烯蓖麻油和它们的任意混合物,表面活性剂用量为常规用量。
在上述优选实施方案中,所述组合物的制剂形式优选为片剂。
本发明的目的还在于提供了一种制卡格列净口服固体药物组合物的方法,该方法无需加入造粒溶剂。
在一实施方案中,一种制备本发明的卡格列净口服固体药物组合物的方法,该方法包括以下步骤:
a)将无定型卡格列净微粉化,获得平均粒径为2.5~30µm的颗粒;
b)将微粉化的卡格列净与填充剂、崩解剂和润滑剂混合成均匀的粉末;
c)制粒;
d)加入崩解剂、润滑剂,混均后压片,得卡格列净片成品。
上述本发明的方法,所说步骤a)的平均粒径优选2.5~20µm,更优选2.5-10µm;所说的填充剂为微晶纤维素、乳糖、预胶化淀粉、甘露醇、淀粉、山梨醇中的一种或者它们的任意混合物,优选填充剂为乳糖、微晶纤维素或其混合物;所述的崩解剂为交联羧甲基纤维素钠、低取代羟丙基纤维素、羧甲基淀粉钠、交联聚维酮或它们的任意混合物,优选的崩解剂为交联羧甲基纤维素钠;所述的润滑剂为硬脂酸镁、硬脂酸钙、硬脂酸、硬脂富马酸钠或者它们的任意混合物,优选的润滑剂为硬脂酸镁。润滑剂的用量为0.5%~5%。
上述本发明的方法,所说步骤b)中,还可加入根据需要还可进一步包含表面活性剂,所述表面活性剂选自十二烷基硫酸钠、聚山梨酯、脂肪酸山梨坦、伯洛沙姆、聚氧乙烯蓖麻油和它们的任意混合物,表面活性剂的用量为常规用量,可选为膜剂重量的1-4.5%。
原料体积平均粒径大小对无定型形态卡格列净可压性的影响研究
本发明通过将无定型形态卡格列净进行微粉化处理,收集了体积平均粒径为1µm、2.5µm、10µm、20µm、30µm、50µm等6种不同平均粒径的卡格列净原料药颗粒,通过干法制粒工艺制备6种含有不同平均粒径的卡格列净的片剂,考察压片压力与片剂硬度的关系,并测定片剂的脆碎度。结果表明:卡格列净原料药颗粒的平均粒径为2.5µm、10µm、20µm、30µm时,其片剂的压片压力与片剂硬度相关性良好,且片剂的脆碎度符合中国药典2010年版相关规定;而卡格列净原料药颗粒的平均粒径为1µm和50µm时,其片剂的压片压力与片剂硬度相关性差,且片剂的脆碎度不符合中国药典2010年版相关规定。
上述研究结果表明,将卡格列净原料药的颗粒平均粒径控制在2.5~30µm之间时,制备的片剂具有良好地可压性,且脆碎度符合中国药典2010年版相关规定。优选的,将卡格列净原料药颗粒的平均粒径控制在2.5~20µm之间,进一步优选为2.5~10µm。
本发明所述的卡格列净片剂的制备采用干法制粒压片工艺,即经过原辅料混合、制粒、颗粒混合、压片的工艺过程。
原料体积平均粒径大小对卡格列净片剂外观性状的影响研究
将上述6种不同颗粒粒径原料制备的卡格列净片剂,进行外观性状对比考察。结果表明:卡格列净原料药颗粒的平均粒径为2.5µm、10µm、20µm、30µm时,其片剂的片面光洁,基本无细粉,外观性状良好;而卡格列净原料药颗粒的平均粒径为1µm 和50µm时,其片剂的片面有凹凸感,且易掉粉,外观性状较差。
通过对比研究考察,卡格列净原料的体积平均粒径控制在2.5~30µm之间,可有效确保产品的外观性状。
原料体积平均粒径大小对卡格列净片剂体外溶出的影响研究
将上述6种不同颗粒粒径原料制备的卡格列净片剂,进行及溶出曲线的对比考察。研究不同颗粒粒径的卡格列净片剂与溶出度的关系。
本发明所述的溶出测定方法采用中国药典2010年版二部附录XC溶出度测定法中的第二法,以0.75%的十二烷基硫酸钠水溶液作为溶出介质,溶出介质体积为900ml、转速为75r/min、温度为37±0.5℃。
通过对比研究发现,卡格列净原料的体积平均粒径控制在2.5~30µm之间,可有效确保产品的溶出。
附图说明
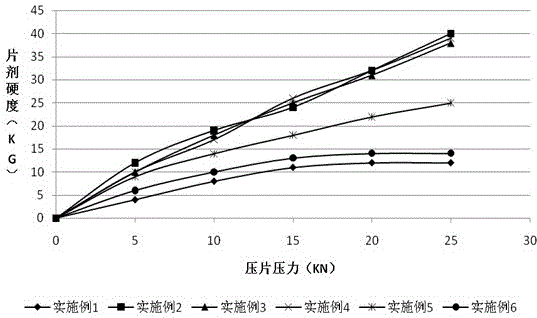
图1:实施例1~6制备的卡格列净片剂的硬度与压片压力的曲线
图2:实施例7~13制备的卡格列净片剂的硬度与压片压力的曲线
图3:实施例1~6制备的卡格列净片剂在0.75%十二烷基硫酸钠水溶液中的溶出曲线
图4:实施例7~13制备的卡格列净片剂在0.75%十二烷基硫酸钠水溶液中的溶出曲线
具体实施例方式:
下述实施例用于进一步解释本发明,但并不表示本发明的范围仅限于以下实施例。
实施例1~6
实施例1~6的处方见表1:
制备工艺:
1. 取上述体积平均粒径的无定型形态卡格列净原料备用;
2. 分别将乳糖、微晶纤维素、交联羧甲基纤维素钠和硬脂酸镁过80目筛备用;
3. 将各实施例的原料药、乳糖、微晶纤维素、70%处方量的交联羧甲基纤维素钠和70%处方量的硬脂酸镁混合均匀,混匀的物料加入干法制粒机中干法制粒,制得的颗粒与30%处方量的硬脂酸镁和30%处方量的交联羧甲基纤维素钠混匀,得到半成品;
4. 测定半成品含量,计算片重。依据半成品含量调节片重压片,压片过程中测定各压片压力下的片剂硬度,并测定6种片剂的脆碎度。
以上实施例1~6的处方及制备工艺完全一致,仅是原料药卡格列净的体积平均粒径不同。
实施例1~6制备的卡格列净片剂在不同压片压力下的片剂硬度测定结果见表2,片剂硬度在不同压片压力下的变化曲线见图1。
表2的结果表明,原料体积平均粒径为2.5µm、10µm、20µm、30µm的卡格列净片剂的硬度随压片压力增加而增加,相关性良好;而原料体积平均粒径为50µm的卡格列净片剂的硬度与压片压力基本不具相关性,且片剂的硬度很小,无法满足正常生产要求;令人意外的是,当原料体积平均粒径为1µm时,片剂的硬度与压片压力也不具相关性,片剂的硬度变变小,且在压片过程中出现粘冲现象,无法满足正常生产要求。
取以上实施例1~6在25 KN压片压力下制备的片剂测定脆碎度,测定结果见表3。
表3的结果表明,实施例2~5的卡格列净片剂脆碎度符合中国药典2010年版二部片剂脆碎度的相关规定;实施例1和实施例6的卡格列净片剂不符合中国药典2010年版二部片剂脆碎度的相关规定。
综上,本发明确定的卡格列净体积平均粒径控制在2.5~30μm范围内可有效确保产品的可压性和硬度。
实施例7~13
实施例7~13的处方见表4:
制备工艺:
1、取上述体积平均粒径的卡格列净原料备用;
2、分别将乳糖、甘露醇、淀粉、预胶化淀粉、微晶纤维素、交联羧甲基纤维素钠、交联聚维酮、羧甲基淀粉钠、十二烷基硫酸钠、二氧化硅和硬脂酸镁过80目筛备用;
3、将以上各处方中除二氧化硅和硬脂酸镁外的所有辅料分别混合均匀,混匀的物料加入干法制粒机中干法制粒,制得的颗粒加入各自处方量的二氧化硅和硬脂酸镁混匀,得半成品;
4、测定半成品含量,计算片重。依据半成品含量调节片重压片,即得实施例6~12的卡格列净片剂。
实施例7~13制备的卡格列净片剂在不同压片压力下的片剂硬度测定结果见表5,片剂硬度在不同压片压力下的变化曲线见图2。
以上表5的结果表明,将原料体积平均粒径控制在选定的2.5~30µm范围内,制备的卡格列净片剂的硬度随压片压力增加而增加,相关性良好,片剂硬度可通过压片压力有效控制。
取以上实施例7~13在25 KN压片压力下制备的片剂测定脆碎度,测定结果见表6。
以上表6的结果表明,实施例7~13的卡格列净片剂脆碎度符合中国药典2010年版二部片剂脆碎度的相关规定。
实施例1~6的卡格列净片剂的溶出曲线对比考察
对比考察的目的是确定本发明所述片剂的最佳体积平均粒径控制范围。
对比考察选用的片剂为实施例1~实施例6制得的卡格列净片剂。
本发明所述的溶出测定方法采用中国药典2010年版二部附录XC溶出度测定法中的第二法,溶出介质采用0.75%十二烷基硫酸钠水溶液、溶出介质体积为900ml、转速为75r/min、温度为37±0.5℃。
在5、10、15、30、60min取样,取出的溶出液用0.45µm的微孔滤膜过滤后,采用紫外分光光度法检测其中的卡格列净含量,并计算各时间点的溶出度,测定结果见表7,溶出曲线见图3。
表7的结果表明:实施例2~实施例5制得的卡格列净片剂的溶出度较好,30min的溶出度均达到90%以上;实施例6制得的卡格列净片剂溶出度较差,30min的溶出度还未达到75%,无法满足药品质量要求。令人意外的是,实施例1制得的卡格列净片剂前期溶出也十分缓慢,虽然60min的溶出度达到了90%以上的溶出度,但仍无法满足药品质量要求。可见,将原料药的体积平均粒径控制在2.5~30μm范围内可有效保证产品的溶出度。
实施例7~13的卡格列净片剂的溶出曲线考察
卡格列净片剂体外溶出的测定:
本发明体外溶出测定方法采用中国药典2010年版二部附录XC溶出度测定法中的第二法,溶出介质采用0.75%十二烷基硫酸钠水溶液、溶出介质体积为900ml、转速为75r/min、温度为37±0.5℃。
在5、10、15、30、60min取样,取出的溶出液用0.45µm的微孔滤膜过滤后,采用紫外分光光度法检测其中的卡格列净含量,并计算个时间点的溶出度,测定结果见表8,溶出曲线见图4。
表8的实施例7~实施例13的体外溶出结果表明:卡格列净原料在选定的体积平均粒径范围内,采用相同(或不同)处方制备的片剂,在相同溶出条件下,体外溶出行为十分接近。
综合以上实施例1-13的溶出曲线考察结果可知:本发明确定的卡格列净体积平均粒径控制在2.5~30μm范围内可有效确保产品的溶出。
在本发明的实质的基础上进行变通的修饰或简单的修改也属于本发明的权利要求的范围。
- 还没有人留言评论。精彩留言会获得点赞!