用于治疗巨噬细胞相关病症的方法和组合物与流程
本申请要求提交于2014年5月16日的美国临时申请号61/994,736以及提交于2014年9月17日的美国临时申请号62/051,849的权益,每个申请均通过引用而整体并入本文。
背景技术:
巨噬细胞是由单核细胞的分裂产生的白细胞。单核细胞和巨噬细胞是吞噬细胞,并在先天性免疫(非特异性免疫防御)中发挥作用以及帮助启动适应性免疫(特异性防御机制)。这些细胞以固定或运动细胞的形式吞噬(吞没并随后消化)细胞碎片和病原体。当被病原体或其他机制激活时,巨噬细胞刺激并募集淋巴细胞以及其他免疫细胞从而对损伤作出响应。活化的巨噬细胞与多种疾病和病症的进展有关。活化的巨噬细胞引起大量白细胞浸润并使周围组织中充满炎症介质、促凋亡因子和基质降解蛋白酶。这些作用可以导致炎症,该炎症可以瓦解组织使其达到遭受严重损伤的程度。由巨噬细胞诱导的炎症所导致的组织破坏与退行性疾病、肿瘤、自身免疫病症以及其他病况的发展相关。
诸如亚氯酸盐等氧化剂可使巨噬细胞回到其非活化状态。亚氯酸盐已被用于治疗多种疾病或病况。例如,亚氯酸盐已用于治疗巨噬细胞相关疾病,如肌萎缩性侧索硬化症(ALS)和阿尔茨海默病(AD)。然而,亚氯酸盐对所有患有疾病的患者的治疗效果可能不同。本发明提供了采用亚氯酸盐治疗患有巨噬细胞相关疾病和相关病况的患者亚群以及监测亚氯酸盐治疗的方法。
技术实现要素:
本发明提供了治疗患有巨噬细胞相关疾病的受试者的方法。所述方法可以包括以下步骤:(a)如果患有巨噬细胞相关疾病的受试者具有选自LPS、IL-6、IL-8、IL-18、IFN-g和CRP的一种或多种炎症因子的升高的血浆水平,则选择所述受试者;以及(b)向所述受试者施用治疗有效量的包含亚氯酸盐的药物组合物。
本发明提供了治疗患有巨噬细胞相关疾病的受试者的方法。所述方法可以包括以下步骤:(a)如果患有巨噬细胞相关疾病的受试者具有选自LPS、IL-6、IL-8、IL-18、IFN-g和CRP的一种或多种炎症因子的升高的血浆水平,则选择所述受试者;以及(b)向所述受试者施用治疗有效量的包含亚氯酸盐的药物组合物。
在一个方面,所述一种或多种炎症因子是IL-18。IL-18的血浆水平在所述施用前可以为至少约60pg/ml。所述受试者的IL-18的血浆水平在所述施用后可降低。
在另一个方面,所述受试者可以进一步具有选自LPS、IL-6、IL-8、IFN-g和CRP的一种或多种炎症因子的升高的血浆水平。在一些情况下,所述一种或多种炎症因子是LPS。在另一种情况下,所述受试者可进一步具有选自IL-18、IL-6、IL-8、IFN-g和CRP的一种或多种炎症因子的升高的血浆水平。
在一些情况下,LPS的血浆水平在所述施用前为至少约0.05、0.1、0.15或0.2EU/ml。在一些情况下,LPS的血浆水平在所述施用前为至少约0.05EU/ml。在又一种情况下,LPS的血浆水平可以高于正常水平。所述受试者的LPS的血浆水平在所述施用后可以降低。在一些情况下,所述受试者的LPS的血浆水平在所述施用后可降低至无法检测到的水平。
在一些情况下,所述受试者具有IL-6和IFN-g的升高的血浆水平。在实施如本文所述的任何方法时,IL-6的血浆水平可以为至少约6pg/ml。IFN-g的血浆水平可以为至少约20pg/ml。CRP的血浆水平可以为至少约1000ng/ml。所述受试者可以具有选自LPS、IL-6、IL-8、IL-18、IFN-g和CRP的至少两种炎症因子的升高的血浆水平。
在另一个方面,所述巨噬细胞相关疾病可以选自肌萎缩性侧索硬化症(ALS)、阿尔茨海默病(AD)、帕金森病(PD)和HIV相关神经认知障碍(HAND)。所述巨噬细胞相关疾病可以是肌萎缩性侧索硬化症(ALS)。在一些情况下,所述受试者在所述施用前不到3年内被诊断为患有巨噬细胞相关疾病。在一些情况下,所述受试者在所述施用后至少6个月内没有显示疾病进展。
在一个方面,所述亚氯酸盐可以以至少约1mg/kg体重或至少约2mg/kg体重的量施用。所述组合物可以静脉内施用。所述组合物可以每个月施用至少两次、三次或五次。所述组合物可以施用至少2、3、4、5或6个月。
在实施如本文所述的任何方法时,所述亚氯酸盐的纯度可以大于95%、99%或99.5%。所述包含亚氯酸盐的组合物可以进一步包含pH调节剂。所述组合物可以是与不含所述pH调节剂的相同组合物相比显示出低25%的pH漂移的液体。所述pH调节剂可以是磷酸盐缓冲液。
在一些情况下,所述亚氯酸盐是亚氯酸钠。在一些情况下,所述亚氯酸盐为WF10的形式。
本发明还提供了监测受试者的巨噬细胞相关疾病的炎症进展的方法。所述方法可以包括以下步骤:(a)向所述受试者施用包含亚氯酸盐的药物组合物;(b)测量选自HLA-DR和CD16的至少一种单核细胞活化标志物的血浆水平;(c)将测得的所述单核细胞活化标志物的血浆水平与所述施用步骤前所述受试者的所述单核细胞活化标志物的血浆水平进行比较;以及(d)如果与所述施用前所述单核细胞活化标志物的血浆水平相比所述单核细胞活化标志物的血浆水平降低,则继续向患者施用所述药物组合物。在一些情况下,所述单核细胞活化标志物的血浆水平高于所述施用前的正常水平。在一些情况下,所述单核细胞活化标志物的血浆水平在所述施用后降低。
可以在所述施用前24小时或所述施用后24小时测量至少一种单核细胞活化标志物的血浆水平。所述单核细胞活化标志物可以是HLA-DR。在一些情况下,所述受试者的HLA-DR的血浆水平高于所述施用前的正常水平。在一些情况下,所述受试者的HLA-DR的血浆水平在所述施用后降低。所述方法可以进一步包括测量CD14的血浆水平。在一些情况下,所述受试者的CD14的血浆水平可以高于所述施用前的正常水平。在一些情况下,CD14的血浆水平在所述施用后降低。
所述单核细胞活化标志物可以是CD16。在一些情况下,CD16的血浆水平高于所述施用前的正常水平。在一些情况下,CD16的血浆水平在所述施用后降低。
单核细胞活化标志物的血浆水平可与所述单核细胞相关疾病的进展率相关。在一些情况下,HLA-DR和CD16的升高的血浆水平提高了所述巨噬细胞相关疾病的进展率。施用所述组合物可以降低所述巨噬细胞相关疾病的进展。在一些情况下,在使用ALSFRS-R评分量表的情况下,施用所述组合物将所述巨噬细胞相关疾病的进展降低至少1.0单位/月。在一些情况下,在使用ALSFRS-R评分量表的情况下,所述进展降低至少0.5单位/月。在一些情况下,在使用ALSFRS-R评分量表的情况下,所述患有巨噬细胞相关疾病的受试者具有至少1.0单位/月的进展率。
在另一个方面,所述巨噬细胞相关疾病可以选自肌萎缩性侧索硬化症(ALS)、阿尔茨海默病(AD)、帕金森病(PD)和HIV相关神经认知障碍(HAND)。所述巨噬细胞相关疾病可以是肌萎缩性侧索硬化症(ALS)。
援引并入
本说明书中提到的所有出版物、专利和专利申请均通过引用而并入本文,其程度如同特别地且单独地指出每个单独的出版物、专利或专利申请通过引用而并入。如果通过引用并入的出版物与本说明书之间存在任何不一致,则以本说明书为准。
附图说明
本发明的新颖特征在所附的权利要求书中具体阐述。通过参考以下对利用本发明原理的说明性实施方案加以阐述的详细描述以及附图,将获得对本发明的特征和优点的更好的理解,在这些附图中:
图1示出了临床试验的总体设计。
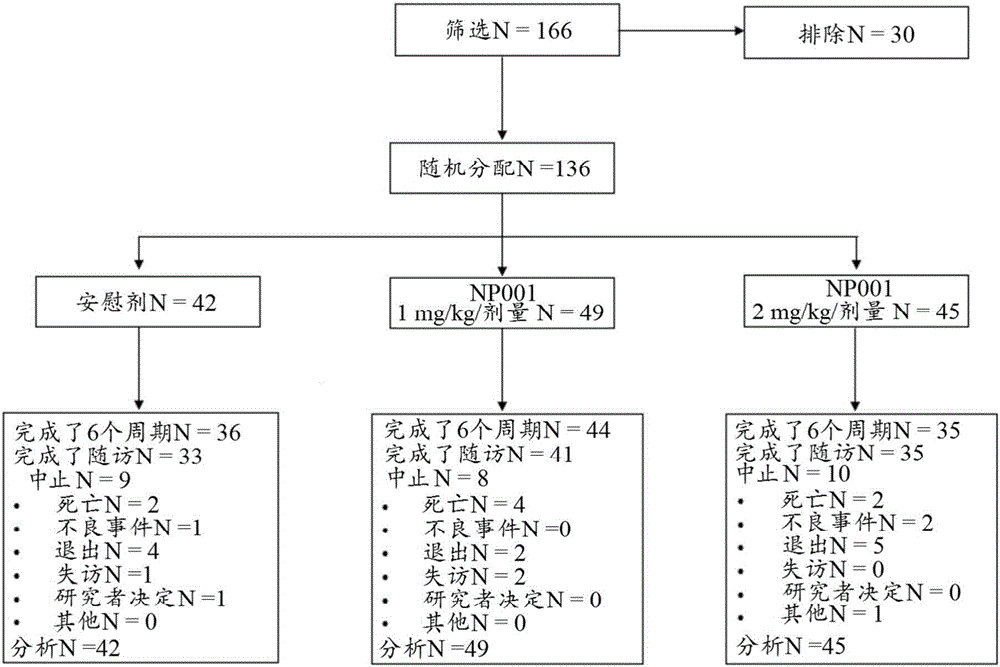
图2描绘了用于评估亚氯酸盐在治疗ALS中的效果的临床研究流程和患者处置的示意图。
图3A示出了在没有(左)和有(右)历史对照的情况下治疗6个月后的ALSFRS-R斜率。
图3B示出了在没有(左)和有(右)历史对照的情况下在第25周的ALSFRS-R评分相对于基线的平均变化。
图3C示出了在基线wrCRP大于或等于基线中值wrCRP的患者中,治疗6个月后的ALSFRS-R斜率。
图4示出了炎症和ALS的工作机理。LPS诱导巨噬细胞活化和NF-kB调节因子的产生。巨噬细胞功能恢复正常后,血浆LPS将会消失。
图5示出了亚氯酸盐在治疗巨噬细胞相关疾病中的工作机理。
图6示出了响应者和无响应者在6个月治疗过程中的ALSFRS-R评分。“响应者”是对亚氯酸钠治疗具有积极响应(respond positively)的受试者亚群。“无响应者”是就ALSFRS-R评分而言对亚氯酸钠治疗没有积极响应的受试者亚群。
图7示出了在6个月的治疗后ALSFRS-R评分相对于基线的变化稳定或有所改善的患者的百分比。
图8是显示响应者与无响应者中炎症血浆因子的归一化基线水平的差异的图表。响应者在基线具有升高的血浆炎症标志物。
图9是显示响应者、安慰剂组和无响应者中炎症血浆因子的归一化基线水平的差异的图表。安慰剂组显示了一致的炎症中间水平。
图10是用于比较每种标志物预测响应者的能力的曲线下面积的ROC曲线。
图11是显示用2mg/kg亚氯酸钠治疗的响应者与无响应者中的炎症血浆因子基线水平的表。
图12示出了响应者、无响应者和安慰剂无进展者在基线和第25周时的一些炎症因子的血浆水平。“安慰剂无进展者”是指在研究持续期间没有表现出疾病进展的安慰剂组的亚群。
图13示出了响应者和安慰剂无进展者在基线和第25周时的IL-18、CRP、IL-8、wrCRP、IFN-g和IL-6的血浆水平。
图14示出了高剂量“响应者”与“无响应者”在基线和6个月治疗期后(第25周)的平均IL-18血浆水平。误差线表示标准差。
图15示出了响应者、无响应者和安慰剂组在基线和第25周时的IL-18水平。
图16是IL-18的对数分布的盒须图(box and whisker plot),显示在基线时的IL-18水平可以区分响应者和无响应者。
图17是显示基线炎症因子血浆基线值相互关系的表。
图18示出了用1mg/kg或2mg/kg亚氯酸盐/NP001治疗的所有患者在基线和6个月治疗期后(第25周)的平均血浆LPS。误差条(error bar)表示标准差。LPS的检测限(LOD)为0.05。
图19示出了安慰剂“响应者”和“无响应者”在基线和6个月治疗期后(第25周)的平均LPS。误差条表示标准差。LPS的检测限(LOD)为0.05。
图20示出了参与研究的每个受试者在基线和第25周时的IL-18血浆水平。
图21指示了在基线处的IL-18血浆水平的截止阈值。
图22示出了在基线处的LPS阳性和阴性患者和ALS疾病进展率。
图23指示ALS LPS阴性患者具有更高的基线ALSFRS-R评分。
图24显示ALS LPS阴性安慰剂患者在6个月内变成LPS阳性。
图25示出了ALS LPS阴性患者的ALSFRS-R评分在6个月内的降低。
图26示出了通过ALS的ALSFRS-R疾病进展率的平均每月变化(每月ALSFRS-R评分降低)评估,基线单核细胞炎症活化相关标志物与历史ALS疾病进展率之间的关系。图26A显示由CD14与HLA-DR共表达所界定的基线单核细胞活化水平与ALS疾病进展率直接相关(r=0.4310,p=0.0138;n=32)。图26B显示在单核细胞的CD16明亚组中表达的CD16的基线水平与ALS疾病进展率之间观察到正相关性(r=0.4499,p=0.0098;n=32)。
图27显示NP001治疗改变了HLA-DR的CD14单核细胞表达,其为基线处单核细胞HLA-DR表达程度的函数。
图28示出了具有升高的基线单核细胞HLA-DR水平的ALS患者与基线单核细胞HLA-DR范围较低的ALS患者之间的NP001治疗反应的比较。
图29示出了具有最高疾病进展率的ALS患者的单核细胞HLA-DR水平的最大变化。
图30示出了NP001诱导的在单核细胞的CD16明亚组中表达的CD16水平相对于基线的按照剂量依赖性方式的变化。
图31示出了接受1.6mg/kg剂量NP001的患者与健康对照相比,单核细胞CD16明亚组中的CD16表达的比较。
具体实施方式
本文使用的术语仅用于描述特定的实施方案的目的,而并非意在限制本发明。除非上下文另有明确说明,否则如本文所用的单数形式“一个”、“一种”和“该/所述”意在也包括复数形式。此外,在术语“包括”、“包含”、“具有”、“含有”、“有”或它们的变型用于具体实施方式和/或权利要求书时,这些术语意在表示与术语“包括”相似地是包含性的。
术语“约”或“大约”意指在如由本领域普通技术人员所确定的特定值的可接受误差范围内,其将部分依赖于如何测量或确定该值,即,测量系统的限制。例如,根据本领域的实践,“约”可以意指在1个标准差内或者大于1个标准差。或者,“约”可以意指给定值的最高20%、最高10%、最高5%或最高1%的范围。或者,特别是对于生物系统或过程而言,该术语可以意指在数值的一个数量级以内,优选在数值的5倍至1/5以内,并且更优选在数值的2倍至1/2以内。在本申请和权利要求书中描述了特定值的地方,除非另有说明,否则应当假定术语“约”意指在该特定值的可接受误差范围内。
如本文所使用的“处理”、“治疗”、“缓解”和“改善”可以互换使用。这些术语是指用于获得有益的或期望的结果的方法,包括但不限于治疗益处和/或预防益处。治疗益处是指消除或改善正在治疗的潜在病症。另外,治疗益处也可以如下实现:消除或改善与潜在病症相关的一种或多种生理症状,从而观察到患者的改善,虽然该患者可能仍然遭受潜在病症的折磨。对于预防益处而言,可将所述组合物施用于处于发展特定疾病的风险下的患者或报告疾病的一种或多种生理症状的患者,即使这种疾病可能还没有确诊。
如本文所使用的“药剂”是指生物学、药学或化学化合物或其他部分。非限制性实例包括简单或复杂的有机或无机分子、肽、蛋白质、寡核苷酸、抗体、抗体衍生物、抗体片段、维生素衍生物、碳水化合物、毒素或化疗化合物。可以合成多种化合物,例如,小分子和寡聚物(如寡肽和寡核苷酸),以及基于各种不同的核心结构的有机合成化合物。此外,各种天然来源可提供供筛选的化合物,如植物或动物提取物等。技术人员可以容易地认识到,对于本发明的药剂的结构性质没有限制。
通常,关于将两个或更多个施用对象,如组分、药剂、物质、材料、组合物和/或类似物,施用于受试者体内的术语“同时施用”、“共施用”或“联合施用”,是指进行施用所采用的剂量和时间间隔使得施用对象一起以低于最低作用量(de minimus quantity)在一定时间间隔内存在于受试者体内或受试者体内的作用部位。该时间间隔可以是任何合适的时间间隔,如适当的数分钟、数小时、数天或数周的间隔。施用对象可以例如作为单个组合物的一部分或例如以其他方式一起施用。施用对象可基本同时施用(例如在彼此间小于或等于约5分钟、约3分钟或约1分钟内)或者在彼此间短时间间隔内(例如在彼此间小于或等于约1小时、30分钟或10分钟内,或在彼此间多于约5分钟到约1小时内)施用。如此施用的施用对象可被认为是基本上同时施用的。本领域普通技术人员将能够确定施用于受试者身体的施用对象的适当的剂量和时间间隔,以使得施用对象以超过最低作用水平(de minimus level)存在于受试者体内和/或以有效浓度存在于受试者体内。当施用对象同时施用于受试者身体时,任何这样的施用对象的有效量可低于该施用对象单独施用时可以使用的有效量。
本文进一步描述的术语“有效量”、“治疗量”或“治疗有效量”,既包含了更少的有效量也包含了常用的有效量,并且实际上还包含了有效引起特定状况、作用和/或反应的任何量。同样,任何这样的同时施用的对象的剂量可低于该对象单独施用时可以使用的剂量。任何这样的施用对象的一种或多种作用可以是累加或协同作用。任何这样的施用对象可以施用超过一次。有效量可以变化,取决于预期的应用(体外或体内)或正在治疗的受试者及疾病状况,例如受试者的体重和年龄、疾病状况的严重程度、施用方式等,这些可以由本领域普通技术人员容易地确定。该术语也适用于在靶细胞中将会诱导特定反应如减少增殖或下调靶蛋白活性的剂量。根据选择的特定化合物、遵循的给药方案、是否与其他化合物联合施用、施用的时机、施用的组织以及携带该化合物的身体递送系统,具体剂量将会不同。
如本文所使用的“治疗效果”包括如上文所述的治疗益处和/或预防益处。预防效果包括延迟或消除疾病或病况的出现,延迟或消除疾病或病况的症状的发作,减慢、中止或逆转疾病或病况的进展,或上述的任何组合。
术语“药学上可接受的盐”是指本领域熟知的来源于各种有机和无机抗衡离子(counter ion)的盐。药学上可接受的酸加成盐可由无机酸和有机酸形成。可得到盐的无机酸包括,例如,盐酸、氢溴酸、硫酸、硝酸、磷酸等。可得到盐的有机酸包括,例如,乙酸、丙酸、羟基乙酸、丙酮酸、草酸、马来酸、丙二酸、琥珀酸、富马酸、酒石酸、柠檬酸、苯甲酸、肉桂酸、扁桃酸、甲磺酸、乙磺酸、对甲苯磺酸、水杨酸等。药学上可接受的碱加成盐可由无机碱和有机碱形成。可得到盐的无机碱包括,例如,钠、钾、锂、铵、钙、镁、铁、锌、铜、锰、铝等。可得到盐的有机碱包括,例如,伯胺、仲胺和叔胺,取代胺,包括天然产生的取代胺,环胺,碱性离子交换树脂等,具体例如异丙胺、三甲胺、二乙胺、三乙胺、三丙胺和乙醇胺。在一些实施方案中,药学上可接受的碱加成盐选自铵盐、钾盐、钠盐、钙盐和镁盐。
“药学上可接受的载体”或“药学上可接受的赋形剂”包括任何和所有的溶剂、分散介质、涂料、抗细菌和抗真菌剂、等渗剂和吸收延迟剂等。这些介质和试剂用于药物活性物质的应用是本领域熟知的。除非任何常规介质或药剂与活性成分不相容,否则考虑将其用于本发明的治疗性组合物。补充性活性成分也可并入该组合物中。
“受试者”是指动物,如哺乳动物,例如人类。本文所述方法可用于人类治疗(临床前)和兽医应用两者。在一些实施方案中,受试者是哺乳动物;而在一些实施方案中,受试者是人类。
术语“体内”是指在受试者的身体内发生的事件。
A.氧化剂
一方面,本发明提供了治疗患有巨噬细胞相关疾病的受试者的方法,所述方法包括向有需要的受试者施用有效量的氧化剂。另一方面,本发明提供了监测针对患有巨噬细胞相关疾病的受试者的氧化剂治疗的方法。该氧化剂可以是亚氯酸盐或包含亚氯酸盐的组合物。
I.亚氯酸盐和其他氧化剂
具有氧化其他物质的能力的物质通常被称为具有氧化性,并且被称为氧化剂、氧化试剂或氧化性试剂,这些名称在本文中可互换使用。氧化剂(也称为氧化试剂、氧化性试剂)也可如下定义:一种易于转移氧原子的化学化合物,或在氧化还原化学反应中得到电子的物质。在这两种情况下,氧化剂在该过程中均被还原。各种常见的氧化剂都含有氧(如KClO4)因而可以被认为是氧的储存形式。或者,术语“氧化剂”也包括其中形式电荷增加(失电子)的任何情况,并且适用于不含氧的物质,其通常含卤素,包括氟(F)、氯(Cl)、溴(Br)、碘(I)和砹(At)等,以及富含这些元素的物质。
可在本发明的方法中使用的常见氧化剂或氧化性试剂包括但不限于:硝酸钾(KNO3)、次氯酸盐和其他次卤酸盐化合物、碘和其他卤素、亚氯酸盐、氯酸盐、高氯酸盐和其他类似的卤素化合物、高锰酸盐、硝酸铈(IV)铵和相关铈(IV)化合物、六价铬化合物(如铬酸和重铬酸以及三氧化铬、氯铬酸吡啶(PCC))以及铬酸盐/重铬酸盐化合物;过氧化物化合物、Tollens试剂、亚砜、过硫酸、臭氧、四氧化锇(OsO4)、硝酸和氧化亚氮(N2O)。氧化剂在生理学有效浓度下可以对单核细胞或巨噬细胞没有毒性。
本发明的氧化剂可以是含有易于转移的氧和卤素原子的化合物,包括但不限于:次氯酸盐和其他次卤酸盐化合物、亚氯酸盐、氯酸盐、高氯酸盐和其他类似卤素化合物以及氯铬酸吡啶(PCC)。如本文所使用的此类化合物被称作活性氧活性卤素化合物。
或者,氧化剂可以是不含氧的物质,其通常含卤素,包括氟(F)、氯(Cl)、溴(Br)、碘(I)和砹(At)。如本文所使用的此类化合物被称为非氧活性卤素化合物。
已经证明许多氧化化合物具有保护和抗炎活性,这可能是因为内源性防御途径的诱导。例如,应激诱导的酶血红素氧合酶1(HO-1)的代谢物,如一氧化碳(CO)和胆绿素,发挥强抗炎作用(Otterbein L E等人.Nat.Med.6(2000)422-428)。HO-1的催化产物,包括氧化剂CO、Fe2+和胆绿素,能够下调炎症反应。已描述了氧化还原反应活性分子硫氧还蛋白的相似的细胞保护特性(Hirota K等人.J.Biol.Chem.274(1999)27891-27897)。亚氯酸盐用于治疗不同的疾病和病症的应用描述于以下文献中:美国专利号4,725,437;美国专利号4,851,222;McGrath等人,Development of WF10,a novel macrophage-regulating agent,Curr Opin Investig Drugs,3(3):365-73(2002年3月);美国专利号6,086,922;美国专利号7,105,183;美国专利号8,029,826;美国专利号8,501,244;美国专利号8,231,856;美国专利号8,252,789;美国专利号8,067,035;以及美国专利申请号13/388,411,所有这些文献均通过引用而整体并入本文。
本文公开了使用亚氯酸盐治疗患有巨噬细胞相关疾病的受试者的组合物和方法。亚氯酸根离子是ClO2-。亚氯酸盐(化合物)是含有该基团的化合物,其中氯的氧化态为+3。亚氯酸盐也被称为亚氯酸的盐。在通常已知的相应的阴离子Cl-、ClO-、ClO2-、ClO3-或ClO4-内,氯可呈现-1、+1、+3、+5或+7的氧化态,并分别被称为氯化物、次氯酸盐、亚氯酸盐、氯酸盐和高氯酸盐。
II.四氯十氧化物(TCDO)和WF10
本发明还提供了使用一种或多种含有亚氯酸盐的药剂的方法。根据本发明,用于亚氯酸盐施用的亚氯酸根离子的来源可以以多种形式提供。例如,亚氯酸根可以作为亚氯酸盐,如碱金属盐(例如,亚氯酸钠、亚氯酸钾等)或亚氯酸盐的混合物进行施用,其中该亚氯酸盐优选为药学上可接受的。另外或者备选地,亚氯酸盐可以作为亚氯酸根离子的基体(matrix)进行施用,如在美国专利号4,507,285中描述的。在一个实施方案中,如在组合物中提供的亚氯酸根离子具有以下通式:
ClO2–x nO2
其中“n”可以是约0.1-0.25的值。这些药剂可以具有在拉曼光谱中在1562cm-1处的O2带和123pm的氧--氧间隔。这些药剂的生产是本领域已知的,参见例如,美国专利号4,507,285。
在一个实施方案中,所述治疗方法包括液体组合物的施用,该液体组合物包含被称为“四氯十氧阴离子复合物”(通常被称为TCDO)的产品的水溶液。TCDO的生产是公知的,参见例如,美国专利号4,507,285的实施例1。在一些实施方案中,可在本发明的用于治疗糖尿病或相关病症的方法中使用的含有亚氯酸盐的药剂包括但不限于:亚氯酸盐如碱金属盐、亚氯酸钠、亚氯酸钾等,亚氯酸盐的基体,亚氯酸根离子的基体,例如通式为ClO2xnO2的合成物,其中“n”可以是约0.1-0.25的值。一个实例是TCDO。一种水性TCDO制剂是WF10。WF10是药物OXO-K993的水性制剂。Oxoferin是同一药物的局部制剂,并作为伤口愈合剂在欧洲和亚洲注册并销售。WF10是OXO-K993的无菌、无热原、10%(w/v)水溶液(没有额外的非活性成分),并且用于静脉内输注。TCDO在分析上被表征为含有4.25%亚氯酸盐、1.9%氯化物、1.5%氯酸盐、0.7%硫酸盐并以钠作为阳离子的溶液。活性成分由亚氯酸根离子含量限定。在一个实施方案中,WF10溶液含有约63mmol/l的亚氯酸盐。
四氯十氧化物(TCDO)是一种用于创伤敷料、免疫调节并作为防辐射剂的含亚氯酸根的药物。虽然TCDO不被视为抗生素,但由于其氧化性质,其可以破坏大多数病原体。然而其用于创伤敷料的主要原因并不是因为其杀菌活性。这种药物被认为是免疫调节剂,即它通过刺激身体的免疫系统而起作用。四氯十氧化物与血红蛋白、肌红蛋白以及过氧化物酶的血红素部分结合,从而形成TCDO-血红素复合物。这继而激活巨噬细胞并加速吞噬过程(吞没存在于创伤表面上的大部分病原体和细胞碎片),从而清洁创伤表面并有助于再生过程。四氯十氧化物还具有促有丝分裂性和趋化性。促有丝分裂刺激导致两种因子,即MDGF(巨噬细胞衍生生长因子)和WAF(创伤血管生成因子)。MDGF使成纤维细胞沉积并合成填充创伤间隙的胶原纤维,WAF有助于形成新的毛细血管,从而进一步加速愈合过程。趋化性刺激作用于肌细胞(肌肉细胞)并使其收缩,从而使创伤边缘更靠近并减小创伤表面。所有这些因子的同时影响加速创伤愈合并几乎无瘢痕形成。
WF10是被配制用于静脉内注射的四氯十氧化物(TCDO)的1:10稀释剂。WF10特异性靶向巨噬细胞。WF10在体内和体外均潜在地调节与疾病相关的免疫应答上调。因此,免疫应答以下调不适当的炎症反应的方式而受到影响(Arzneimittelforschung.2001;51(7):554-62.Schempp H等人)。目前正在研究将WF10用于治疗晚期HIV疾病,以及复发性前列腺癌、晚期放射后膀胱炎、自身免疫疾病和慢性活动性丙型肝炎疾病。WF10以商品名IMMUNOKINE在泰国被批准用于放射后慢性炎性疾病患者,包括膀胱炎、直肠炎和粘膜炎患者。
体内研究已经研究了WF10对单核细胞、巨噬细胞和淋巴细胞,对体液免疫和细胞免疫以及对局部或全身辐射反应的影响(综述于McGrath M S等人,Current Opinion in Investigational Drugs 2002 3(3))。在人创伤愈合模型中,WF10增加了浸润皮肤水泡的巨噬细胞的数目(Hansel M等人.Skin Pharmacol 1988 1:64)。在大鼠中,WF10增加了粒细胞、外周血单核细胞(PBMC)和大颗粒淋巴细胞(LGL)的比例,并在全身X射线照射后刺激红细胞生成(Ivankovic S等人.OXO Study Report 1988年3月;Ivankovic S等人.Radiat Res 1988 115:115-123)。在小鼠中,WF10刺激接受亚致死剂量的J射线照射的造血干细胞的再生(Mason K A等人.Radiat Res 1993 136:229-235)。在其他研究中,WF10显示出对辐射诱导的、危险品诱导的(hemical-induced)和转移性恶性和良性肿瘤的直接抗肿瘤作用(Kempf S R等人.International Symposium on Tissue Repair 1990Thailand;Milas L.OXO Study Report 1991年9月;Kempf S R等人.Radiat Res 1994 139:226-231)。WF10改变了脾和胸腺中辅助性T细胞和抑制性细胞/细胞毒性细胞的比例,并增强了体液和细胞免疫应答(Gillissen G等人.OXO Study Report 1993)。
不受理论的约束,已经提出WF10导致与T细胞增殖相关的诱导型基因的显著抑制,并且在体外引起巨噬细胞中炎症基因表达的可再现的上调,这被认为有助于活化的巨噬细胞中的更高的凋亡率。这些数据以及早期关于WF10抑制T细胞活化的报道(McGrath M S等人.Transplant Proc.1998 30:4200-4202)表明,WF10通过调节巨噬细胞活化而引起T细胞功能的深刻变化。WF10氧/亚氯酸盐基体在与血红素缔合的铁相互作用之前是稳定的,随即通过Michealis-Menten反应和反应性化合物I的中间产生而转化为活性亚氯酸盐分子。亚氯酸盐是被认为介导巨噬细胞中的免疫效应的药物的活性形式。
从1993年到1994年,在44名具有<500个CD4+T细胞/mm的HIV阳性患者中进行了剂量范围的临床研究(Raffanti S P等人.Infection 1998 26:201-206)。该研究确定,当以5天为一个周期施用四个周期(每个周期后停止治疗16天)时,WF10的最大耐受剂量为0.5ml/kg/天。在该剂量下没有观察到明显的不良事件或临床实验室毒性。在四个周期的WF10施用中,血浆CD8+T细胞计数以剂量依赖性方式增加。这项研究表明,在0.5ml/kg的剂量下,WF10与持续的免疫应答,即CD8+T细胞数目的持续升高相关,这与所提出的作用机制是一致的。此外,在1997年进行了单中心I/II期研究,以评估WF10的安全性和对红细胞(RBC)存活动力学、HIV疾病的选择性免疫标志物、巨噬细胞活化和病毒动力学的影响(Herndier B等人.Keystone Symposia on Molecular and Cellular Biology.1998)。来自HIV+患者的响应于WF10治疗的细胞免疫参数的变化汇总于McGrath M S等人.Current Opinion in Investigational Drugs 2002 3(3)的表1中,包括CD3+CD4+细胞的增加、CD3+CD8+细胞的增加、CD3+CD4+CD38-细胞的增加、CD3+CD8+CD38-细胞的增加、CD3+CD8+CD28-细胞的增加、CD3+CD8+CD28+细胞的减少、CD3+CD4+CD38+细胞的减少、所有CD14+细胞的减少以及CD20+HLR-DR+细胞的减少。结果表明WF10减少了抗原呈递,而同时诱导功能受损的巨噬细胞中的吞噬作用。在试验过程中WF10对HIV载量没有影响。没有检测到WF10和安慰剂组之间血液学和血液化学值(包括与溶血特别相关的参数)的显著差异。
适当时,提供亚氯酸根离子来源的药剂可以以游离碱或游离酸形式,即作为游离化合物而不是作为盐来施用。在一些实施方案中,亚氯酸盐制剂含有约150μM亚氯酸盐。
另外,也可以使用所述化合物的任何药学上可接受的盐。药学上可接受的盐是那些保留游离化合物的生物活性并且在生物学或其他方面令人满意的盐。适当时,在本发明中也可以使用所公开的化合物的立体异构体,包括非对映异构体和对映异构体,以及立体异构体的混合物,包括但不限于外消旋混合物。除非明确指出结构的立体化学,否则该结构意在包括所述化合物的所有可能的立体异构体。
如本文所述的氧化化合物或亚氯酸盐可以是WF10。WF10是一种基于亚氯酸根的化合物。与血红素蛋白相互作用后,WF10的亚氯酸根基体获得氧化和氯化特性(Schempp H等人,1999)。已经表明WF10最有可能通过产生生理氧化化合物即氯胺而发挥有效的免疫调节作用。据报道,氯胺具有细胞保护和抗炎活性(Choray M等人.Amino Acids 23(2002)407-413)。
促氧化物质也可以对NFAT类转录因子的转录活性产生直接影响。NFAT的核转位需要钙/钙调蛋白依赖性丝氨酸/苏氨酸磷酸酶即钙调磷酸酶(calcineurin)将其去磷酸化。钙调磷酸酶的磷酸酶活性是氧化还原敏感的。WF10能够抑制抗原受体驱动的淋巴细胞增殖。NFAT调控基因的表达被WF10强烈抑制,并且NFATc的核转位受到抑制。活化T细胞中NFAT调控基因的WF10相关抑制与若干单核细胞相关促炎基因的诱导的协合(in concert)表明,先天骨髓功能的激活伴随适应性增殖性淋巴细胞应答的失活。这种方法代表了靶向氧化还原调节来治疗炎性病症的新方法。在一些实施方案中,可以使用本发明的方法治疗的巨噬细胞相关疾病是炎性疾病。
III.亚氯酸盐纯度和pH
在提交于2006年12月21日,且标题为“Chlorite Formulations,and Methods of Preparation and Use Thereof”的美国专利公开号20070145328中,已经描述了制备亚氯酸盐的方法,该申请通过引用而整体并入本文。这样的制剂适合于各种不同的施用方式,包括但不限于非局部、肠胃外、全身或静脉内施用。
本发明描述了使用在水溶液中配制的亚氯酸盐的组合物和方法,其中亚氯酸盐的纯度大于95%。在一些情况下,亚氯酸盐的纯度可以大于97%、99%、99.5%或99.9%。在一些情况下,亚氯酸盐的纯度可以为至少95%、97%、99%、99.5%或99.9%。如本文所使用,样品中亚氯酸盐的“纯度”计算为亚氯酸盐相对于样品总重量的重量百分比。在确定溶液中亚氯酸盐的纯度时,不包括溶剂(例如水溶液中的水)的重量。可以使用离子色谱法和离子检测器通过相应峰的校准积分来评估纯度;所述峰例如化合物或制剂中的亚氯酸根、氯离子、氯酸根、磷酸根和硫酸根。例如,可商购获得作为工业级、纯度为80%的亚氯酸钠(目录号244155,Sigma-Aldrich)的亚氯酸盐。
或者,提供纯度大于95%、大于96%、大于97%、大于98%、大于99%、大于99.5%或大于99.9%的晶体亚氯酸钠。除了一种或多种药物赋形剂之外,固体药物制剂还包含纯度大于95%、大于96%、大于97%、大于98%、大于99%、大于99.5%或大于99.9%的晶体亚氯酸钠。
用于本发明的亚氯酸盐制剂可以包含少量的氯酸盐、硫酸盐或氯化物。如本文所用的,如果在制剂中每1000重量份的非溶剂分子中包含不超过1份的某分子,则制剂“基本不含”该分子。在某些实施方案中,亚氯酸盐与氯酸盐的重量比大于100∶1.5、大于100∶0.5、大于100∶1或大于100∶0.1。在一个实施方案中,所述组合物基本不含氯酸盐。在另一个实施方案中,亚氯酸盐与氯化物的重量比大于100∶45.5或大于100∶8.5。在一个实施方案中,所述组合物基本不含氯化物。在又一个实施方案中,亚氯酸盐与硫酸盐的重量比大于100:16.4或大于100∶1.6。在一个实施方案中,所述组合物基本不含硫酸盐。
用于本发明的亚氯酸盐制剂的pH可以调节到约7至约11.5之间。在一些实施方案中,使用不将制剂暴露于高局部酸度的pH调节化合物将亚氯酸盐制剂的pH降低到约7至约11.5之间。在一些实施方案中,该pH调节化合物是磷酸二氢钠、磷酸氢二钠或乙酸中的任意一种或多种。
本文还描述了制备亚氯酸盐制剂和药物制剂(包括但不限于本文具体描述的亚氯酸盐制剂)的方法。本文还描述了本文所述的制剂和药物制剂的药剂盒和施用方法。本发明的各种示例性方面和变型描述于“发明内容”及本文其他地方,包括但不限于实施例。还应理解,本发明包括含有、基本上由和/或由如本文所述的一种或多种要素组成的实施方案。
在一些实施方案中,本发明使用包含亚氯酸盐的水性制剂。在一些实施方案中,亚氯酸盐制剂包括水性溶剂以及任选的一种或多种用于亚氯酸盐的其他溶剂。在一些实施方案中,所述制剂包含亚氯酸盐和用于亚氯酸盐的水性溶剂,并且具有约7至约11.5的pH。
用于本文所述制剂的溶剂或溶剂组合可通过本领域已知的多种方法来确定。一个非限制性实例包括(1)用本领域标准方程在理论上估计溶剂溶解度参数值并选择与亚氯酸盐匹配的溶剂;和(2)实验测定亚氯酸盐在溶剂中的饱和溶解度,以及(3)选择具有所需溶解度的一种或多种溶剂,以及(4)选择不降低亚氯酸盐活性,或者不与亚氯酸盐反应或仅与亚氯酸盐有最低程度反应的一种或多种溶剂。在一些实施方案中,本文所述的液体制剂包含多种溶剂。
在一些实施方案中,亚氯酸盐制剂包含水性溶剂。在一些变化方案中,水是水性制剂的主要溶剂。在一些变化方案中,水占水性制剂的溶剂组分的至少约50体积%。在一些变化方案中,水占水性制剂的至少约50体积%。在一些变化方案中,水占溶剂组分的以下任意体积百分比:约50%至约60%、约60%至约70%、约70%至约80%、约80%至约90%、约90%至约99%、至少约50%、至少约60%、至少约70%、至少约80%、至少约90%、或至少约95%、约50%、约60%、约70%、约80%、约90%或约95%。在一些变化方案中,水占水性制剂的以下任意体积百分比:约50%至约60%、约60%至约70%、约70%至约80%、约80%至约90%、约90%至约99%、至少约50%、至少约60%、至少约70%、至少约80%、至少约90或至少约95%。在一些变化方案中,水占水性制剂的至少约95体积%。在一些变化方案中,水占水性制剂的约80体积%至约90体积%。在一些变化方案中,水占水性制剂的约90体积%至约99体积%。
所述制剂可具有不同的亚氯酸盐浓度。在一些实施方案中,所述制剂中亚氯酸盐的浓度很高,随后在施用前稀释成较低浓度形式。在一些实施方案中,将本文所述的制剂稀释约、至少约或小于约2.5x、约5x、约7.5x、约10x、约20x、约25x、约50x、约100x、约200x、约250x、约300x、约500x或约1000x。在一些实施方案中,将本文所述的制剂稀释约2x至约10x、约10x至约50x、约50x至约100x、约100x至约500x、或约500x至约1000x。在一些实施方案中,将如本文所述的制剂稀释约2x至约10x。在一些实施方案中,将如本文所述的制剂稀释约10x至约50x。在一些实施方案中,将如本文所述的制剂稀释约7.5x。在一些实施方案中,将如本文所述的制剂稀释约25x。在一些实施方案中,将如本文所述的制剂稀释约200x。
在一些实施方案中,本文所述制剂中亚氯酸盐的浓度为约1μM至约1.5M。在另一个实施方案中,本文所述制剂中亚氯酸盐的浓度为以下的任何浓度:约1M至约1.5M;约1μM至约100mM;约10μM至约100mM;约0.1mM至约10mM;约0.1mM至约500mM;约0.1mM至约200mM;约1mM至约100mM;约0.1mM至约5mM;约50mM至约100mM;约55mM至约70mM;约60mM至约65mM;约100mM至约500mM;约200mM至约400mM;约300mM至约700mM;约1mM;约1.5mM;约2mM;约2.5mM;约3mM;约3.5mM;约4mM;约5mM;约10mM;约20mM;约30mM;约40mM;约50mM;约60mM;约62mM;约65mM;约70mM;约80mM;约90mM;约100mM;至少约0.1mM;至少约1mM;至少约2mM;至少约5mM;至少约10mM;至少约20mM;至少约30mM;至少约40mM;至少约50mM;至少约60mM;至少约70mM;至少约80mM;至少约90mM;或至少约100mM。在优选的实施方案中,本文所述制剂中亚氯酸盐的浓度为约或至少约60mM。
在一些实施方案中,本文所述制剂中氯酸盐的浓度为约50mM至约100mM。在一些实施方案中,本文所述制剂中氯酸盐的浓度为约55mM至约75mM。在一些实施方案中,本文所述制剂中氯酸盐的浓度为约0.1mM至约10mM。在一些实施方案中,本文所述制剂中氯酸盐的浓度为约1mM至约5mM。
在一些实施方案中,亚氯酸盐制剂具有不大于约12.0的pH。在一些实施方案中,制剂的pH为以下的任意值:不大于约11.5、约11.0、约10.5、约10.0、约9.5、约9.0、约8.5、约8.0、约7.5、约7.0、约6.5或约6.0。在一些实施方案中,制剂的pH不大于约11.5。在一些实施方案中,制剂的pH不大于约10.5。在一些实施方案中,制剂的pH不大于约8.5。在一些实施方案中,制剂的pH不大于约7.5。在一些实施方案中,制剂的pH为以下的任意一个或多个:约7至约12;约7至约11.5;约7至约10.5;约7至约10;约7至约9.5;约7至约9.0;约7至约8.5;约7至约8.0;约7至约7.5;约7.5至约8;约7.5至约8.5;约7至约8;约8至约9;约7.0至约8.5;约8至约8.5;约8.5至约9;约7.1至约7.7;约7.2至约7.6;约7.3至约7.4;约7.0;约7.1;约7.2;约7.3;约7.4;约7.5;约7.6;约7.7;约7.8;约7.9;约8.0;约8.1;约8.2;约8.3;约8.4;约8.5;约8.6;约8.7;约8.8;或约8.9。在一些实施方案中,亚氯酸盐制剂具有约7.0至约9.0的pH。在一些实施方案中,亚氯酸盐制剂具有约7.0至约8.5的pH。在一些实施方案中,亚氯酸盐制剂具有约6.0至约8.5的pH。在一些实施方案中,亚氯酸盐制剂具有约7.0至约8.0的pH。在一些实施方案中,亚氯酸盐制剂具有约7.4的pH。亚氯酸盐制剂可以具有处于生理水平的pH。
在一些实施方案中,亚氯酸盐制剂具有如上所述的pH,并且被配制用于肠胃外、全身或静脉内施用中的任意一种或多种。在一些实施方案中,亚氯酸盐制剂具有如上所述的pH,并且具有如本文所述的亚氯酸盐纯度百分比。
在一些实施方案中,本文所述的制剂具有如上所述的pH,并且具有如本文所述的亚氯酸盐浓度。在一些实施方案中,本文所述的水性制剂具有以下的pH:约7至约11.5、或约7.0至约10、或约7.0至约9.0、或约7.0至约8.5、或约7.1至约7.7,并且具有约1至约100mM的亚氯酸盐浓度。在一些实施方案中,本文所述的水性制剂具有以下的pH:约7至约11.5、或约7.0至约10、或约7.0至约9.0、或约7.0至约8.5、或约7.1至约7.7,并且具有约1至约5mM的亚氯酸盐浓度。在一些实施方案中,本文所述的水性制剂具有以下的pH:约7至约11.5、或约7.0至约10、或约7.0至约9.0、或约7.0至约8.5、或约7.1至约7.7,并且具有约50至约80mM的亚氯酸盐浓度。
在一些实施方案中,本文所述的水性制剂具有以下的pH:约7至约11.5、或约7.0至约10、或约7.0至约9.0、或约7.0至约8.5、或约7.1至约7.7,其中使用pH调节剂来调节pH,该pH调节剂是磷酸盐或乙酸中的任意一种或多种。
在一些实施方案中,本文所述的制剂在以下的任意时间段内就pH或亚氯酸盐降解中的一种或多种而言是稳定的:至少约1天、至少约2天、至少约3天、至少约4天、至少约5天、至少约6天、至少约1周、至少约2周、至少约3周、至少约4周、至少约5周、至少约6周、至少约7周、至少约8周、至少约1个月、至少约2个月、至少约3个月、至少约4个月、至少约5个月或至少约6个月。在一些实施方案中,本文所述的制剂在至少约1周的任意时间段内就pH或亚氯酸盐降解中的一种或多种而言是稳定的。在一些实施方案中,所述制剂在至少约1个月的任意时间段内就pH或亚氯酸盐降解中的一种或多种而言是稳定的。在一些实施方案中,本文所述的制剂在室温、冷藏条件或大约4℃中的一种或多种条件下就pH或亚氯酸盐降解中的一种或多种而言是稳定的。在一些实施方案中,本文所述的制剂在减少光或储存在限制制剂所经受的光的量的容器中的条件下就pH或亚氯酸盐降解中的一种或多种而言是稳定的。在一些实施方案中,本文所述的制剂在暗处储存时就pH或亚氯酸盐降解中的一种或多种而言是稳定的。如本文所使用的稳定pH的实例意指相对于最初制备的制剂的pH,该制剂的pH变化小于以下的任意值:约0.1、约0.2、约0.3、约0.4、约0.5、约0.6、约0.7、约0.8、约0.9或约1。在一些实施方案中,相对于最初制备的制剂的pH,该制剂的pH变化小于约0.2。可以使用例如pH计来测量pH。稳定的亚氯酸盐制剂的实例包括相对于最初制备的制剂中存在的亚氯酸根的量,少于以下任意比例的亚氯酸根降解成非亚氯酸根离子的那些亚氯酸盐制剂:约0.1%、少于约0.2%、少于约0.3%、少于约0.4%、少于约0.5%、少于约0.6%、少于约0.7%、少于约0.8%、少于约0.9%、少于约1%、少于约2%、少于约3%、少于约4%、少于约5%、少于约6%、少于约7%、少于约8%、少于约9%或少于约10%。在一些实施方案中,相对于最初制备的制剂中存在的亚氯酸盐的量,少于约2%的亚氯酸盐降解成非亚氯酸盐化合物。在一些实施方案中,相对于最初制备的制剂中存在的亚氯酸盐的量,少于约0.5%的亚氯酸盐降解成非亚氯酸盐化合物。可以例如使用气相色谱(GC)、质谱或本领域技术人员已知的其他方法测量非亚氯酸盐组分的存在。
在一些实施方案中,本文所述的亚氯酸盐制剂包含不大于约5重量%的其他市售制剂中的有害非亚氯酸盐组分。在一些实施方案中,本文所述的亚氯酸盐制剂包含以下任意重量百分比的其他市售制剂中的有害非亚氯酸盐组分:不大于约4%、约3%、约2%、约1%、约0.5%、约0.3%、约0.25%、约0.2%、约0.1%、约0.05%或约0.02%。在一些实施方案中,本文所述的亚氯酸盐制剂包含不大于约4重量%的任意比例的其他市售制剂中的有害非亚氯酸盐组分。在一些实施方案中,本文所述的亚氯酸盐制剂包含不大于约2重量%的任意比例的其他市售制剂中的有害非亚氯酸盐组分。在一些实施方案中,本文所述的亚氯酸盐制剂包含不大于约0.5重量%的任意比例的其他市售制剂中的有害非亚氯酸盐组分。在一些实施方案中,本文所述的亚氯酸盐制剂包含不大于约0.05重量%的任意比例的其他市售制剂中的有害非亚氯酸盐组分。在一些实施方案中,本文所述的亚氯酸盐制剂基本不含其他市售制剂中的有害非亚氯酸盐成分。检测非亚氯酸盐组分的方法的非限制性实例包括:HPLC;SPCS,例如使用以3.6mM碳酸钠作为流动相的Novosep A2柱,5μ,250×4.0mm,流速0.8mL/min;DS-Plus抑制剂,例如使用以3.6mM碳酸钠作为流动相的Novosep A2柱,5μ,250×4.0mm,流速0.8mL/min;使用2.1mM NaHCO3/1.6mM Na2CO3作为流动相的Allsep A-2阴离子柱,100×4.6mm,流速2.0mL/min;使用在10%甲醇中的2.8mM NaHCO3:2.2mM Na2CO3作为流动相的阴离子HC柱,150×4.6mm,流速1.4mL/min;或使用2.1mM NaHCO3/1.6mM Na2CO3作为流动相的Allsep A-2阴离子柱,5μ,100×4.6mm,流速1.0mL/min。例如,详细参见Alltech Associates公司的Grace Davison系列产品和产品信息。在一些实施方案中,本文所述的制剂包含以与配制成WF10或其稀释剂的亚氯酸根相等的重量/体积百分比存在的不大于约10%、约20%、约30%、约40%、约50%、约60%、约70%、约75%、约80%、约85%、约90%或约95%的量的氯酸根离子和硫酸根离子中的成员或氯酸根离子和硫酸根离子中的任意一种或多种。也就是说,在一些实施方案中,当如本文所述的非WF10制剂包含一定w/v百分比的亚氯酸盐时,这样的制剂具有不大于约所述百分比的量的WF10或其稀释剂中的一种或多种特定非亚氯酸盐组分,其中所述WF10或其稀释剂包含与所制备的非WF10制剂相同的亚氯酸盐w/v百分比。在一些实施方案中,本文所述的制剂包含以与配制成WF10的亚氯酸根相等的重量/体积百分比存在的不大于约75%的量的氯酸根离子和硫酸根离子中的成员或氯酸根离子和硫酸根离子中的任意一种或多种。在一些实施方案中,本文所述的制剂包含以与配制成WF10的亚氯酸根相等的重量/体积百分比存在的不大于约85%的量的氯酸根离子和硫酸根离子中的成员或氯酸根离子和硫酸根离子中的任意一种或多种。在一些实施方案中,本文所述的制剂包含以与配制成WF10的亚氯酸根相等的重量/体积百分比存在的不大于约50%的量的氯酸根离子和硫酸根离子中的成员或氯酸根离子和硫酸根离子中的任意一种或多种。
从WF10的产品插页可以理解,所报道的WF10包括100∶35.7(4.25%∶1.5%)的亚氯酸盐与氯酸盐之比,100∶45.5(4.25%∶1.9%)的亚氯酸盐与氯化物之比,以及100∶16.4(4.25%∶0.7%)的亚氯酸盐与硫酸盐之比。
有害非亚氯酸盐组分的实例包括向生理系统施用时引起不良反应的非亚氯酸盐组分。在一些变化方案中,有害非亚氯酸盐组分与本领域已知的一种或多种体外或体内测定中的一种或多种毒性指标相关,或与向生理系统施用时的一种或多种毒性指标相关,该生理系统包括但不限于受试者,包括但不限于人类受试者。有害非亚氯酸盐组分包括但不限于硫酸盐、二氧化氯、氯酸盐和硼酸盐。在一些实施方案中,本文所述的亚氯酸盐制剂基本不含WF10中的有害非亚氯酸盐成分。在一些变化方案中,本文所述的亚氯酸盐制剂基本不含硫酸根和氯酸根离子。
在一些实施方案中,本文所述的亚氯酸盐制剂包含少于约1.9%的氯离子。在一些实施方案中,亚氯酸盐制剂包含以下任意重量百分比的氯离子:少于约1.9%;少于约1.8%;少于约1.5%;少于约1.0%;少于约0.5%;少于约0.3%;少于约0.1%;少于约0.05%;少于约0.01%;少于约0.001%;约0.001%至约0.1%;约0.1%至约0.5%;约0.5%至约1.0%;约1.0%至约1.5%;或约1.5%至约1.8%。在一些实施方案中,亚氯酸盐制剂包含少于约0.5重量%的氯离子。在一些实施方案中,亚氯酸盐制剂包含少于约0.24重量%的氯离子。在一些实施方案中,亚氯酸盐制剂包含少于约0.2重量%的氯离子。在一些实施方案中,亚氯酸盐制剂包含少于约0.1重量%的氯离子。在一些实施方案中,亚氯酸盐制剂基本不含氯离子。在一些实施方案中,氯离子的水平低于采用HPLC的检测水平。
在一些实施方案中,亚氯酸盐制剂包含少于约1.5%的氯酸根离子。在一些实施方案中,亚氯酸盐制剂包含以下任意比例的氯酸根离子:少于约1.4%;少于约1.3%;少于约1.0%;少于约0.5%;少于约0.3%;少于约0.1%;少于约0.01%;少于约0.001%;约0.001%至约0.1%;约0.001%至约0.01%;约0.01%至约0.1%;约0.1%至约0.5%;约0.5%至约1.0%;或约1.0%至约1.4%。在一些实施方案中,亚氯酸盐制剂基本不含氯酸根离子。在一些实施方案中,亚氯酸盐制剂包含少于约0.5重量%的氯酸根离子。在一些变化方案中,亚氯酸盐制剂基本不含氯酸根离子。在一些实施方案中,亚氯酸盐制剂包含少于约0.19重量%的氯酸根离子。在一些实施方案中,亚氯酸盐制剂包含少于约0.1重量%的氯酸根离子。在一些实施方案中,氯酸根离子的水平低于采用HPLC的检测水平。
在一些实施方案中,亚氯酸盐制剂包含少于约0.7%的硫酸根离子。在一些实施方案中,亚氯酸盐制剂包含以下任意比例的硫酸根离子:少于约0.65%;少于约0.6%;少于约0.5%;少于约0.4%;少于约0.3%;少于约0.2%;少于约0.1%;少于约0.08%;少于约0.07%;少于约0.06%;少于约0.05%;少于约0.005%;少于约0.0005%;约0.001%至约0.1%;约0.01%至约0.1%;约0.01%至约0.5%;约0.06%至约0.08%;或约0.5%至约0.65%。在一些实施方案中,亚氯酸盐制剂包含约0.5%至约0.65%的硫酸根离子。在一些实施方案中,亚氯酸盐制剂基本不含硫酸根离子。在一些实施方案中,亚氯酸盐制剂包含少于约0.5重量%的硫酸根离子。在一些实施方案中,亚氯酸盐制剂基本不含硫酸根离子。在一些实施方案中,亚氯酸盐制剂包含少于约0.08重量%的硫酸根离子。在一些实施方案中,硫酸根离子的水平低于采用HPLC的检测水平。
在一些实施方案中,本文所述的亚氯酸盐制剂包含磷酸根离子。在一些实施方案中,本文所述的亚氯酸盐制剂包含钠离子。在一些实施方案中,亚氯酸盐制剂包含亚氯酸根、水性溶剂、钠和磷酸根离子。在一些变化方案中,水性溶剂基本由水组成。在一些实施方案中,亚氯酸盐制剂基本由亚氯酸盐、水、钠和磷酸盐组成,并且基本不含氯酸盐。在一些实施方案中,亚氯酸盐制剂基本由亚氯酸盐、水、钠和磷酸盐组成,并且基本不含氯酸盐,并进一步包含药学上可接受的稀释剂。在一些实施方案中,钠和磷酸盐全部或部分以磷酸二氢钠或磷酸氢二钠提供。在一些实施方案中,药学上可接受的稀释剂是盐溶液。
在一些实施方案中,本文所述的亚氯酸盐制剂包含不大于约10重量%的市售工业级亚氯酸盐中存在的副产物或杂质。市售工业级亚氯酸盐中存在的副产物或杂质的非限制性实例包括氯酸盐、硫酸盐、二氧化氯、氯化物、碳酸氢钠和碳酸钠。在一些实施方案中,本文所述的亚氯酸盐制剂包含不大于约以下任意重量百分比的市售工业级亚氯酸盐中存在的一种或多种降解产物或杂质(包括但不限于氯酸盐或硫酸盐中的一种或多种):15%、约12%、约10%、约9%、约8%、约7%、约6%、约5%、约4%、约3%、约2%、约1%、约0.5%、约0.3%、约0.1%、约0.1%至约5%、约5%至约10%、或约10%至约15%。在一些实施方案中,本文所述的亚氯酸盐制剂包含不大于约0.5重量%的市售工业级亚氯酸盐中存在的降解产物或杂质,包括但不限于氯酸盐或硫酸盐中的一种或多种。在一些实施方案中,本文所述的亚氯酸盐制剂包含不大于约5重量%的市售工业级亚氯酸盐中存在的降解产物或杂质,包括但不限于氯酸盐或硫酸盐中的一种或多种。在一些实施方案中,本文所述的亚氯酸盐制剂基本不含市售工业级亚氯酸盐中存在的降解产物或杂质,包括但不限于氯酸盐或硫酸盐。
在一些实施方案中,当通过本文所述的至少一种施用途径(包括但不限于非局部、全身、肠胃外或静脉内施用)进行施用时,在相同的亚氯酸盐浓度下,本文所述的制剂比先前报道的亚氯酸盐制剂对受试者的毒性更小。在一些实施方案中,使用体内或体外毒性测定(包括众所周知的毒性测定)来分析亚氯酸盐制剂的毒性。在一些实施方案中,使用非特异性体外毒性测定来分析亚氯酸盐制剂。
在另一变化方案中,根据与施用其他市售亚氯酸盐制剂相比施用本文所述的亚氯酸盐制剂后受试者不同的毒性反应指标来测量毒性。在一些变化方案中,相对于全身施用配制为WF10的亚氯酸盐来测量毒性。在另一变化方案中,相对于将配制为WF10的亚氯酸盐静脉内施用于受试者来测量毒性。在一些变化方案中,向哺乳动物受试者(包括但不限于人类受试者)施用之后测量毒性。在一些变化方案中,毒性被测量为对亚氯酸盐制剂所暴露的表面的一种或多种刺激,包括但不限于胃肠道、恶心、呕吐、腹泻、腹痛、溶血、高铁血红蛋白症、发绀、无尿、昏迷、抽搐、肝损伤、肾损伤、食欲不振或体重减轻。在一些变化方案中,毒性被测量为虚弱、注射部位疼痛、头痛、鼻炎或腹泻中的一种或多种。参见例如,McGrath M S,"Development of WF10,A Novel Macrophage-Regulating Agent,"Curr Opin Investig Drugs,3(3):365-73(2002年3月),该文献通过引用而全文并入本文。在另一变化方案中,毒性被测量为贫血。参见例如,Kempf等人,"Comparative Study on the Effects of Chlorite Oxygen Reaction Product TCDO(Tetrachlorodecaoxygen)and Sodium Chlorite Solution(NaClO2)With Equimolar Chlorite Content on Bone Marrow and Peripheral Blood of BDIX Rats,"Drugs Under Experimental and Clinical Research,19(4):165-1(1993)。在一些变化方案中,毒性被测量为虚弱。在一些变化方案中,毒性被测量为注射部位反应。在一些变化方案中,毒性被测量为注射部位疼痛。
IV.调节对pH敏感的制剂的pH的方法
可以使用不同的方法来调节包含亚氯酸盐的制剂和药物制剂的pH。目的在于使本文所述的方法可用于制备本文所述的用于本发明的制剂或药物制剂。然而,本文所述的制剂和药物制剂也可以通过其他方法进行制备,并且本文所述的制剂和药物制剂不限于通过本文所述的方法制备的那些。
一些化合物或制剂对高局部酸度或碱度敏感,需要适当的方法来调节这些化合物或制剂的pH。优选的pH调节剂或pH调节化合物是pKa为约4至约9、约5至约9、或约5至约8、或约6至约7.5的弱酸或弱碱。实例包括但不限于本领域熟知的pKa为约4至约9的磷酸盐缓冲液,例如酸式磷酸盐或磷酸二氢钠和/或磷酸氢二钠以及低级链烷酸,例如乙酸或丙酸。在一些实施方案中,使用不将制剂暴露于酸度(包括但不限于在pH调节化合物周围区域中的高局部酸度)的pH调节化合物将对酸度敏感的制剂的pH降低至约7至约11.5之间。在一些实施方案中,使用不将制剂暴露于酸度(包括但不限于在pH调节化合物周围区域中的高局部酸度)的pH调节化合物将对酸度敏感的制剂的pH降低至约7至约10之间。在一些实施方案中,使用不将制剂暴露于酸度(包括但不限于在pH调节化合物周围区域中的高局部酸度)的pH调节化合物将对酸度敏感的制剂的pH降低至约7至约9.5之间。在一些实施方案中,使用不将制剂暴露于酸度(包括但不限于在pH调节化合物周围区域中的高局部酸度)的pH调节化合物将对酸度敏感的制剂的pH降低至约7至约9.0之间。在一些实施方案中,使用不将制剂暴露于酸度(包括但不限于在pH调节化合物周围区域中的高局部酸度)的pH调节化合物将对酸度敏感的制剂的pH降低至约7至约8.5之间。在一些实施方案中,使用不将制剂暴露于酸度(包括但不限于在pH调节化合物周围区域中的高局部酸度)的pH调节化合物将对酸度敏感的制剂的pH降低至约7.1至约7.7之间。
如本文所使用的“高局部酸度”是指相对于例如使用pH计测量的溶液的总酸度,亚氯酸盐分子局部的一个或多个分子的pKa。为了确定pH调节剂是否将使亚氯酸盐经受高局部酸度,可以使用例如,the CRC Handbook of Chemistry and Physics(第86版,David R.Lide编著,CRC Press,2005)来鉴定pH调节剂的pKa。
降低亚氯酸盐制剂的pH一直充满挑战,因为许多pH调节剂使化合物或制剂暴露于pH调节化合物分子的局部区域中的高酸度。在高局部酸度存在下会产生一定量的非亚氯酸盐化合物,如氯酸盐和/或二氧化氯。参见,例如,Ullmann's Encyclopedia of Industrial Chemistry,Vol.A6,Wolfgang Gerhartz编著,第5版(1986),该文献通过引用而本文并入本文。这样的降解产物在用于肠胃外或全身施用于生理系统的制剂中可能是不希望的,例如因为它们在生理系统中并非是无活性的。一些这样的降解产物导致毒性,包括但不限于多种毒性,包括但不限于本文所述的非特异性毒性。
除非上下文清楚说明,否则本文所述的任何制剂或药物制剂的pH均可使用本文所述的方法进行调节。
在一些变化方案中,包括但不限于亚氯酸盐的治疗剂的活性通过暴露于高局部酸度而减弱。如本文所使用的“减弱的活性”是指治疗剂的活性在性质上或数量上低于治疗剂暴露于高局部酸度之前的活性。作为一个实例,改变的活性(即在性质上或数量上低于治疗剂暴露于高局部酸度之前的活性)将具有较低的创伤愈合功效,或在治疗本文所述的一种或多种疾病或病况方面具有较低的功效。在一些变化方案中,改变的活性比暴露于高局部酸度之前治疗剂的活性低以下任意比例:至少约3%、至少约5%、至少约10%、至少约15%、至少约20%或至少约25%。在一些变化方案中,改变的活性比暴露于高局部酸度之前治疗剂的活性低至少约5%。
在一些实施方案中,将亚氯酸盐制剂的pH调节至制剂部分或本文别处所述的任意一种或多种pH水平。在一些实施方案中,所述亚氯酸盐制剂的pH为约7至约11.5。在一些实施方案中,所述方法包括使用不将亚氯酸盐暴露于高局部酸度的pH调节剂,将含有亚氯酸盐的制剂的pH降低到以下任意值:约7至约11;约7至约10.5;约7至约10;约7至约9.5;约7至约9;约7至约8.5;约7至约8.0;约7至约7.5;约7.5至约8;约7.5至约8.5;约7至约8;约7.1至约7.7;约7.2至约7.6;约7.3至约7.5;约8至约9;约8至约8.5;约8.5至约9;约7.0;约7.1;约7.2;约7.3;约7.4;约7.5;约7.6;约7.7;约7.8;约7.9;约8.0;约8.1;约8.2;约8.3;约8.4;约8.5;约8.6;约8.7;约8.8;或约8.9。在一些实施方案中,所述方法包括将含有亚氯酸盐的制剂的pH降低到约7至约8.5。在一些实施方案中,所述方法包括将含有亚氯酸盐的制剂的pH降低到约7至约8.0。在一些实施方案中,所述方法包括将含有亚氯酸盐的制剂的pH降低到约7.1至约7.7。在一些实施方案中,所述方法包括将含有亚氯酸盐的制剂的pH降低到约7.4。
在一个非限制性实例中,使用pH调节剂调节含有亚氯酸盐的混合物的pH,当暴露于含有亚氯酸盐的混合物时,该pH调节剂不使亚氯酸盐经受低于7的局部pH。在一些实施方案中,该pH调节剂是磷酸二氢钠、磷酸氢二钠或它们的混合物。在一些实施方案中,磷酸二氢钠和/或磷酸氢二钠作为固体或在溶液中使用。在一些实施方案中,该pH调节剂是乙酸。
在一些实施方案中,通过将亚氯酸盐或含有亚氯酸盐的水性混合物添加到含有缓冲液的溶液中来调节亚氯酸盐的pH。在一些实施方案中,通过将亚氯酸盐或含有亚氯酸盐的水性混合物添加到磷酸盐缓冲液的水溶液中来调节亚氯酸盐的pH。
在一些变化方案中,使用一种或多种pH调节剂来调节亚氯酸盐溶液或混合物的pH,并分析所得溶液或混合物中亚氯酸盐降解产物的存在,包括但不限于由高局部酸度产生的降解产物的存在。在一些变化方案中,使用诸如乙酸、磷酸二氢钠和/或磷酸氢二钠等pH调节剂来调节亚氯酸盐溶液或混合物的pH,并分析所得溶液或混合物中氯酸盐或二氧化氯的存在。
在一些实施方案中,使用例如HPLC、质谱等熟知的分析方法来分析所得溶液或混合物中的降解产物。在一些实施方案中,使用毒性测定(包括众所周知的毒性测定)来分析所得溶液或混合物中的降解产物。在一些实施方案中,使用非特异性毒性测定来分析所得溶液或混合物中的杂质。
在一些实施方案中,在亚氯酸盐纯化步骤后调节亚氯酸盐制剂的pH。在一些实施方案中,将亚氯酸盐制剂的pH调节到约7至约11.5,而不产生由于高局部酸度而产生的亚氯酸盐降解产物。在一些实施方案中,将亚氯酸盐制剂的pH调节到约7至约8.0,而不产生由于高局部酸度而产生的亚氯酸盐降解产物。在一些实施方案中,将亚氯酸盐制剂的pH调节到以下任意值:约7至约11、约7至约10.5、约7至约10、约7至约9.5、约7至约9、约7至约8.5、约7至约8、约7至约7.5、约7.5至约8、约7.5至约8.5、约7至约8、约8至约9、约8至约8.5、或约8.5至约9,而不产生由于高局部酸度而产生的亚氯酸盐降解产物。
V.药物制剂
除非上下文另有明确说明,否则本文所述的任何制剂可用于本文所述的任何药物制剂。在优选的实施方案中,药物组合物可以包含:(a)亚氯酸盐;以及(b)药学上可接受的赋形剂。该药物组合物可以进一步包含pH调节剂。在一些实施方案中,该pH调节剂包含磷酸二氢钠和/或磷酸氢二钠。该pH调节剂可以包含磷酸盐缓冲液。所述组合物的pH可以在约7.1至约7.7之间,例如7.4。所述制剂可具有低水平的有害氯酸盐(例如,亚氯酸盐:氯酸盐的重量比可以大于100:1.5),或基本不含氯酸盐。此类制剂可以配制成静脉内施用。
本文所述的药物制剂可适于向受试者施用。“适于向受试者施用”是指当从新打开的瓶中获得药物制剂并通过期望途径施用时,该药物制剂造成的有害副作用不大于临床可接受的水平。
本文所述的制剂或药物制剂可以进一步包含盐溶液。如本文所使用的盐溶液是指具有生理上可接受水平的氯化钠的生理上可接受的溶液。在一些实施方案中,该盐溶液是等渗的。
用于本发明的亚氯酸盐制剂可以是包含一种或多种药学上可接受的赋形剂的药学上可接受的亚氯酸盐制剂。如本文所使用的赋形剂是指药物制剂中的任何非亚氯酸盐、非水或非盐成分。赋形剂包括但不限于技术人员已知并在本文进一步描述的载体、佐剂、稀释剂、稳定剂、润湿剂、乳化剂、缓冲剂、防腐剂、调味剂、非活性成分、凝胶制剂、可侵蚀和不可侵蚀的聚合物、微球、脂质体等,包括前述的组合。在一些实施方案中,赋形剂的重量相对于制剂或药物制剂的总体积的百分比不大于以下的任意百分比:约10%、约9%、约8%、约7%、约6%、约5%、约4%、约3%、约2%、约1%、约0.5%、约0.4%、约0.3%、约0.2%、约0.1%或约0.05%。在一些实施方案中,赋形剂的重量相对于制剂或药物制剂的总体积的百分比不大于约1%。在一些实施方案中,赋形剂的重量相对于制剂或药物制剂的总体积的百分比不大于约3%。
以下是制药领域中常用的赋形剂的非限制性且非穷尽式列表。这些赋形剂通常用于不同类型的制剂,包括那些配制用于静脉内、口服、肌内或肠胃外施用的制剂。鉴于亚氯酸盐的反应性,可能以下所列的一些赋形剂不适于给定的药物制剂。特定赋形剂是否不适用于给定的药物制剂可能依赖于添加到药物制剂中的赋形剂的量。将任何赋形剂中的一种或多种(包括但不限于本文所述的赋形剂)添加到亚氯酸盐的药物制剂之前,重要的是考虑赋形剂与亚氯酸盐的反应性。常用作赋形剂的一些有机分子与亚氯酸盐反应使得赋形剂变化,包括但不限于相比赋形剂暴露于亚氯酸盐之前导致药物制剂的毒性增加的变化。在一些实施方案中,本文所述的药物制剂包含一种或多种不与亚氯酸盐反应的药学上可接受的赋形剂。优选地,本文所述的药物制剂包含一种或多种药学上可接受的赋形剂,相对于暴露于赋形剂之前,该赋形剂不降低药物制剂的治疗效果。
本文所述的亚氯酸盐制剂可以包含一种或多种药学上可接受的赋形剂,该赋形剂不产生其他市售亚氯酸盐制剂中的一种或多种有害非亚氯酸盐成分。在一些实施方案中,本文所述的亚氯酸盐制剂包含赋形剂,并且基本不含其他市售亚氯酸盐制剂中的一种或多种有害非亚氯酸盐成分。本文所述的亚氯酸盐制剂可以包含赋形剂,并且可以基本不含如本文所述的其他市售亚氯酸盐制剂中的降解产物或杂质中的一种或多种。
亚氯酸盐制剂可以包含稳定剂。稳定剂包括但不限于将具有以下任何作用的试剂:(1)改善赋形剂与容器(包括玻璃瓶或如明胶等封装材料)的相容性,(2)改善亚氯酸盐的稳定性(例如防止降解),(3)改善制剂的稳定性,或上述作用的组合。稳定剂可以选自例如脂肪酸、脂肪醇、醇、长链脂肪酸酯、长链醚、脂肪酸的亲水性衍生物、聚乙烯吡咯烷酮、聚乙烯醚、聚乙烯醇、烃、疏水聚合物、吸水性聚合物以及它们的组合。也可以使用稳定剂的酰胺类似物。选定的稳定剂可以改变制剂的疏水性(如油酸、蜡),或改善制剂中不同组分的混合(如乙醇),控制制剂中的水分含量(如PVP或聚乙烯吡咯烷酮),控制相的流动性(熔点高于室温的物质例如长链脂肪酸、醇、酯、醚、酰胺等或它们的混合物;蜡),和/或改善制剂与封装材料的相容性(如油酸或蜡)。这些稳定剂中的一些可以用作溶剂/助溶剂(例如乙醇)。稳定剂可以以足以抑制亚氯酸盐降解的量存在。
本文所述的制剂可以包含胶凝剂或释放改变剂中的一种或多种。
本文所述的制剂可以包含一种或多种适用于所述施用途径的佐剂。再次,在向本文所述制剂中添加任何赋形剂之前,应当就所得到的药物制剂是否适用于通过期望的施用途径进行施用来考虑亚氯酸盐的反应性。可以与治疗剂混合的佐剂包括但不限于乳糖、蔗糖、淀粉粉末、链烷酸的纤维素酯、硬脂酸、滑石、硬脂酸镁、氧化镁、磷酸和硫酸的钠盐和钙盐、阿拉伯胶、明胶、藻酸钠、聚乙烯吡咯烷酮和/或聚乙烯醇。当需要溶解的制剂时,治疗剂可以在溶剂中,该溶剂包括但不限于不同分子量的聚乙二醇、不同分子量的丙二醇、羧甲基纤维素胶体溶液、甲醇、乙醇、DMSO、玉米油、花生油、棉籽油、芝麻油、黄蓍胶和/或各种缓冲液。其他佐剂和施用方式是制药领域所熟知的,并且可用于实施本文所述的方法和制剂。载体或稀释剂可以包括延时材料,例如单独的或与蜡或本领域熟知的其他材料组合的单硬脂酸甘油酯或双硬脂酸甘油酯。如本文所述使用的制剂还可以包含凝胶制剂、可侵蚀和不可侵蚀的聚合物、微球和脂质体。
常用于制药领域的添加剂和稀释剂可以任选地添加到药物组合物和液体制剂中。这些添加剂和稀释剂包括增稠剂、成粒剂、分散剂、调味剂、甜味剂、着色剂和稳定剂(包括pH稳定剂)、其他赋形剂、抗氧化剂(如生育酚、BHA、BHT、TBHQ、乙酸生育酚、抗坏血酸棕榈酸酯、抗坏血酸没食子酸丙酯等)、防腐剂(如对羟基苯甲酸酯)等。示例性的防腐剂包括但不限于苄醇、乙醇、苯扎氯铵、苯酚、氯丁醇等。一些抗氧化剂提供氧或过氧化物抑制剂,并且可以在本文所述的制剂中使用,包括但不限于丁基羟基甲苯、丁基羟基苯甲醚、没食子酸丙酯、抗坏血酸棕榈酸酯、α-生育酚等。如果需要,例如为了改善制剂的一种或多种性质例如质地,可以使用增稠剂,如卵磷脂、羟基丙基纤维素、硬脂酸铝等。
在一些变化方案中,用于本发明的亚氯酸盐制剂是无菌的。可以通过与亚氯酸盐兼容的任何方法进行灭菌。在一些实施方案中,通过不产生大量亚氯酸盐降解产物的方法进行灭菌。在一些实施方案中,通过不会造成亚氯酸盐结构改变的方法进行灭菌。在一些实施方案中,本文所述的制剂是用于肠胃外或静脉内施用的无菌药物制剂。在一些实施方案中,本文所述的亚氯酸盐制剂是无菌过滤的,例如通过0.22微米无菌过滤器。
所述制剂或药物制剂可以是可无菌过滤的。在一些实施方案中,将本文所述的亚氯酸盐制剂配制用于通过本文所述的一种或多种施用途径进行施用。如本文所使用的“配制用于通过特定施用途径施用”的制剂是不包含被相关领域技术人员认为不适于该施用途径的药物赋形剂的制剂。作为一个实例,适合静脉内施用的制剂不包含旨在用于局部施用的牙膏赋形剂或载体,其中该赋形剂或载体被相关领域的技术人员认为不适合用于该特定的施用途径。
本文公开的任何形式的含亚氯酸盐的药剂可以以任何合适的制剂提供,其可以根据如本文公开的期望的施用途径来选择。在一个实施方案中,药物产品的制剂包含纯化的亚氯酸钠,其可以包含一定量的水、诸如磷酸氢二钠等缓冲剂和作为媒介物的无菌注射用水(USP)。在一个实施方案中,纯化的亚氯酸钠的量为约5.6mg/mL(包括反映批次水含量的批次因子),磷酸氢二钠的量为约0.107mg/mL,并添加无菌水以使体积达到1mL。在某些实施方案中,根据本发明的制剂基本上由纯化的亚氯酸钠、缓冲剂和作为媒介物的无菌注射用水(USP)组成。在某些实施方案中,配制的药物产品在25℃/60%相对湿度和/或40℃/75%相对湿度条件下稳定长达3个月。
美国专利号4,725,437描述了化学上稳定的亚氯酸盐基体的水溶液,其适于静脉内施用,在人类和非人类动物中的给药量为每千克体重约6.2×10-6摩尔的ClO2至9.3×10-5摩尔的ClO2。该溶液含有浓度为每ml约12至72微摩尔ClO2的亚氯酸盐基体。更多关于亚氯酸盐制剂的内容描述于美国专利号4,507,825和4,725,437中,这些专利通过引用而全文并入本文。
本发明还提供了治疗疾病或并发症的方法,该方法包括在受试者中施用有效量的TCDO。本申请提供了TCDO的制剂。在一个实例中,该TCDO制剂是WF10。WF10也称为Oxoferin.RTM,并且可以商购获得。在另一个实例中,亚氯酸盐制剂包含亚氯酸盐。TCDO或亚氯酸盐的其他制剂包含在本发明的范围内。或者,在一些实施方案中,可以部分或全部排除TCDO和/或WF10。
含亚氯酸盐的组合物诸如TCDO可以配制用于肠胃外或肠内施用,通常为肠胃外施用。因此,亚氯酸盐制剂或含亚氯酸盐的药剂如TCDO和WF10适于肠胃外、局部或经皮施用,通常为静脉内、肌内或皮下施用,并且可以适于通过团注(bolus injection)、持续释放(包括控制释放)、输注等施用。关于施用途径的更多细节在下文中公开。在一些实施方案中,含亚氯酸盐的药剂的施用通过输注,例如通过皮下或静脉内输注,或以栓剂的形式进行。
VI.亚氯酸盐或含亚氯酸盐药剂的施用和给药
除非上下文另有说明,本文所述的所有制剂和药物制剂可以通过以下任何途径进行施用:全身,肠胃外(例如肌内、腹膜内、静脉内、ICV、脑池内注射或输注、皮下注射或植入),通过吸入喷雾,使用气雾剂推进剂喷雾或雾化,经鼻,阴道,直肠,舌下,尿道(如尿道栓剂),通过输注、动脉内、鞘内、支气管内、皮下、皮内、静脉内、宫颈内、腹内、颅内、肺内、胸内、气管内、鼻途径、全身递送治疗剂的口服给药,药物递送装置,或通过全身、经皮或经口递送治疗剂的皮肤贴片。在一些变化方案中,所述制剂被配制用于除口服或经颊以外的施用。
在一些变化方案中,本文所述的制剂不局部施用。
在一些实施方案中,本文所述的制剂、药物制剂以及施用和治疗的方法适用于任何脊椎动物,例如温血动物或冷血动物。在一些实施方案中,本文所述的制剂、药物制剂以及施用和治疗方法适用于哺乳动物,包括用于兽医领域,包括家养宠物(如猫、狗、兔、鸟、马等)和农用牲畜(如牛、绵羊、家禽等)。在一些变化方案中,本文所述的制剂、药物制剂以及施用和治疗的方法适用于灵长类动物,包括但不限于人类。
亚氯酸盐制剂通常对应于受试者的体重进行体内给药。由于活性剂在血液中的连续分解,通常以规律间隔施用药剂。本领域技术人员将容易理解,实际的剂量和方案将随着药剂、制剂、症状的严重程度、受试者对治疗的易感性和/或副作用等而变化。本领域技术人员通过多种方法容易地且常规地确定剂量。
含亚氯酸盐制剂的示例性剂量可以分别在约0.1ml/kg体重至约1.5ml/kg体重之间,以及每升约40至约80mmol的ClO2-的浓度之间变化。例如,含亚氯酸盐制剂的剂量可以分别包括约0.5ml/kg体重和通常每升约60mMol的ClO2。在TCDO的情况下,例如,WF10以大约0.5ml/kg体重的最大剂量静脉内施用于患有糖尿病或糖尿病相关疾病或并发症的患者。其他合适的剂量可以是大约0.25ml/kg体重。
施用方案例如剂量以及施用频率通常包括以一定量和频率施用从而提供期望的效果,例如,施用有效提供患有糖尿病或糖尿病相关疾病或并发症(例如心血管疾病、代谢疾病例如代谢综合征和黄斑变性症状)的患者的一种或多种症状的改善的量。例如,亚氯酸盐或含亚氯酸盐药剂可以连续施用2、3、4、5、6、7、8、9、10天或更多天,该施用期可以在最后一次给药后1、2、3周或更多周之后重新开始。
施用方案例如剂量以及施用频率通常包括以一定量和频率施用从而提供期望的效果,例如,施用有效提供患有巨噬细胞相关疾病或并发症(例如炎症、损伤、肌肉变性和肥胖)的患者的一种或多种症状的改善的量。巨噬细胞相关疾病的一些实例可以包括但不限于阿尔茨海默病(AD)、帕金森病(PD)、亨廷顿病(HD)、多发性硬化和肌萎缩性侧索硬化症(ALS)、癌症和慢性肉芽肿病。例如,亚氯酸盐或含亚氯酸盐药剂可以连续施用2、3、4、5、6、7、8、9、10天或更多天,该施用期可以在最后一次给药后1、2、3周或更多周之后重新开始。
根据本发明的亚氯酸盐可以每天施用。在一些实施方案中,亚氯酸盐以约0.2mg/kg/天的亚氯酸盐至约3.3mg/kg/天的亚氯酸盐的剂量每天施用。在一些实施方案中,亚氯酸盐以约0.2mg/kg/天的亚氯酸盐、约0.4mg/kg/天的亚氯酸盐、约0.5mg/kg/天的亚氯酸盐、约0.6mg/kg/天的亚氯酸盐、约0.7mg/kg/天的亚氯酸盐、约0.8mg/kg/天的亚氯酸盐、约0.9mg/kg/天的亚氯酸盐、约1.0mg/kg/天的亚氯酸盐、约1.1mg/kg/天的亚氯酸盐、约1.2mg/kg/天的亚氯酸盐、约1.3mg/kg/天的亚氯酸盐、约1.4mg/kg/天的亚氯酸盐、约1.5mg/kg/天的亚氯酸盐、约1.6mg/kg/天的亚氯酸盐、约1.7mg/kg/天的亚氯酸盐、约1.8mg/kg/天的亚氯酸盐、约1.9mg/kg/天的亚氯酸盐、约2.0mg/kg/天的亚氯酸盐、约2.1mg/kg/天的亚氯酸盐、约2.2mg/kg/天的亚氯酸盐、约2.3mg/kg/天的亚氯酸盐、约2.4mg/kg/天的亚氯酸盐、约2.5mg/kg/天的亚氯酸盐、约2.6mg/kg/天的亚氯酸盐、约2.7mg/kg/天的亚氯酸盐、约2.8mg/kg/天的亚氯酸盐、约2.9mg/kg/天的亚氯酸盐、约3.0mg/kg/天的亚氯酸盐、约3.1mg/kg/天的亚氯酸盐、约3.2mg/kg/天的亚氯酸盐、约3.3mg/kg/天的亚氯酸盐、约3.4mg/kg/天的亚氯酸盐或约3.5mg/kg/天的亚氯酸盐的剂量每天施用。
在一些实施方案中,在本发明的方法中使用的药物组合物可以进一步以周期施用。示例性周期由以下组成:a)第一时间段,其中所述药物组合物以第一剂量施用第一次数;以及b)第二时间段,其中所述药物组合物以第二剂量施用第二次数。在一些实施方案中,第一时间段为约一周,第一次数为约五次,第二时间段为约两周,并且第二次数为零次。在其他实施方案中,第一时间段为约一周,第一次数为约三次,第二时间段为约一周,并且第二次数为零次。第一剂量可以是约0.4mg/kg/天的亚氯酸盐至约3.3mg/kg/天的亚氯酸盐。例如,第一剂量可以是约2.1mg/kg/天的亚氯酸盐。该周期可以进行多次,例如约2、3、4、5、6、7、8、9、10次或10次或更多次。在一些实施方案中,该周期进行约2-4次。
在一些实施方案中,给药时间表由交替的施用期和非施用期组成。例如,亚氯酸盐可以以三周的周期进行施用,该周期包括在一周内给药亚氯酸盐最多5次,而在随后的两周不进行治疗。必要时可以重复该周期以获得期望的结果。在另一个实施方案中,亚氯酸盐以两周的周期进行施用,例如,在一周内施用最多3次,而在随后的一周不施用。在一些实施方案中,进行了总共2-4个周期。在示例性实施方案中,给药方案包括施用2.1mg/kg/天的亚氯酸盐总共2-4个三周的周期。
B.巨噬细胞活化
一方面,本发明提供了治疗患有巨噬细胞相关疾病的受试者的方法,该方法包括向有需要的受试者施用有效量的氧化剂。该巨噬细胞相关疾病可能与活化的巨噬细胞相关。患有巨噬细胞相关疾病的受试者的一种或多种炎症因子的血浆水平可高于阈值水平、其正常水平或其疾病水平。所述氧化剂可以包括但不限于亚氯酸盐。术语“正常水平”是指在没有罹患将通过施用氧化剂治疗的巨噬细胞相关疾病的受试者中测量的因子的平均浓度。术语“疾病水平”是指在患有将通过施用氧化剂治疗的巨噬细胞相关疾病的受试者中测量的因子的平均浓度。
巨噬细胞作为未成熟的单核细胞从骨髓中释放,在血流中循环,并且最终可以迁移到组织中从而最终分化为定居巨噬细胞。定居巨噬细胞包括肝中的枯否细胞(Kupffer cell)、肺中的肺泡巨噬细胞和骨骼中的破骨细胞。单核细胞和巨噬细胞是吞噬细胞,在脊椎动物的先天性免疫中发挥作用以及辅助脊椎动物的适应性免疫。它们的作用是作为固定或运动细胞吞噬(吞没并随后消化)细胞碎片和病原体,并刺激淋巴细胞和其他免疫细胞以对病原体做出反应。它们可以通过多种蛋白质的特异性表达,利用流式细胞术或免疫组织化学染色进行鉴定,该蛋白质包括CD14、CD11b、F4/80(小鼠)/EMR1(人)、溶菌酶M、MAC-1/MAC-3和CD68(Khazen W等人.2005FEBS Lett.579(25):5631-4)。当单核细胞穿过血管内皮进入受损组织(称为白细胞外渗的过程)时,该单核细胞经历一系列变化从而变成巨噬细胞。单核细胞通常通过趋化作用被化学物质吸引到受损部位,该趋化作用由一系列刺激(包括受损细胞、病原体和已经在该部位处的巨噬细胞释放的细胞因子)触发。在诸如睾丸的一些部位,已经显示巨噬细胞通过增殖进入器官。不同于短期中性粒细胞,巨噬细胞在体内存活时间更长,最长达几个月。
巨噬细胞执行对组织重塑、炎症和免疫所必需的众多功能,包括但不限于吞噬作用,细胞毒性,以及多种细胞因子、生长因子、溶菌酶、蛋白酶、补体成分、凝血因子和前列腺素的分泌。巨噬细胞的一个重要作用是去除肺中坏死的细胞碎片。去除死细胞物质在慢性炎症中是重要的,因为炎症的早期阶段主要由中性粒细胞控制,如果达到一定阶段这些中性粒细胞将被巨噬细胞摄取。坏死组织的去除在更大程度上由固定的巨噬细胞处理,该固定的巨噬细胞通常停留在诸如肺、肝、神经组织、骨、脾和结缔组织等关键部位,在这些部位可能会出现微生物入侵或灰尘积累,从而使固定的巨噬细胞摄取外来物质如病原体,如果需要则募集额外的巨噬细胞。巨噬细胞可以在其对该器官的功能具有特异性的器官内呈现出旁分泌功能。例如在睾丸中,巨噬细胞已经显示出能够通过分泌25-羟基胆固醇(一种可通过相邻的睾丸间质细胞转化为睾酮的氧甾酮)与睾丸间质细胞相互作用。此外,睾丸巨噬细胞可参与创建睾丸中的免疫免除(immune privileged)环境,并且在睾丸炎症期间介导不育。表1示出了组织中不同类型的巨噬细胞的列表。
表1:组织中的不同类型的巨噬细胞
巨噬细胞作为清除剂,从身体中去除死亡细胞和其他碎片。它们是一类在启动免疫应答中起关键作用的抗原呈递细胞。作为分泌细胞,单核细胞和巨噬细胞对于免疫应答的调节和炎症的进展至关重要,因为它们产生单核因子,包括酶、补体蛋白以及诸如白介素-1等调节因子。巨噬细胞还携带用于淋巴细胞活化的淋巴因子受体,淋巴细胞活化对于杀死微生物和肿瘤细胞是重要的。消化病原体后,巨噬细胞将II类MHC分子上的抗原呈递给相应的辅助T细胞。最终,抗原呈递导致产生与病原体抗原结合的抗体,从而导致由巨噬细胞产生的吞噬作用或抗体依赖性细胞毒性。在淋巴结中感染的巨噬细胞表面上的抗原呈递(在II类MHC的情况下)刺激TH1(1型辅助T细胞)增殖(主要由于巨噬细胞分泌的IL-12引起)。当淋巴结中的B细胞用其表面结合的抗体识别微生物上相同的未加工的表面抗原时,该抗原被内吞并被加工。加工的抗原随后在B细胞表面上的MHCII中呈递。已经增殖的TH1受体识别抗原-MHCII复合物(具有共刺激因子-CD40和CD40L),并使B细胞产生有助于抗原调理作用的抗体,从而使得巨噬细胞可以更好地清除病原体。
巨噬细胞提供了针对被真菌或寄生虫感染的肿瘤细胞和体细胞的另一道防线。一旦T细胞已经识别出其在异常细胞表面上的特定抗原,则T细胞变成产生淋巴因子(包括白介素、趋化因子和干扰素家族)的活化的效应细胞,该淋巴因子进一步刺激并激活巨噬细胞。这些活化的巨噬细胞随后可以更有效地吞没并消化受感染的细胞。巨噬细胞不产生对抗原特异的应答,而是攻击存在于其被激活的局部区域中的细胞。
巨噬细胞也在肌肉再生中发挥作用。先前的研究已经表明巨噬细胞对小鼠的比目鱼肌修复的影响(Tidball J G,Wehling-Henricks M,2007,The Journal of Physiology 578:327-336)。在发育期,巨噬细胞的损耗也会减少肌肉的生长。
Ⅰ.经典活化的巨噬细胞
在一个方面,本发明提供了调节巨噬细胞聚集或活化的方法,该方法包括施用有效量的氧化剂(例如,亚氯酸盐)。该氧化剂可以是亚氯酸盐或WF10。该氧化剂可以通过由巨噬细胞表达的受体来调节巨噬细胞的刺激,该受体包括但不限于干扰素(IFN)-γ受体、CD14/LPS受体、MHCⅡ分子、或诸如IL-4和IL-13受体等白介素受体。在一些实施方案中,该氧化剂调节巨噬细胞的趋化因子的释放。在一些实施方案中,该氧化剂调节巨噬细胞释放诸如IL-1、IL-6、IL-18、INF-g、CRP和TNF-α等促炎细胞因子,或诸如IL-10和TGF-β等抗炎细胞因子。在一些实施方案中,该氧化剂或免疫调节剂调节巨噬细胞的蛋白酶释放。在一些实施方案中,该氧化剂或免疫调节剂调节巨噬细胞的细胞外基质(ECM)相关分子释放。
已经开发了两种主要巨噬细胞类型的模型(Gordon,S.(1999)Fundamental Immunology,第4版,Paul,W.E编著,Lippincott-Raven Publishers,Philidelphia,pp.533-545;Stein,M等人(1992)J.Exp.Med.176:287)。经典活化的巨噬细胞通常显示出Th1-样表型,促进炎症、细胞外基质(ECM)破坏和坏死,而替代性活化的巨噬细胞通常显示Th2-样表型,促进ECM构建、细胞增殖和血管生成。尽管这两种表型都是先天和适应性免疫系统的重要组分,但经典活化的巨噬细胞倾向于引发慢性炎症和组织损伤,而替代性活化的巨噬细胞倾向于消除炎症并促进创伤愈合(参见,reviews:Duffield,J.S.(2003)Clin.Sci.104:27;Gordon,S.(2003)Nat.Rev.Immunol 3:23;Ma,J等人(2003)Cell.Mol.Life Sci.60:2334;Mosser,D.M.(2003)J.Leukoc.Biol.73:209)。
一般而言,经典活化的巨噬细胞的分化需要通过IFN-γR产生的IFN-γ形式的引发信号(Dalton,D.K等人(1993)Science 259:1739;Huang,S等人(1993)Science 259:1742)。当引发的巨噬细胞随后遭遇适当的刺激,例如细菌LPS时,其变成经典活化的。LPS首先被可溶性LBP结合,随后被可溶性或膜结合的CD14结合。CD14将LPS递送到LPS识别复合物(Janeway,C.A.&R.Medzhitov(2002)Annu.Rev.Immunol 20:197),该LPS识别复合物由至少TLR410和MD-2组成(Nagai,Y等人(2002)Nat.Immunol.3:667)。病原体和病原体组分随后通过吞噬作用被摄取(Honey,K.&A.Y.Rudensky(2003)Nat.Rev.Immunol.3:472)并被递送至溶酶体,在溶酶体中它们暴露于多种降解酶,包括几种组织蛋白酶半胱氨酸蛋白酶。合适的抗原被加工并装载到晚期内吞室中的II类MHC分子上,并且抗原/MHCII复合物以及协同刺激的B7族成员被呈递给T细胞(Harding,C.V等人(2003)Curr.Opin.Immunol15:112)。
这些事件后,紧接着出现细胞形态的明显变化和细胞分泌情况的显著改变。包括IL-8/CXCL8、IP-10/CXCL10、MIP-1α/CCL3、MIP-1β/CCL4和RANTES/CCL5的多种趋化因子作为中性粒细胞、不成熟的树突细胞、自然杀伤细胞和活化的T细胞的化学引诱物被释放(Luster,A.D.(2002)Curr.Opin.Immunol.14:129)。此外,包括IL-1β/IL-1F2、IL-6和TNF-α/TNFSF1A的数种促炎细胞因子被释放。TNF-α也有助于经典活化的巨噬细胞的促凋亡活性(Boyle,J.J等人(2003)Arterioscler.Thromb.Vasc.Biol.23:1553;Duffield,J.S等人(2001)Am.J.Pathol.159:1397;Song,E等人(2000)Cell.Imunol.204:19)。由于iNOS上调,TNF-α伴随着Fas配体/TNFSF6的分泌和NO的释放(Hesse,M等人(2001)J.Immunol.167:6533;Thomassen,M.J.&M.S.Kavuru(2001)Int.Immunopharmacol.1:1479;Duffield,J.S等人(2000)J.Immunol 164:2110;Munder,M等人(1998)J.Immunol 160:5347)。此外,经典活化的巨噬细胞释放包括MMP-1、MMP-2、MMP-7、MMP-9和MMP-12的蛋白酶,该蛋白酶降解胶原蛋白、弹性蛋白、纤连蛋白和其他ECM组分(Chizzolini,C等人(2000)J.Immunol.164:5952;Gibbs,D.F等人(1999)Am.J.Respir.Cell Mol.Biol.20:1136;Gibbs,D.F等人(1999)Am.J.Respir.Cell Mol.Biol.20:1145)。
尽管这些分子的释放对宿主防御和适应性免疫系统的引导非常重要,但它们的不受控制的释放可能对微环境造成显著的附带损伤。通过引发大量的白细胞浸润并使周围组织充满炎症介质、促凋亡因子和基质降解蛋白酶,经典活化的巨噬细胞能够分解组织,达到造成严重损伤的程度。慢性炎症造成的组织破坏与肿瘤的进展、1类自身免疫疾病和肾小球性肾炎以及其他病理学相关(Gordon,S.(2003)Nat.Rev.Immunol.3:23;Mosser,D.M.(2003)J.Leukoc.Biol.73:209)。
在一些实施方案中,本发明的方法包括施用例如亚氯酸盐的氧化化合物来治疗巨噬细胞相关疾病。在一些实施方案中,本发明提供了使用氧化剂通过调节至少一种IFN-γ受体来治疗巨噬细胞相关疾病的方法。在一些实施方案中,本发明提供了使用氧化剂通过调节LPS、调节MHC II抗原呈递途径、调节趋化因子(包括但不限于IL-18、IL-6、CRP和/或IFN-g)的释放来治疗巨噬细胞相关疾病的方法。
II.替代性活化的巨噬细胞
替代性活化的巨噬细胞的分化不需要任何引发。IL-4和/或IL-13可用作足够的刺激物(Stein,M等人(1992)J.Exp.Med.176:287;Doherty,T.M等人(1993)J.Immunol.151:7151)。这些因子与其各自受体结合,随后为可溶性抗原的液相胞饮(Brombacher,F.(2000)BioEssays 22:646;Montaner,L.J等人(1999)J.Immunol 162:4613;Conner,S.D.&S.L.Schmid(2003)Nature 422:37)。然后可溶性抗原被装载到II类MHC分子上,并且随后对T细胞展示抗原/MHCII复合物和协同刺激的B7族成员(Harding,C.V等人(2003)Curr.Opin.Immunol 15:112)。
与经典活化的巨噬细胞相似,替代性活化的巨噬细胞由于适当的刺激而改变其细胞形态及分泌模式。巨噬细胞通过其释放趋化因子来吸引白细胞,该趋化因子包括MDC/CCL22(Andrew,D.P等人(1998)J.Immunol 161:5027;Imai,T等人(1999)Int.Immunol 11:81)、PARC/CCL18(Kodelja,V等人(1998)J.Immunol.160:1411;Goerdt,S等人(1999)Pathobiology 67:222)和TARC/CCL17。诸如IL-1ra/IL-1F3(Mantovani,A等人(2001)Trends Immunol 22:328)、Ym1、Ym2、RELMa(Raes,G等人(2002)J.Leukoc.Biol.71:597;Loke,P等人(2002)BMC Immunol.3:7)、IL-10和TGF-β等因子的释放消解了炎症。TGF-β也通过诱导附近的成纤维细胞产生ECM组分而间接起到促进ECM构建的作用。替代性活化的巨噬细胞自身分泌ECM组分、纤连蛋白和bIG-H3(Gratchev,A等人(2001)Scand.J.Immunol 53:386)、ECM交联酶、谷氨酰胺转氨酶(Haroon,Z.A等人(1999)Lab.Invest.79:1679)和骨桥蛋白,其参与ECM的细胞粘附(Murry,C.E等人(1994)Am.J.Pathol.145:1450)。
此外,替代性活化的巨噬细胞上调精氨酸酶I,精氨酸酶I参与脯氨酸以及多胺生物合成。脯氨酸促进ECM的构建,而多胺参与细胞增殖(Hesse,M等人(2001)J.Immunol.167:6533)。由替代性活化的巨噬细胞分泌的促进细胞增殖的其他因子包括PDGF、IFG和TGF-β(Song,E等人(2000)Cell.Imunol.204:19;Cao,B等人(2000)Chin.Med.J.113:776)。这些因子与碱性FGF、TGF-α和VEGF一起也参与血管生成(Cao,B等人(2000)Chin.Med.J.113:776;Sunderkotter,C等人(1991)Pharmac.Ther.51:195)。
替代性活化的巨噬细胞分泌的分子由于其抗炎性、纤维化、增殖性和血管生成活性而致力于消除炎症和促进创伤修复。这种巨噬细胞在对抗寄生虫感染如血吸虫病方面也特别有效。除了其有益的活性,替代性活化的巨噬细胞还牵涉数种病理学,其中最突出的是过敏和哮喘(Duffield,J.S.(2003)Clin.Sci.104:27;Gordon,S.(2003)Nat.Rev.Immunol.3:23)。
本发明还包括使用靶向信号传导途径的氧化剂调节巨噬细胞聚集或活化的方法,该信号传导途径包括但不限于脂多糖(LPS)、toll样受体(TLR)、前列腺素E2(PGE2)、干扰素(IFN)-a、IFN-b、IFN-g、白介素(IL)-1、IL-4、IL-6、sIL1Ra、IL-10、IL-12、IL-12p40、IL-13、IL-18、CRP、IP10、II类MHC(主要组织相容性复合体)分子(MHCII)、TNF-a、巨噬细胞炎性蛋白1α(MIP1-a)、IFN-g-诱导因子(IGIF)、巨噬细胞刺激蛋白(MSP)、细胞间粘附分子1(ICAM-1)、集落刺激因子1(CSF-1R)、L-精氨酸和氮氧化物信号传导途径。本发明的氧化剂可靶向或影响任意受体、细胞质或细胞核中间信号传导分子,或本文公开的参与任何一条信号传导途径的转录因子。可由本发明的氧化剂调节的作为一个或多个信号传导途径的一部分的重要信号传导分子的实例包括但不限于TLR2、TLR4、CAT2、ICSBP、IL1-R、Tie-2、TRIF/IRF3、IFNR-I、IFNR-II、IRF1、IRF2、Raf-1、MEK1、MEK2、ERK1、ERK2、p38、MAPKK4、MAPKK6、PKC、JAK1、JAK2、STAT1、STAT3、Elk1、JNK/SAPK、AP1、Pu1、NFkB、NFAT、iNOS、USF1、ISGF3、SP1、Bc16、ATF2、c-Jun和COX-2。可由本发明的氧化剂直接或间接调节的对于巨噬细胞活化或作用至关重要的分子包括属于转录因子、细胞表面受体、细胞因子、趋化因子、细胞因子或趋化因子受体、生长因子、干扰素、干扰素受体、和粘附分子的那些分子。特别地,本发明的氧化剂可以调节分子,该分子包括但不限于TLR-2、TLR-4、mkp-1、COX-2、SOCS-3、Fc.γ.R1、IFN-a、IFN-b、IFN-g、CRP、IL-4、IL-6、IL-18、IL-1Ra、IGIF、IL-b、MHCI、MHCII IAA、MHCII IAB、MHCII IEB、IP10、IL-10、组织蛋白酶H、溶菌酶、CathB、stk、TNF-a、IL-12p35、IL-12p40、MIP-1a、ICAM-1、INOS、mig、Cat-2、CIITA、ICSBP、CathL、CSF1R、GM-CSF、IRF1、IRF-2、c-fos、VEGF、IL-8、bFGF、CSF-1、EGF、MMP-2、MMP-7、MMP-9、MMP-12、EMAPII、内皮素2、HIF-1、HIF-2、CXCL8、TGF-b、PGE2和/或MDF。
III.单核细胞
单核细胞被认为是一种类型的白细胞。单核细胞在免疫系统中有两个主要功能:(1)补充正常状态的定居巨噬细胞和树突细胞;以及(2)响应炎症信号,单核细胞可以快速移动到组织中的感染部位,并且分裂/分化成巨噬细胞和树突细胞以引起免疫应答。单核细胞由称作成单核细胞的骨髓造血干细胞前体的骨髓产生。单核细胞在血流中循环约1至3天,然后通常进入全身的组织。在组织中单核细胞成熟,在不同解剖学位置成为不同类型的巨噬细胞。从血流中迁移到其他组织中的单核细胞随后分化为组织定居巨噬细胞或树突细胞。巨噬细胞负责保护组织免受外来物质侵害,但也疑似是参与引发动脉粥样硬化的主要细胞。巨噬细胞是拥有大的光滑细胞核、大面积的细胞质和用于加工外来物质的多个内部囊泡的细胞。
在人类血液中存在两种类型的单核细胞:a)经典单核细胞,其特征是CD14细胞表面受体(CD14++单核细胞)的高水平表达,以及b)非经典促炎单核细胞,其具有较低水平的CD14表达,并且具有与CD16受体的额外共表达(CD14+CD16+单核细胞)。在被微生物产物刺激后,CD14+CD16+单核细胞产生了大量促炎细胞因子,例如肿瘤坏死因子(TNF-α)和白介素-12。
已在不同的疾病中指示CD14+CD16+单核细胞数目的增加或减少(Loems Ziegler-Heitbrock,Journal of Leukocyte Biology,Vol 81,2007)。这些CD14+CD16+单核细胞可能在产生导致疾病的炎症的巨噬细胞方面发挥作用。CD14+CD16+单核细胞牵涉许多炎性疾病,包括但不限于类风湿性关节炎、糖尿病、血液渗析、动脉粥样硬化、川崎病以及细菌性感染和病毒性感染,其中更多细节在下文公开。在一些实施方案中,本发明提供了治疗巨噬细胞相关疾病的方法,该方法包括向有需要的受试者施用有效量的氧化剂和/或免疫调节剂,其中所述药剂调节或影响CD14+CD16+单核细胞。单核细胞是来源于骨髓的组织巨噬细胞的前体,该前体是创伤愈合、细菌和细胞碎片清除以及炎症的诱导和清除的关键效应物。与经典炎症有关的巨噬细胞称为M1,并且这些细胞产生诸如TNF-α、IL-1和其他促炎症因子等因子。与炎症逆转和免疫应答的抑制有关的巨噬细胞称为M2。在ALS发病机理的情况下,脊髓内的M2巨噬细胞表型与正常功能有关,而脊髓内的新M1型巨噬细胞的出现与疾病进展有关(Henkel等人,(2009)J Neuroimmune Pharmacol 4(4):389-398)。
近来的研究已表明ALS小鼠G93A种的疾病进展与炎性单核细胞迁移至脊髓直接相关(Butovsky等人,(2012)The Journal of Clinical Investigation,122(9):3063-3087)。对小鼠G93A SOD1同源品种中NP001的初步研究显示了与对照小鼠相比,进行治疗的小鼠的存活率的显著改善(McGrath等人,(2010)21st internation al symposium on ALS/MND,Clinical Work in Progress 11-13)。炎症相关疾病的进展可能以与ALS小鼠模型中看到的相似的方式受到影响。
IV.肿瘤相关巨噬细胞(TAM)
在一些实施方案中,本发明提供了调节肿瘤相关巨噬细胞的方法,该方法包括向受试者施用氧化剂。巨噬细胞在几乎所有类型的恶性肿瘤的基质室中都占重要地位。巨噬细胞对于肿瘤不同部位刺激物存在的响应为释放不同的整套(repertoire)生长因子,细胞因子,趋化因子,以及调节肿瘤生长、血管生成、入侵和/或转移的酶。肿瘤相关巨噬细胞(TAM)作用的不同微环境包括:1)TAM促进癌细胞运动性的入侵区域;2)TAM促进转移的基质和血管周区域;以及3)缺氧性TAM刺激血管生成的无血管和周围坏死区域(Lewis C E等人综述,Cancer Res.2006(66)605-612)。TAM的表型相对不成熟,特征为分化相关巨噬细胞抗原、羧肽酶M和CD51的低表达,IL-1和IL-6的高组成型表达,以及TNF-a的低表达。
TAM浸润与如由乳腺癌中的MIB-1水平、子宫内膜癌中的Ki67水平或肾细胞癌中的有丝分裂指数测量的肿瘤细胞增殖正相关(Lewis C E等人综述,Cancer Res.2006(66)605-612)。各种研究表明,TAM表达刺激肿瘤细胞增殖和存活的多种因子,包括表皮生长因子(EGF)(Goswami S等人.Cancer Res 2005;65;5278-83;Lewis C E等人.Lancet1993;342;148-9)、血小板源生长因子(PDGF)、TGF-h1、肝细胞生长因子、MMP-9和碱性成纤维细胞生长因子(bFGF)。TAM还在调节血管生成方面发挥重要作用。TAM释放多种有效的促血管生成细胞因子和生长因子,例如血管内皮生长因子(VEGF)、TNF-a、IL-8和bFGF。此外,TAM表达一系列的血管生成调节酶,包括MMP-2、MMP-7、MMP-9、MMP-12和环氧化酶-2(COX-2)(Sunderkotter C等人.Pharmacol Ther1991;51:195-216;Klimp A H等人.Cancer Res.2001;61:7305-9)。TAM通过上调缺氧诱导转录因子HIF-1和HIF-2来响应肿瘤缺氧。响应于缺氧,巨噬细胞还上调VEGF和其他促血管生成因子。例如,当暴露于人类肿瘤的体外和无血管区域的缺氧环境时,巨噬细胞合成升高水平的MMP-7。cDNA阵列研究已鉴定了在暴露于缺氧环境的原代巨噬细胞中编码超过30种其他促血管生成基因的信息的上调,包括CXCL8、血管生成素、COX-2和其他因子(White J R等人.Genomics 2004;83:1-8)。
TAM也牵涉对转移的调控。原发性肿瘤中大量的TAM与多种肿瘤类型的转移的早期建立相关(Hanada T等人.Int J.Urol 2000;7:263-9)。TAM在原发性肿瘤的转移细胞的释放以及远位点处继发性肿瘤的建立方面都发挥作用。
TAM还在肿瘤免疫抑制中发挥作用。与健康组织中能够呈递肿瘤相关抗原、裂解肿瘤细胞以及刺激T细胞和NK细胞的抗肿瘤功能的巨噬细胞不同,肿瘤微环境中的TAM缺少这些活性,只留下没有增加有效抗肿瘤免疫反应能力的宿主。多种研究已经表明肿瘤来源的分子,例如细胞因子、生长因子、趋化分子和蛋白酶,影响TAM的功能(Elgert K D等人.J Leukoc Biol 1998;64:275-90)。例如,肿瘤细胞分泌可以抑制TAM的细胞毒性活性的蛋白质,例如IL-4、IL-6、IL-10、MDF、TGF-h1和PGE 2(Ben-Baruch,Semin Cancer Biol 2005)。此外,TGF-h1、IL-10和PGE 2可通过肿瘤微环境以及诸如脾和腹膜等远部位中的巨噬细胞来抑制II类MHC分子的表达。该作用可能会限制这些区域中TAM将肿瘤相关抗原有效呈递给T细胞的能力。TAM涉及抗肿瘤免疫机制的另一个重要方面是这些细胞释放免疫刺激性细胞因子的能力。例如,IL-12(一种已知刺激T细胞和NK细胞的增殖和细胞毒性的细胞因子)的巨噬细胞表达在肿瘤中受到显著抑制,这可能是通过暴露于IL-10、PGE 2和TGF-h1(Mitsuhashi M等人.J Leuko Biol 2004;76:322-23)。肿瘤微环境中的缺氧有可能促成抑制TAM的抗肿瘤活性,因为它刺激有效的免疫抑制因子PGE 2和IL-10的释放。它们作用于TAM以减少其对肿瘤细胞的细胞毒性活性。缺氧还抑制巨噬细胞吞噬已死亡的或将死亡的细胞以及向T细胞呈递抗原的能力。可达到该目的的一个机理是通过减少CD80的表面表达,CD80是一种T细胞响应抗原肽而完全活化所需要的辅刺激分子。
许多信号传导途径对于TAM的功能非常重要。调节TAM功能的示例性信号传导途径包括但不限于NFkB途径,TLR途径(特别是TLR/IL-1R信号传导、TLR2和TLR4信号传导)Tie-2/Ang-2途径,TRIF/TBK1/IRF3途径以及缺氧诱导的途径。NFkB是调节巨噬细胞的炎性反应的最重要的转录因子之一,特别是它们响应于不同环境诱因(例如,应激信号、炎性细胞因子、病原体和缺氧)对促炎细胞因子、辅刺激分子和其他活化标志物的表达。TLR/IL-1R信号传导是巨噬细胞中NFkB活化的重要上游组分。在炎症诱导的癌症中,基质巨噬细胞上TLR/IL-1R的活化可通过以下作用来触发:1)在慢性感染的位点与细菌直接相互作用(例如,结肠炎相关结肠癌中的肠细菌或胃癌中的幽门螺杆菌)(Karin M等人,Cell 2006 124:823-835);或2)与肿瘤细胞来源的促炎细胞因子例如IL-1相互作用;和/或3)识别坏死的肿瘤细胞碎片例如HMGB1(高迁移率组盒1)或S100(综述于Biswas S K等人,J.Immunol.2008 180:2011-2017)。人类肺癌细胞中TLR4的活化促进免疫抑制性细胞因子TGF-β和促血管生成因子VEGF和CXCL8的产生,以及赋予对TNF-α诱导的凋亡和肿瘤细胞存活的抗性(He W等人.Mol.Immunol 2007 44:2850-2859)。已报道了TLR2活化在通过ERK/MAPK磷酸化触发树突细胞和巨噬细胞中M2(免疫抑制性)样细胞因子谱(IL-12低,IL-10高)中的优先作用(Dillon S等人.J Immunol 2004 172:4733-4743)。
表达Tie-2的单核细胞(TEM)存在于人类和鼠肿瘤中(De Palma等人2005Cancer Cell 8:211-226)。已知内皮细胞以及肿瘤细胞上调Ang-2,即肿瘤中Tie-2的一种配体。已有人提出肿瘤衍生的Ang-2可促进Tie-2单核细胞/巨噬细胞募集入肿瘤(Murdoch C等人.J Immunol 178:7405-7411)。重要的是,Ang-2也显著抑制Tie-2单核细胞体外释放促炎细胞因子如TNF-α和IL-2(Biswas S K等人.J.Immunol.2008 180:2011-2017)——一种在缺氧中更加明显的作用。这些发现表明Ang-2/Tie-2轴可代表TAM中抑制抗血管生成表型和促进免疫抑制性表型的另一个潜在机理,尤其是在肿瘤的缺氧区域中。
已在鼠纤维肉瘤的TAM中证明了TRIF依赖性IRF3/STAT1途径(其中TRIF为诱导IFN-β的含有TLR/IL-1R结构域的衔接子,TBK是结合TANK的激酶,而IRF是IFN调控因子)的优先活化(Biswas S等人.Blood 107:2112-2122)。这在基础条件和LPS活化条件下的TAM中STAT1的组成型活化和I型IFN诱导型基因(包括CCL5、CXCL9和CXCL10)的上调中显而易见(Biswas S K等人.J.Immunol 2008 180:2011-2017)。还表明IL-10转录经由TRAF3和I型IFN通过TRIF/IRF3途径进行调节(Chang E Y等人.J Immunol 178:6705-6709)。总之,诸如TBK1和IRF3的TRIF途径成员可能在介导TAM的作用中起作用,并且可能代表潜在的治疗靶标。
如上文提到的,缺氧对巨噬细胞功能(包括它们向肿瘤的迁入以及基因表达的模式,特别是编码促血管生成细胞因子和酶的基因)具有深远影响。缺氧通过上调转录因子即缺氧诱导因子(HIF)1和2(HIF-1和HIF-2)来诱导这些细胞中的基因表达。巨噬细胞上调人类肿瘤中缺氧/坏死区域的HIF和随后的一系列HIF靶基因(Murdoch C等人.2005Int J Cancer,117:701-708)。最重要的是,缺氧是TAM中的VEGF和MMP7两者的有效诱导条件,已知VEGF和MMP7均支持肿瘤血管生成、入侵和转移。此外,缺氧上调M2巨噬细胞标志物,如IL-10、精氨酸酶和PGE 2的表达。它还调节促炎基因如TNF-a、IL-1、迁移抑制因子(MIF)、CCL3和COX2的表达。
在一些实施方案中,本发明提供了治疗癌症的方法,该方法包括施用氧化剂。在一些实施方案中,巨噬细胞的活化或功能通过本发明的氧化剂或免疫调节剂来调节,使得抗肿瘤活性得以增强。在一些实施方案中,本发明的氧化剂调节参与巨噬细胞的活化或功能的一种或多种途径,其中所述途径包括但不限于NFkB途径、TLR途径、Tie-2/Ang-2途径、TRIF/TBK1/IRF3途径、缺氧诱导的途径以及涉及本文公开的任何分子的任何途径。
C.巨噬细胞相关疾病的患者选择和治疗监测
在一个方面,本发明提供了治疗患有巨噬细胞相关疾病的受试者的方法,该方法包括向有需要的受试者施用有效量的氧化剂。本发明还提供了对患有巨噬细胞相关疾病的受试者亚群进行鉴定。所述受试者亚群可以比患有巨噬细胞相关疾病的其他受试者亚群更有效地响应氧化剂的施用。本发明还提供了通过测量一种或多种炎症因子的血浆水平来鉴定患有巨噬细胞相关疾病的受试者的方法。患有巨噬细胞相关疾病的受试者的一种或多种炎症因子的血浆水平可以高于阈值水平、其正常水平或其疾病水平。所述氧化剂可以包括但不限于亚氯酸盐。
巨噬细胞相关疾病可以是异质的。在一些情况下,巨噬细胞相关疾病也可能部分地与基因突变相关。根据疾病进展的程度或疾病的病因,受试者对氧化剂治疗响应的有效性可能不同。本发明还提供了通过施用氧化剂来监测巨噬细胞相关疾病的治疗的方法。为了防止不必要的治疗以及较好地诊断患有该疾病的亚群,正确鉴定可以积极响应氧化剂(例如,亚氯酸盐)治疗的患有巨噬细胞相关疾病的受试者是重要的。
I.炎症因子筛选
在一些情况下,可以积极响应施用氧化剂的治疗的患有巨噬细胞相关疾病的受试者可以通过测量受试者血浆或血液中的一种或多种炎症因子的水平来进行鉴定并随后进行治疗。所述受试者的一种或多种炎症因子的血浆水平分别高于所述一种或多种炎症因子的阈值水平、正常水平或疾病水平。可以连续监测不具有高于阈值水平、正常水平或疾病水平的炎症因子的血浆水平的受试者的一种或多种炎症因子的水平,以确定该治疗的正确时机。
所述炎症因子可以包括但不限于IL-18、LPS、IL-6、INF-g、CRP、IL-8、wrCRP和它们的组合。在优选实施方案中,可以通过测量至少一种炎症因子例如IL-18、LPS或二者的血浆水平,来鉴定并治疗受试者。在优选实施方案中,可以通过测量IL-6和INF-g的血浆水平来鉴定并治疗受试者。
患有巨噬细胞相关疾病的可以积极响应施用氧化剂治疗的受试者的IL-18血浆水平可以高于阈值水平、正常水平或疾病水平。在血浆中的阈值水平可以为约或至少约或大于约30、40、50、60、70、80、90、100pg/ml。阈值水平可以为约、至少约或大于约60pg/mL。测得的水平可以比阈值水平、正常水平或疾病水平高至少10%、15%、20%、25%、30%、35%、40%、45%、50%、60%、70%、80%、90%、1倍、2倍、3倍、4倍或5倍。
IL-18的血浆水平可用作筛选患有巨噬细胞相关疾病的受试者的标志物。在一些情况下,施用如本文公开的包含亚氯酸盐的组合物之前和之后的IL-18血浆水平的比较可以指示所述巨噬细胞相关疾病治疗的功效。
本文提供了治疗患有巨噬细胞相关疾病的受试者的方法,所述方法包括:a)如果患有巨噬细胞相关疾病的受试者具有选自LPS、IL-6、IL-8、IL-18、IFN-g和CRP的一种或多种炎症因子的升高的血浆水平,则选择所述受试者;以及b)向所述受试者施用治疗有效量的包含亚氯酸盐的药物组合物。
本文进一步提供了将患有巨噬细胞相关疾病的受试者诊断为可以采用包含亚氯酸盐的药物组合物治疗的方法,该方法包括:a)测量选自LPS、IL-6、IL-8、IL-18、IFN-g和CRP的一种或多种炎症因子的血浆水平;以及b)如果所述受试者具有所述一种或多种炎症因子的升高的血浆水平,则将所述受试者诊断为患有可以采用包含亚氯酸盐的药物组合物治疗的巨噬细胞相关疾病。如果IL-18的血浆水平为至少约60pg/mL,则患有巨噬细胞相关疾病的受试者可以采用包含亚氯酸盐的药物组合物进行治疗。如果LPS的血浆水平为至少约0.05EU/mL,则患有巨噬细胞相关疾病的受试者可以采用包含亚氯酸盐的药物组合物进行治疗。如果IL-6的血浆水平为至少约6pg/mL,则患有巨噬细胞相关疾病的受试者可以采用包含亚氯酸盐的药物组合物进行治疗。如果INF-g的血浆水平为至少约20pg/mL,则患有巨噬细胞相关疾病的受试者可以采用包含亚氯酸盐的药物组合物进行治疗。如果CRP的血浆水平为至少约1000ng/mL,则患有巨噬细胞相关疾病的受试者可以采用包含亚氯酸盐的药物组合物进行治疗。
本文进一步提供了用于选择针对采用包含亚氯酸盐的药物组合物治疗的响应者的方法,该方法包括:a)在患有巨噬细胞相关疾病的受试者中测量选自LPS、IL-6、IL-8、IL-18、IFN-g和CRP的一种或多种炎症因子的血浆水平;以及b)如果所述受试者具有一种或多种炎症因子的升高的血浆水平,则选择所述受试者来施用亚氯酸盐的药物组合物。
巨噬细胞相关疾病可以是神经退行性疾病,包括但不限于肌萎缩性侧索硬化症(ALS)、阿尔茨海默病和帕金森病或HIV相关神经认知障碍(HAND)。巨噬细胞相关疾病还可以是选自以下的婴儿期或儿童期神经退行性疾病:软骨发育不全及变体(DE)、急性小脑性共济失调(SE)、急性延迟性麻疹脑炎(Lyon)(PE)、急性播散性脑脊髓炎(LE)、急性出血性坏死性脑白质炎(LE)、肾上腺脑白质营养不良及变体(LE)、肾上腺脊髓神经病(LE)、屈肌痉挛爱尔卡迪综合征、胼胝体发育不全、视神经发育不全(andoptic hypoplasia)(DE)、具有退行性特征的白化病及变体(DE)、Albright遗传性骨营养不良症(CE)、酒精中毒性脑病(DE)、Alexander类纤维素性脑白质营养不良及变体(LE)、Alpers灰质营养不良(PE)、α-氨基脂肪酸尿症(DE)、α-酮己二酸尿症(DE)、α-甲基-β-羟丁酸尿症(DE)、Angleman快乐木偶综合征(DE)、精氨酸血症(Arginemia)(DE)、精氨琥珀酸尿症(DE)、天冬氨酰葡糖胺尿症(DE)、共济失调毛细血管扩张症(CE)、伴随脑灰质病的孤独症(PE)、Balo同心性轴周性脑炎(Balo encephalitis periaxialis concentrica)(LE)、Bassen-Kornzweig病(SE)、Behget综合征(CE)、Behr视觉-脊髓小脑变性(SE)、Biemond后柱性共济失调(SE)、Bloch-Sulzberger病(色素失调症)(DE)、蓝尿布综合征(DE)、Canavan海绵状脑白质营养不良(LE)、氨甲酰磷酸合成酶缺乏症(DE)、一氧化碳脑病(Carbon monoxide encephalopathy)(CE)、肉碱缺乏症(DE)、肌肽血症(高肌肽血症)(DE)、脑桥中央髓鞘溶解症(LE)、脑肝肾综合征(Zellweger病)(DE)、脑腱黄瘤病(DE)、腓骨肌萎缩症及变体(SE)、Chediak-Higashi病(DE)、慢性先天性"torch"脑病(PE)、伴随晚期变性的慢性先天性弓形体病(PE)、慢性巨细胞病毒感染(PE)、伴随肝功能不全的慢性脑病(CE)、伴随肺功能不全的慢性脑病(DE)、慢性遗传性脊髓小脑变性(SE)、慢性淋巴细胞性脑膜炎(DE)、慢性锰毒性脑病(Chronic manganese encephalopathy)(CE)、伴随肌阵挛的慢性"torch"脑病(CE)、慢性中毒性脑病(PE)、瓜氨酸血症(DE)、Cockayne综合征(LE)、伴随间质性角膜炎、眩晕和耳聋的Cogan综合征(SE)、伴随脑病的胶原-血管综合征(DE)、先天性脱髓鞘脑病(Mackay)(DE)、先天性痛觉丧失(CE)、先天性肌磷酸化酶缺乏症(SE)、Conradi先天性钙化性软骨发育不良(Conradi chondrodystrophia calcificans congenita)(DE)、颅缝早闭(DE)、Crigler-Najjar核黄疽及变体(CE)、皮肤脑膜性黑变病(Cutaneous meningeal melanosis)(DE)、胱硫醚尿症(DE)、胱氨酸病(DE)、胱氨酸尿症(DE)、胞质酪氨酸氨基转移酶缺乏症(DE)、Delange-Brachmann综合征(LE)、Delange先天性肌肉肥大及锥体束外失调(Delange congenital muscle hypertrophy and extrapyramidal disturbances)(CE)、Devic视神经脊髓炎(LE)、糖尿病性脑病(DE)、伴随空腔形成的播散性脑软化(Stevenson,Ford)(LE)、播散性肉瘤脑白质病(LE)、Vogt双侧手足徐动症(status demyelinasatus)(CE)、伴随痴呆的唐氏综合征(DE)、变形性肌张力障碍及变体(CE)、Fabry弥漫性躯体性血管角化瘤(DE)、Fahr病(CE)、家族性钙化脑灰质病(Geylin,Penfield)(PE)、家族性变性锥体束外综合征(CE)、家族性肥厚性间质神经炎(Dejerine-Sottas)(SE)、家族性肥厚性副蛋白多神经炎(Gibberd,Gabrilescu)(SE)、家族性高铁血红蛋白症(DE)、家族性多腔脑软化症(Crome,Williams)(LE)、家族性橄榄体脑桥小脑变性及变体(Konigsmark,Weiner)(SE)、家族性阵发舞蹈-手足徐动-肌张力障碍(Familial paroxysmal chorea-athetosis-dystonia)(CE)、家族性蛋白质不耐受(DE)、家族性纹状体变性(CE)、家族性Werdnig-Hoffmann进行性脊髓性肌萎缩(SE)、法伯脂肪肉芽肿病(Farber lipogranulomatosis)(LE)、Fazio-Londe家族性肌萎缩性侧索硬化症(SE)、伴随脑病的头骨纤维结构发育不良(DE)、局灶性真皮发育不全(Gorlin)(DE)、Ford“312”基底神经节综合征(CE)、Ford“312”脊髓小脑综合征(SE)、Friedreich共济失调(SE)、额颞痴呆、额颞叶变性、果糖不耐症及变体(DE)、半乳糖血症及变体(DE)、GM神经节苷脂沉积症及变体(PE)、GM2神经节苷脂沉积症及变体(Tay-Sachs病)(PE)、Gaucher病及变体(DE)、遗传性呆小症(DE)、巨轴突神经病(SE)、谷氨酸脱氢酶缺乏症(脊髓小脑变性)(SE)、谷氨酰半胱氨酸合成酶缺乏症(DE)、戊二酸尿症及变体(DE)、谷胱甘肽血症(DE)、甘油激酶缺乏症(Guggenheim)(DE)、Glycopeptidosis(DE)、伴随铜代谢缺陷的Haas伴性疾病(CE)、Hallervorden-Spatz病(CE)、伴随脉络膜炎、白癜风和耳聋的Harada综合征(SE)、Hartnup病(SE)、Heller痴呆(PE)、Hematosidosis(GM3合成代谢神经节苷脂沉积症)(PE)、血红蛋白脑病(DE)、血友病脑病及变体(DE)、遗传性眼球萎缩(Fazio-Londe)(SE)、遗传性小脑共济失调(Menzel,Holmes)(SE)、伴随智力缺陷的遗传性小脑共济失调(Norman,Jervis)(SE)、遗传性出血性毛细血管扩张症(DE)、伴随脑病的遗传性黄斑营养不良(DE)、遗传性运动感觉神经病(England,Denny-Brown)(SE)、遗传性肌阵挛脑病(CE)、遗传性灰质营养不良(PE)、遗传性感觉神经病(Hicks,Denny-Brown)(SE)、遗传性痉挛性截瘫(SE)、家族遗传性臂丛神经炎(Taylor)(SE)、伴随脊髓病的带状孢疹(SE)、Hippel-Lindau成血管细胞瘤(DE)、组氨酸血症及变体(DE)、组织细胞增多症及变体(DE)、Holmes-Logan婴儿CNS变性(CE)、羧化全酶缺乏(Biotin)(DE)、Homocarnosinuria(DE)、高胱氨酸尿症及变体(DE)、亨廷顿病(CE)、Hunt青少年型震颤麻痹(家族性)(CE)、Hunt青少年型震颤麻痹(散发性)(CE)、伴随弥散性脑病的高血氨症(DE)、Hyper-B-贫血(DE)、坏死性脑病高脑肽啡综合征(Brandt)(CE)、高甘氨酸血症(非酮症)(DE)、伴随丙戊酸盐治疗的高甘氨酸血症(DE)、高赖氨酸血症(DE)、高蛋氨酸血症(DE)、高苯丙氨酸血症及变体(DE)、高呱啶酸血症(Hyperpipecolatemia)(DE)、高脯氨酸血症及变体(DE)、高色氨酸血症(DE)、高缬氨酸血症(DE)、低磷酸酯酶症(DE)、伴随婴儿痉挛的缺氧退行性脑病(DE)、缺氧退行性脑灰质病(CE)、伴随婴儿痉挛的缺氧退行性脑灰质病(PE)、特发性退行性脑病(DE)、特发性痴呆/孤独症(PE)、伴随脑灰质病的特发性痴呆(PE)、特发性甲状旁腺功能减退(DE)、特发性散发性脑灰质病(PE)、特发性皮层下变性(CE)、伴随脑病的免疫缺陷综合征(遗传性)(DE)、伴随脑病的免疫缺陷综合征(散发性)(DE)、婴儿神经元变性(Steiman,Radermacher)(CE)、婴儿多发性阵挛(CE)、异戊酸血症(DE)、伴随慢性脑病的Jervis胆固醇沉积(DE)、I型Joseph病(SE)、青少年Creutzfeldt-Jakob病(CE)、青少年播散性硬化(LE)、青少年张力障碍性脂沉积症(CE)、青少年神经轴突营养不良(CE)、毛囊角化病(DE)、核黄疽(CE)、Krabbe球形细胞脑白质病及变体(LE)、库鲁病(CE)、乳酸血症(DE)、乳糖神经酰胺过多症(Lactosyl-ceramidosis)(PE)、Laurence-Moon-Biedl综合征(DE)、慢性铅中毒脑病(DE)、Leber遗传性视神经病(DE)、Leigh亚急性坏死性脑脊髓炎及变体(CE)、Lennox-Gastaut综合征(PE)、矮怪病(Donohue)(DE)、麻风病痴呆(DE)、Lesch-Nyhan病(CE)、Economo昏睡性脑炎(CE)、Letterer-Siwe组织细胞增多症(DE)、伴随不整红纤维的脑白质病(LE)、伴随脑病的Jadassohn线性皮脂腺痣(DE)、伴随脑病的脂质营养不良性肌肉肥大(DE)、Lowe眼脑肾综合征(PE)、赖氨酸不耐症(DE)、伴随脑病的吸收障碍综合征(DE)、恶性丘疹(DE)、槭糖尿病及变体(LE)、Marfan病(DE)、Marinesco-Sjogren-Garland综合征(SE)、Menkes毛发灰白营养不良(PE)、代谢性灰质营养不良(PE)、异染性脑白质营养不良及变体(LE)、甲基丙二酸血症及变体(DE)、伴随扑翼样震颤的甲泛葡胺脑病(CE)、Mollaret复发性脑膜炎(SE)、粘脂贮积病及变体(PE)、粘多糖贮积病及变体(PE)、粘硫脂病(Mucosulfatidosis)(DE)、多发性脑脊髓动静脉畸形(Wyborn-Mason)(CE)、伴随慢性脑病的多发性脂肪过多症(DE)、多系统神经元变性(Dyck)(DE)、伴随进行性颅神经麻痹的肌阵挛脑病(Dyken)(CE)、肌阵挛+综合征(Myoclonic-plus syndrome)(Dyken)(CE)、新生儿内毒素脑病(DE)、神经纤维瘤(DE)、伴随痴呆的神经鱼鳞癣(DE)、神经元蜡样脂褐质沉积症及变体(PE)、Nevus unis lateris(DE)、Niemann-Pick神经鞘磷脂沉积病及变体(PE)、Norman-Wood先天性黑蒙性家族性白痴(PE)、伴随脑病的营养缺乏综合征(DE)、忽布房病(Oasthouse urine disease)(DE)、寡糖贮积病及变体(PE)、眼肌麻痹+综合症综合征(Ophthalmoplegia-plus syndrome)(CE)、Opsoclonic脑膜脑炎(CE)、Opticocochlodentatic变性(Opticocochlodentatic degeneration)(DE)、有机汞小脑变性(SE)、鸟氨酸转氨甲酰酶缺乏(Ornithine carbamylase deficiency)(DE)、鸟氨酸血症(HHH综合征)(DE)、正染色脑白质营养不良及变体(LE)、骨硬化病(DE)、5-氧代脯氨酸血症(谷胱甘肽合成酶缺乏症)(DE)、伴随脑病的Parry-Romberg面偏侧萎缩(DE)、Pelizaeus-Merzbacher病及变体(LE)、过氧化物酶缺乏症(Boehme)(CE)、苯丙酮尿症及变体(LE)、苯妥英小脑变性(Phenytoin cerebellar degeneration)(SE)、苯妥英痴呆/变性(PE)、Leri骨化过早症(DE)、先天性皮肤异色病(DE)、Pompe病(SE)、卟啉症及变体(PE)、百日咳后脑病(PE)、接种后脑病(PE)、大脑原发性胶质增生(Primary gliosis of the brain)(DE)、早衰症(Hutchinson-Gilford)(DE)、早衰症(Werner)(DE)、伴随光敏感的渐进性痴呆(Kloepfer)(LE)、进行性遗传性骨干发育不良(Engelmann)(DE)、进行性遗传性神经性耳聋(SE)、进行性苍白球变性症(Winkelman)(CE)、进行性风疹全脑炎(LE)、丙酸血症及变体(DE)、丙酮酸羧化酶缺乏症(CE)、丙酮酸脱氢酶复合物缺乏症(CE)、辐射诱导的脑病(DE)、破碎红线粒体病(Ragged-red mitochondrial disease)(Kearns-Sayre)(CE)、Ramsay-Hunt齿状核红核萎缩(CE)、Refsum病(多神经炎型遗传性共济失调)(SE)、Rendu-Osler-Weber血管瘤病(DE)、Riley-Day家族性自主神经异常(CE)、Roussy-Levy病(SE)、Rubinstein-Taybi综合征(DE)、酵母氨酸尿(DE)、Salta病(PE)、肌氨酸血症(DE)、Schilder弥漫性轴周性脑炎(Schilder encephalitis periaxialis diffusa)(LE)、Segawa遗传性进行性肌张力障碍(每日的)(CE)、Seitelberger婴儿神经轴突性营养不良(CE)、伴随肌阵挛和锥体束外体征的伴性共济失调(CE)、伴性脑白质营养不良(LE)、Sialidoses及变体(PE)、Sotos脑性巨人症(DE)、海绵状脊髓灰质脑病(Spongiform polioencephalopathies)及变型(PE)、散发性克汀病(DE)、散发性青少年肌萎缩性侧索硬化症(SE)、散发性肌阵挛脑病(CE)、散发性橄榄体脑桥小脑变性(Dejerine-Thomas)(SE)、散发性视神经炎、球后视神经炎(LE)、散发性原发性侧索硬化(SE)、散发性渐进性丘脑萎缩(CE)、伴随肌阵挛的散发性海绵状脑病(CE)、大理石样病变(CE)、Sturge-Weber病(CE)、亚急性脊髓视神经病(肠病性肢端皮炎)(DE)、亚急性硬化性全脑炎及变体(LE)、丘脑核变性(Malmud,Denny)(CE)、Sugarman-Reed颅面白斑病(DE)、亚硫酸盐尿症(硫酸盐氧化酶)(DE)、核上性眼肌瘫痪(遗传性)(CE)、西登哈姆舞蹈病(CE)、海蓝组织细胞综合征(SE)、脊髓空洞症(家族性)(SE)、Tourette综合征(CE)、过渡弥散硬化(LE)、磷酸丙糖异构酶缺乏症(CE)、结节性硬化症(DE)、酪氨酸血症(DE)、Unverricht-Lundborg-Lafora病(CE)、Vogt-Koyanagi综合征(SE)、Waardenburg综合征(DE)、Wadia-Swami脊髓小脑变性(SE)、Weill-Marchesani综合征(DE)、Welander-Kugelberg-Wohlfart青少年脊髓性肌萎缩(SE)、West病(伴随变性的特发性婴儿痉挛症)(PE)、West病(非遗传性弥散性脑病)(DE)、Wilson肝豆状核变性及变体(CE)、Wolman脑病(LE)、以及着色性干皮症及变体(SE)。根据以上列表,CE表示核心脑病(corencephalopathies);DE表示弥散性脑病;LE表示脑白质病;PE表示脑灰质脑病(polioencephalopathies);以及SE表示脊髓小脑病(spinocerebellopathies)。
IL-18(整个体系包括IL-18、胱天蛋白酶-1、IL-18R和IL-18BP),是属于IL-1家族的细胞因子。它通过与特定的受体复合物(IL-18R)相结合来发挥作用,并且虽然其初始来源是巨噬细胞,但其表达可以在若干种不同类型的细胞,例如单核细胞、树突细胞(DC)、枯否细胞、角化细胞、软骨细胞、成骨细胞和成纤维细胞中检测到。在脑细胞中,IL-18可主要由小神经胶质细胞、星形胶质细胞、室管膜细胞和神经元表达。大脑IL-18表达可以在神经炎症事件期间在体内得到增强,以响应于不同的外源性或内源性有害刺激如大脑感染、缺氧缺血、高氧和外伤性脑损伤的有害作用。对于神经退行性疾病,尤其是阿尔茨海默病,由应激、受损或其他不正常的脑细胞产生的信号可以通过引发细胞因子的释放来活化先天性免疫系统。先前的研究表明PPR、TLR和NLR中的两个主要家族的组分涉及阿尔茨海默病神经炎症和神经变性。
不适当的TLR响应可以促成神经炎症和神经变性。对AD的先天免疫受体的研究显示了聚合的Aβ与LPS受体CD14之间的相互作用,其可通过TLR4发信号。TLR的触发(通过经由CD14结合的LPS治疗获得),可以导致转录因子NF-kB的活化,这继而调节参与炎症反应激活的一系列基因(包括IL-18)的表达。在小神经胶质细胞中,LPS可诱导IL-18的表达。在神经炎症环境内用于IL-18产生的另一种重要的信号是炎症小体的活化,其触发促炎细胞因子IL-1β和IL-18的加工和释放。此外,炎症小体对使无活性的胱天蛋白酶原-1转化为活性的胱天蛋白酶-1起关键作用,胱天蛋白酶-1能够将无活性的IL-18前体裂解成分泌性活性细胞因子。
不受任何理论约束,通常已知对于成熟的IL-18的产生需要两类事件,即增强的前体合成和前体加工。增强的前体合成主要通过TLR活化以及NF-κB转录进行调节。在另一方面,前体加工主要依赖于炎症小体的参与和胱天蛋白酶-1的活化,这两者显示都发生在阿尔茨海默病患者的大脑中。与此相一致,据观察在阿尔茨海默病患者的大脑额叶中特别地观察到IL-18蛋白和胱天蛋白酶-1的增加的表达。在这种情况下,除了沾满IL-18的星形胶质细胞和神经元,在淀粉样沉积物和神经纤维缠结的附近还观察到小神经胶质细胞。因此,很可能阿尔茨海默病特异性致病刺激,诸如Aβ累积,可以通过PPR活化导致阿尔茨海默病受试者的大脑内的IL-18释放增加。
脂多糖(LPS)(也被称为脂聚糖和内毒素),是由脂质和多糖组成的大分子,该多糖进一步包含由共价键连接的O-抗原、外核和内核。LPS是革兰氏阴性菌外膜的主要成分,对细菌结构的完整性有巨大贡献,并保护膜免受某些类型的化学侵蚀。LPS也可以提高细胞膜的负电荷,并帮助稳定整体膜结构。此外,LPS是一种诱导正常动物免疫系统应答的内毒素。
巨噬细胞相关疾病可能与炎症或小神经胶质细胞活化有关。通常,炎症和小神经胶质细胞活化被认为是多种神经退行性疾病,例如阿尔茨海默病(AD)、帕金森病(PD)、亨廷顿病(HD)、多发性硬化和肌萎缩性侧索硬化症(ALS)的病理的共同组成部分。小神经胶质细胞——大脑中的固有先天免疫细胞,积极地监测其环境,并且可以响应不同的诱因而变得过度活化从而产生细胞毒性因子,例如肿瘤坏死因子α(TNFα)。尽管小神经胶质细胞活化对宿主防御有必要并且至关重要,但小神经胶质细胞的过度活化是神经毒性的。仅在小神经胶质细胞存在时,LPS可以损害多巴胺能(DA)神经元。在体内和体外小神经胶质细胞的LPS活化均可以造成DA神经元随时间的进行性和累积损失。在胚胎发育的关键期,小鼠中母体暴露于低浓度的LPS影响后代的小神经胶质细胞活化和DA神经元的存活(这持续至成年)。而且,若干报道表明LPS激活肝中的细胞以产生TNFα,TNFα分布在血液中并通过TNFα受体转移至大脑,以诱导额外的TNFα和其他促炎症因子的合成,形成诱导成年动物中延迟和渐进性DA神经元损失的持久自推进神经炎症。LPS可以将巨噬细胞转化为活化的巨噬细胞,并可以导致不必要的炎症。
在一些情况下,脂多糖(LPS)可以用作与ALS相关的巨噬细胞功能紊乱的标志物。例如,循环LPS可以是来源于胃肠道的微生物易位的指示物,并且已被用于监测巨噬细胞相关疾病的进展,如由Brenchley等人所示的(Brenchley等人,Nature Med 2006)。LPS在慢性HIV感染的个体和猿猴免疫缺陷病毒(SIV)感染的恒河猴中显著提高(Brenchley等人,(2006)Nature Med,12:1365-1371)。在实验室研究中,循环LPS的水平的升高也可以加快例如ALS等巨噬细胞相关疾病的进展。在腹腔内经单独无毒或重复的1mg LPS/kg注射攻击后,表达与ALS相关的超氧化物歧化酶1(SOD1)的突变型的转基因小鼠加重病情进展达3周并且加重机动轴突退化(Nguyen等人,(2004)J Neuroscience,24(6):1340-1349)。在另一项研究中显示大鼠脊髓中巨噬细胞的LPS活化导致特定的运动神经元损失(Li等人,Brain Res,(2008)1226:199-208)。更新的研究表明,ALS血液单核细胞表达与疾病的严重程度无关的LPS活化基因(Zhang等人,JNI(2011),230:114-123)。在我们的临床试验数据中,与LPS阳性阶段(DPR-0.88U/月)相比早期没有血浆LPS的ALS(DPR-0.55U/月)进展较慢(Neuraltus IIA trial,2014)。
患有巨噬细胞相关疾病的可以积极响应施用氧化剂治疗的受试者的LPS的血浆水平可以高于阈值水平、正常水平或疾病水平。所述阈值水平可以为约或至少约或大于约0.01、0.05、0.1、0.15、0.2EU/ml或血浆中的任何可检测水平。所述阈值水平可以为约、至少约或大于约0.1EU/ml。治疗前测得的水平可以比阈值水平、正常水平或疾病水平高至少10%、15%、20%、25%、30%、35%、40%、45%、50%、60%、70%、80%、90%、1倍、2倍、3倍、4倍或5倍。
LPS的血浆水平可用作筛选患有巨噬细胞相关疾病的受试者的标志物。在一些情况下,施用如本文公开的包含亚氯酸盐的组合物之前和之后的LPS血浆水平的比较可以指示所述巨噬细胞相关疾病治疗的功效。所述巨噬细胞相关疾病可以是神经退行性疾病,包括但不限于肌萎缩性侧索硬化症(ALS)、阿尔茨海默病和帕金森病或HIV相关神经认知障碍(HAND)。例如,与所述治疗前LPS的血浆水平相比,所述治疗后LPS的血浆水平可减少至少10%、15%、20%、25%、30%、35%、40%、45%、50%、60%、70%、80%、90%、1倍、2倍、3倍、4倍或5倍。
患有巨噬细胞相关疾病的可以积极响应施用氧化剂治疗的受试者的IL-6的血浆水平可以高于阈值水平、正常水平或疾病水平。在血浆中的阈值水平可以为约或至少约或大于约1、2、3、4、5、6、7、8、9、10pg/ml。所述阈值水平可以为约、至少约或大于约6pg/ml。治疗前测得的水平可以比阈值水平、正常水平或疾病水平高至少10%、15%、20%、25%、30%、35%、40%、45%、50%、60%、70%、80%、90%、1倍、2倍、3倍、4倍或5倍。
患有巨噬细胞相关疾病的可以积极响应施用氧化剂治疗的受试者的INF-g的血浆水平可以高于阈值水平、正常水平或疾病水平。在血浆中的阈值水平可以为约或至少约或大于约5、10、15、20、25、30、35或40pg/ml。所述阈值水平可以为约、至少约或大于约20pg/ml。治疗前测得的水平可以比阈值水平、正常水平或疾病水平高至少10%、15%、20%、25%、30%、35%、40%、45%、50%、60%、70%、80%、90%、1倍、2倍、3倍、4倍或5倍。
患有巨噬细胞相关疾病的可以积极响应施用氧化剂治疗的受试者的CRP的血浆水平可以高于阈值水平、正常水平或疾病水平。在血浆中的阈值水平可以为约或至少约或大于约100、200、300、400、500、600、700、800、900、1000、1100、1200、1300、1400、1500、1600、1700、1800、1900或2000ng/ml。所述阈值水平可以为约、至少约或大于约1000ng/ml。测得的水平可以比阈值水平、正常水平或疾病水平高至少10%、15%、20%、25%、30%、35%、40%、45%、50%、60%、70%、80%、90%、1倍、2倍、3倍、4倍或5倍。
IL-6和INF-g的血浆水平可用于鉴定、筛选以及随后通过向受试者施用氧化剂来治疗患有巨噬细胞相关疾病的受试者。IL-18的血浆水平可以是用于鉴定、筛选以及随后治疗受试者的强有力的指示标志物。IL-18的血浆水平高于某一水平的受试者可以积极地响应施用氧化剂的治疗。在一些情况下,LPS的血浆水平也可以是用于受试者筛选的另一种标志物。
可收治患有诸如ALS或AD等巨噬细胞相关疾病的受试者,并可测量一种或多种炎症因子的血浆水平。如果发现一种或多种炎症因子的测得的血浆水平高于阈值水平、正常水平或疾病水平,则该受试者随后可以用氧化剂进行治疗,并且可能预期得到正响应。如果发现所述一种或多种炎症因子较低,则可建议受试者寻求替代治疗,或者继续监测所述因子的血浆水平,以确定诸如亚氯酸盐的氧化剂治疗的最佳时机。
Ⅱ.治疗监测
本发明还提供了用于监测用于治疗巨噬细胞相关疾病的氧化剂治疗的方法。通过了一种或多种炎症因子血浆水平筛选的患有巨噬细胞相关疾病的受试者可以用包含诸如亚氯酸盐的氧化剂的组合物进行治疗。然后在治疗期中可测量所述受试者的一种或多种生物标志物的水平。随后可将测得的生物标志物的水平与所述生物标志物的正常和疾病水平,和/或治疗前所述受试者的生物标志物水平相关联。所述生物标志物可以选自IL-18、LPS、IL-6、INF-g、CRP、IL-8、wrCRP以及它们的组合。在一些情况下,用于治疗监测的生物标志物可以是IL-18。在一些情况下,用于治疗监测的生物标志物可以是LPS。
通常,在使用包含本文公开的氧化剂的组合物治疗巨噬细胞相关疾病后,生物标志物的血浆水平降低。用于监测所述治疗的生物标志物的非限制性实例可选自IL-18、LPS、IL-6、INF-g、CRP、IL-8、wrCRP以及它们的组合。在一些情况下,一种或多种生物标志物的血浆水平可降低10%、20%、30%、40%、50%、60%、70%、75%、80%、90%或更多。在一些情况下,与治疗前生物标志物的血浆水平相比,一种或多种生物标志物的血浆水平可降低至低于约50pg/ml、约40pg/ml、约30pg/ml、约20pg/ml、约10pg/ml或约5pg/ml。例如,在施用所述组合物之前,一种或多种生物标志物的血浆水平为至少约50pg/ml、约40pg/ml、约30pg/ml、约20pg/ml、约10pg/ml或约5pg/ml。作为另一个实例,在施用所述组合物之前,一种或多种生物标志物的血浆水平为至多约50pg/ml、约40pg/ml、约30pg/ml、约20pg/ml、约10pg/ml或约5pg/ml。作为另一个实例,在施用所述组合物之前,一种或多种生物标志物的血浆水平为约5pg/ml至约50pg/ml、约8pg/ml至约12pg/ml、约10pg/ml至约30pg/ml、约15pg/ml至约25pg/ml、约25pg/ml至约35pg/ml、约30pg/ml至约45pg/ml、或约40pg/ml至约50pg/ml。在一些情况下,在向受试者施用含有诸如亚氯酸盐的氧化剂的组合物之后,血浆水平可降低至无法检测到的水平。在一些情况下,与治疗前生物标志物的血浆水平相比,一种或多种生物标志物的血浆水平可降低至低于约0.1EU/ml、约0.05EU/ml、约0.01EU/ml或约0.005EU/ml。例如,在施用所述组合物之前,一种或多种生物标志物的血浆水平为至少约0.1EU/ml、约0.05EU/ml、约0.01EU/ml或约0.005EU/ml。作为另一个实例,在施用所述组合物之前,一种或多种生物标志物的血浆水平为至多约0.1EU/ml、约0.05EU/ml、约0.01EU/ml或约0.005EU/ml。作为另一个实例,在施用所述组合物之前,一种或多种生物标志物的血浆水平为约0.005EU/ml至约0.1EU/ml、约0.01EU/ml至约0.05EU/ml、约0.04EU/ml至约0.08EU/ml、或约0.06EU/ml至约0.09EU/ml。
用于监测治疗的生物标志物可以是IL-18。在施用含有诸如亚氯酸盐的氧化剂的组合物之后,IL-18的水平降低。所述治疗可通过IL-18血浆水平的降低率进行监测。在一些情况下,IL-18的血浆水平可降低10%、20%、30%、40%、50%、60%、70%、75%、80%、90%或更多。在一些情况下,与治疗前IL-18的血浆水平相比,IL-18的血浆水平可降低至低于约50pg/ml、约40pg/ml、约30pg/ml、约20pg/ml、约10pg/ml或约5pg/ml。
用于监测治疗的生物标志物可以是LPS。在施用含有诸如亚氯酸盐的氧化剂的组合物之后,LPS的水平降低。所述治疗可通过LPS血浆水平的降低率进行监测。在一些情况下,LPS的血浆水平可降低10%、20%、30%、40%、50%、60%、70%、75%、80%、90%或更多。在向受试者施用含有诸如亚氯酸盐的氧化剂的组合物之后,LPS血浆水平可降低至无法检测到的水平。在一些情况下,与治疗前LPS的血浆水平相比,LPS的血浆水平可降低至低于约0.1EU/ml、约0.05EU/ml、约0.01EU/ml或约0.005EU/ml。
本发明还提供了通过将至少一种单核细胞活化标志物的血浆水平与施用所述组合物之前和之后的受试者的所述单核细胞活化标志物的血浆水平进行比较,来监测巨噬细胞相关疾病的炎症进展的方法。所述方法还提供了如果与所述施用前所述单核细胞活化标志物的血浆水平相比,所述施用后所述单核细胞活化标志物的血浆水平发生变化则确定治疗继续的指示。通过了一种或多种炎症因子血浆水平筛选的患有巨噬细胞相关疾病的受试者可以用包含诸如亚氯酸盐的氧化剂的组合物进行治疗。然后在治疗期中可测量所述受试者的一种或多种生物标志物的水平。随后可将测得的生物标志物的水平与所述生物标志物的正常和疾病水平,和/或治疗前所述受试者的生物标志物水平相关联。所述生物标志物可以选自IL-18、LPS、IL-6、INF-g、CRP、IL-8、wrCRP、CD16、HLA-DR、CD14以及它们的组合。在一些情况下,用于监测炎症进展的生物标志物可以是单核细胞活化标志物,例如CD16。在一些情况下,用于监测炎症进展的生物标志物可以是单核细胞活化标志物,例如HLA-DR。在一些情况下,用于监测炎症进展的生物标志物可以是单核细胞活化标志物,例如CD14。任选地,用于监测炎症进展的生物标志物可以是选自HLA-DR、CD14和CD16的单核细胞活化标志物。可以在施用本发明组合物之前至少约24小时,或之后至少24小时测量受试者的至少一种单核细胞活化标志物的血浆水平。在所述施用之前,至少一种单核细胞活化标志物的血浆水平可升高并高于或为至少约正常水平。在一些情况下,单核细胞活化标志物的血浆水平的升高可以与巨噬细胞相关疾病的进展率相关联,例如单核细胞活化标志物的血浆水平的升高可以提高巨噬细胞相关疾病的进展率。通常,在所述施用之后至少一种单核细胞活化标志物的血浆水平可降低例如至少约5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、60%、70%、80%、90%、1倍、2倍、3倍、4倍、5倍或更多倍。在一些情况下,一种或多种单核细胞活化标志物的血浆水平可降低至无法检测到的水平。
施用如本文公开的包含亚氯酸盐的组合物可以减少巨噬细胞相关疾病的进展,该疾病例如肌萎缩性侧索硬化症(ALS)、阿尔茨海默病(AD)帕金森病(PD)以及HIV相关神经认知障碍(HAND),其他神经退行性病症可以包括亨廷顿病(HD)和多发性硬化。在一些情况下,使用ALSFRS-R评分量表作为指示时,施用所述组合物可将巨噬细胞相关疾病的进展减少至少0.2单位/月、0.4单位/月、0.5单位/月、0.6单位/月、0.8单位/月、1.0单位/月、1.2单位/月、1.5单位/月、1.8单位/月、2单位/月、3单位/月、4单位/月、5单位/月或更多。例如,巨噬细胞相关疾病的进展可减少至少0.5单位/月。作为又一个实例,巨噬细胞相关疾病的进展可减少至少1.0单位/月。
实施例
实施例1-具有不同LPS基线水平的ALS的治疗
在六个周期内执行随机、双盲、以安慰剂为对照的亚氯酸盐/NP001试验。对根据E1Escorial标准被诊断为可能的、很可能的或确定的ALS的年龄为21至80岁的136位男性和女性进行登记(图1)。要求所有参与者在施用第一剂量的研究药物前ALS相关虚弱发作少于3年,根据年龄和身高预测的FVC≥70%,且预期寿命>6个月。接受利鲁唑的参与者必须施用稳定剂量>30天。在12周的随机选择内处于CPAP或BiPAP、患有正在治疗的活动性肺病以及接受免疫治疗剂的参与者被排除在外。在随机选择后需要BiPAP、CPAP或胃造口术的参与者可以留在研究中。
所述研究根据优质临床实践的规则进行,并在每个地点被适当的机构审查委员会和管理机构所批准。从所有患者处获得了知情同意书。所述研究在clinicaltrials.gov进行了注册(NCT01281631)。
以1:1:1的方式分配参与者在6个月的治疗期内接受亚氯酸盐/NP001 2mg/kg、亚氯酸盐/NP001 1mg/kg或安慰剂。研究药物通过输液泵在30分钟内输注。患者预期在25周(6个月)的双盲治疗期内经6个周期接受总共20次输注(图2)。每个周期开始时间之间相隔大约4周。第1周期/第1周包括5次连续的每日输注。第2、3、4、5和6个周期(分别为第5、9、13、17和21周)各自包括3次连续的每日输注。给药方案基于之前HIV群体的数据(McGrath等人,(2002)Curr.Opin.Investig.Drugs 3:365-373)。在最后一次输注之后4周(第25周),对受试者进行治疗期结束拜访。随后每位患者具有12周的随访期,其包括3次连续的每月拜访(第19、33和37周)。在每个给药周期的第一天,以及在第25、29、33和37周测定ALSFRS-R和VC。研究调查员、地方工作人员和ALSFRS-R评估者对整个研究中治疗的分配不知情。在试验过程中IDMC周期性地对数据进行评估。
90(90/136)位患者完成了该研究。考虑到该研究的探究性,在6个月的治疗期中,最终样品大小仅有大约65%的能力检测出ALSFRS-R的估计下降斜率的30%的差异(双侧,α=0.10)。事先限定的次级分析方法包括使用与本研究分析匹配的历史安慰剂数据库的ALSFRS-R数据。对于该分析,通过过滤本研究的基线特征(延髓起源与肢体发病,VC,虚弱症状的持续时间,年龄)的数据制备匹配的历史安慰剂受试者的样品,并将该样品加入到本实验的安慰剂受试者数据中。这允许点评估的能力增强且精确度增加,导致具有87%的能力检测如通过ALSFRS-R斜率评估的疾病进展的30%的改善。
ALSFRS-R斜率分析使用具有随机效应的一般线性混合效应模型来估计从基线到治疗期完成的ALSFRS-R下降率(斜率)(表示为每月的分数)。对斜率终点的次级分析包括添加匹配的历史安慰剂对照。使用ANCOVA分析进行的ALSFRS-R评分的相对于基线变化进行至25周的治疗期的结束,从12周的随访期的开始到结束(从第25周到第37周)进行、以及从基线到随访期的结束(从第1周到第37周)进行。使用了以下的协变量:年龄、种族、性别、利鲁唑使用、持续时间、ALS发作的类型和部位、el Escorial标准、基线ALSFRS-R和肺活量(VC)。对每个剂量组和安慰剂组进行斜率和相对于基线变化的成对比较。分析了相同时期VC相对于基线的变化,以及进行了使用ALSFRS-R域分项得分、性别、发作部位以及基线wrCRP或单核细胞趋化蛋白-1(MCP-1)高于或等于整个参加群体的基线中值的患者的斜率的子集分析。使用相对于基线的描述性统计和百分比变化分析治疗期间的生物标志物浓度。没有指配(impute)缺失数据。对在6个月的治疗期中每个组中病情改善或没有进展的患者(“响应者”)的百分比(如通过ALSFRS-R评分中相对于基线的变化评估的)进行事后(post-hoc)探索性分析。
通过对治疗突发不良事件(TEAE)的计数和列表来评估安全性和耐受性数据,治疗突发不良事件被定义为在第一次给药期间或之后以及在最后一次给药研究药物后30天之内发生的事件以及化验值、生命体征、体检和EKG的相对于基线的变化。没有进行对TEAE的正式统计学分析。
为试验筛选了166位患者,并排除了30位患者。总共136位受试者参加并被随机分配(图1)。进行随机化和接受研究药物和早期终止的受试者没有被替换。
每组中大约95%或更多的患者完成了第1周期计划的所有5次输注。每组中大部分受试者完成了6个给药周期(78%至90%)。所有3个组中大部分受试者完成了随访(78%至84%)。
表2列出了患者群体的基线人口统计数据和临床特征。尽管在数字上安慰剂组ALSFRS-R≥42的患者百分比(28%)高于NP001 1mg/kg组(18%)和NP001 2mg/kg组(20%),但这些组具有相似的人口统计数据和相似的ALS基线特征。组之间的基线平均wrCRP和MCP-1值相似。各组的基线特征之间没有显著差异。
表2基线人口统计数据和疾病特征
an=随机化的患者数目。除非另有说明,所有值均为平均值+/-SD
表3示出了发生在≥5%的患者中的最常见的TEAE。在治疗组之间没有记录生命体征或ECG参数的相对于基线的临床相关平均变化。
表3发生在≥5%的患者中的最常见的TEAE
图3A和3B中示出了在6个月的治疗(第1周到第25周)后,在有和没有匹配的历史安慰剂对照的情况下,ALSFRS-R评分中相对于安慰剂的平均斜率和相对于基线的平均变化。
NP001 2mg/kg与安慰剂相比在减少ALS进展方面具有有数值上的临床益处,如通过以每月点数表示的平均斜率百分比变化(13%)所示的。在NP001 2mg/kg组中平均斜率为-0.77,而在安慰剂组中平均斜率为-0.89。在将匹配的历史安慰剂对照患者添加到并行的安慰剂中时,安慰剂组的平均斜率为-0.95,因此,与结合的安慰剂对照相比,高剂量组在进展率方面有19%的改善(p=0.16)。在添加和不添加匹配的历史安慰剂对照患者的情况下观察到高剂量组中相对于基线的ALSFRS-R变化的相似的临床益处,分别减缓21%(添加)和减缓17%(不添加)。相似的分析证明与安慰剂相比,NP001 1mg/kg剂量是几乎无效或无效的剂量。
图3C示出了基线wrCRP水平处于或高于全部随机化群体的中值的使用NP001治疗的患者与基线wrCRP值也处于或高于该中值的安慰剂患者相比,进展减慢更多。以每月点数表示的估计的斜率下降在NP001 2mg/kg组中为-0.55,在NP001 1mg/kg组中为-0.73,而在安慰剂组中为-0.93。2mg/kg组中进展率的减缓代表与安慰剂相比41%的改善(p=0.2)。在低于基线中值wr-CRP的患者中,2mg/kg、1mg/kg和安慰剂组的估计的斜率分别为-0.87、-1.38和-0.84。斜率差异的仅有趋势存在于NP001 1mg/kg组与安慰剂组的比较中(p=0.09)。
在3个月的停药随访中,ALSFRS-R评分的平均下降在NP001 2mg/kg组中为-3.3,在NP001 1mg/kg组中为-3.7,而在安慰剂组中为-3.7。因此,在停药随访期间与高剂量NP001组的安慰剂相比,到治疗期结束的ALSFRS-R评分的相对于基线的平均变化有剩余11%的功能性下降减缓。NP001 2mg/kg组与安慰剂组相比,ALSFRS-R评分中相对于基线的平均变化更小的临床趋势在所有不同的时间段(治疗期和随访期)中是一致的,尽管没有达到统计学上的显著性。
实施例2–响应者和无响应者的生物标志物的检测
在六个周期内执行随机、双盲、以安慰剂为对照的研究。使用亚氯酸盐2mg/kg/输注或安慰剂来治疗患者。患者预期在25周(或6个月)的治疗期中经6个周期接受总共20次输注。在至少6个月的治疗(第25周)之后ALSFRS-R评分的相对于基线的变化≥0(即,ALSFRS-R评分没有下降或者改善)的患者被定义为“响应者”。在不同的时间点,例如,在基线(第1周期、第1周给药前)、第1个月、2个月、3个月、4个月、5个月和6个月收集并测试来自响应者和无响应者的样品。在选定的时间点测量响应者和无响应者的ALSFRS-R评分和生物标志物水平,并将其与安慰剂组相比较。生物标志物的水平可用本领域已知的技术,例如ELISA或流式细胞术进行测量。
图4和图5示意性地图示了亚氯酸盐治疗巨噬细胞相关疾病的工作机理。通常,亚氯酸盐在单核细胞/巨噬细胞内转化成下调NF-kB表达并且抑制促炎细胞因子IL-1β的产生的生物活性细胞内氯胺。在正常的巨噬细胞功能下,血浆LPS水平将消失,并且NF-kB诱导的炎症因子将减少。
结果表明亚氯酸盐治疗能够在6个月的时间段内减慢接受治疗的患者亚群的疾病进展(图6)。观察患者群体中的两个亚群并将其标记为响应者和无响应者。稳定的ALFRS-R水平表示疾病进展减慢。在响应者中在6个月的治疗期内ALSFRS-R评分保持稳定,表明响应者对该治疗的正响应。而在无响应者中观察到的ALSFRS-R值下降表明该治疗对患者具有很少的作用或没有作用。
图7示出了响应者百分比的剂量依赖性增加。在高剂量组中,27%的患者在6个月的治疗期内病情没有进展(图7)。这是安慰剂组的百分比(11%)的大约2.5倍。在向分析中增加匹配的历史安慰剂对照时,2mg/kg组与安慰剂组相比优势非常明显(p=0.007)。与这些发现相一致,在至少6个月的治疗后,响应者的肺活量(1mg:-8.95+/-10.1;2mg:-3.76+/-5.7)与无响应者(1mg:-17.7+/-17.9;2mg:-14.5+/-13.2)相比有较小的剂量依赖性下降。
发现响应者、无响应者和安慰剂组的四种生物标志物,即IL-18、IL-6、INF-g、CRP的基线水平也不同(图8)。对于每种生物标志物,其在响应者、无响应者和安慰剂组中的基线水平均相对于其在响应者中的基线水平进行归一化。因此,对于所有的生物标志物,它们在响应者中的基线水平均为100。如图所示,与无响应者和安慰剂组相比,响应者所有生物标志物的基线水平都是最高的。此外,图8和图9示出了在高剂量组中,与无响应者相比,响应者的基线IL-18、IL-6、INF-γ和CRP升高。图10示出了ROC曲线,以比较每种标志物预测响应者的能力的曲线下面积。
生物标志物或炎症因子分析显示响应者、无响应者和安慰剂组在基线(第1周期、第1周给药前)和第25周的IL-18水平不相同(图11)。图12示出了响应者、无响应者和安慰剂无进展者的一些炎症因子在基线和第25周的血浆水平。所述“安慰剂无进展者”指安慰剂组中在研究持续时间内没有显示疾病进展的亚群。图13示出了响应者和安慰剂无进展者在基线和第25周的IL-18、IL-6、IL-8、CRP、wrCRP和INF-g的血浆水平。
与无响应者和安慰剂组相比,通过表现减慢的疾病进展而响应亚氯酸盐治疗的患者(响应者)具有较高的IL-18基线水平(图14-15)。在25周后在无响应者和安慰剂组两者中均出现IL-18水平相对于基线的上升,而在25周的治疗后在响应者中发现IL-18水平相对于基线下降,这表明该治疗仅在响应者中有效。图16示出了IL-18的对数分布盒须图,显示IL-18基线水平可以区分出响应者和无响应者。图17示出了炎症因子血浆基线值的相互关系。
还测量了响应者和无响应者在基线和第24周的LPS水平。如图18所示,通过采用有效的治疗,可以治愈巨噬细胞功能障碍并且可以观察到LPS水平的降低。在24周的治疗后在响应者和无响应者中均观察到LPS水平的这样的降低。对于无响应者,LPS水平从在基线的初始值约0.35下降至第24周的最终值约0.2(相对于其基线水平下降了约40%)。而对于响应者,在24周的治疗后,LPS水平从约0.3的基线水平大幅降低至第24周无法检测到的水平。在24周的治疗后如果使用最小检测水平(约为0.075,并且仍高于第24周的最终水平)进行计算,相对于基线水平有约75%的下降,这比无响应者中的下降高几乎两倍。响应者的LPS水平在24周后下降到无法检测到的水平。
所有高剂量响应者血浆中的LPS都呈阳性(图19)。在至少6个月的治疗后,70%的高剂量响应者的LPS下降,并且80%的高剂量响应者的IL-18下降(图14-15,图18)。与低剂量组相似,8位响应者中有7位是LPS阳性的,并且75%的基线IL-18处于或高于所有患者的基线中值。在至少6个月的治疗后,一半患者的LPS下降,并且1位患者的IL-18下降。所有4位安慰剂响应者在基线时都是LPS阴性的;然而4位中有3位的IL-18升高。在6个月的治疗期中所有安慰剂患者(响应者和无响应者)的LPS水平明显升高(图19)。
本实验的结果证明亚氯酸盐/NP001(2mg/kg)能够减慢具有IL-18、IL-6、INF-g和CPR的高血浆水平的ALS患者的疾病进展。通过采用亚氯酸盐治疗该患者亚群的LPS水平也下降到无法检测到的水平。
实施例3–IL-18基线水平的检测
被诊断为患有ALS的32位患者在6个月的疗程中使用亚氯酸盐进行治疗。记录每位患者初始的IL-18血浆浓度并将其设定为基线水平。在6个月的疗程中采用1-2mg/kg亚氯酸盐输注对患者进行治疗。在治疗期间的不同时间点记录ALSFRS-R评分和IL-18水平,并将其与初始浓度相比较。
总的来说,观察到10位响应者和22位无响应者(图20-21)。在使用亚氯酸盐治疗后响应者的IL-18血浆水平下降。大多数响应者的基线IL-18高于60pg/ml(图21)。相比之下,无响应者的IL-18基线水平较低,并且他们中大多数的基线IL-18低于60pg/ml。
本实验的结果证明亚氯酸盐/NP001能够降低IL-18的血浆水平,并且IL-18基线水平高于60pg/ml的患者可以受益于亚氯酸盐/NP001治疗。本实验的结果还暗示基线血浆水平可能是亚氯酸盐/NP001治疗响应者的指标。
实施例4–具有不同IL-18基线水平的ALS患者的治疗
被诊断为患有ALS的患者在6个月的疗程中使用亚氯酸盐进行治疗。基于IL-18在基线处的初始浓度或其基线水平,将患者分为两组。IL-18基线水平高于60pg/ml的患者被分在高IL-18组,而IL-18基线水平低于60pg/ml的患者被分在低IL-18组。两个组都在6个月的疗程中采用1-2mg/kg亚氯酸盐输注进行治疗。在治疗期间的不同时间点记录ALSFRS-R评分和IL-18水平,并将其在高IL-18组和低IL-18组之间进行比较。
稳定的ALSFRS-R值指示疾病进展的减慢并因此指示对该治疗的正响应。ALSFRS-R评分的下降或提高表明对该治疗无有效响应或有消极响应。在整个治疗过程中在高IL-18组中ALSFRS-R水平稳定,这表明具有高IL-18基线水平的患者对亚氯酸盐治疗有正响应。而在低IL-18组中的患者中不能观察到稳定的ALSFRS-R水平,意味着亚氯酸盐治疗对该疾病没有作用或者只有很少的作用。
相似地,如果亚氯酸盐发挥了一定的作用,则可以观察到IL-18基线水平的下降。IL-18基线水平的这一下降表明该治疗的有效性以及对该治疗的正响应(仅在高IL-18组中可观察到)。
对于其他生物标志物(包括IL-6、INF-g和CRP)也发现了相同的结果。通常,在使用亚氯酸盐治疗患者6个月后,仅在生物标志物的初始基线水平比其相应的截止水平(或疾病水平)高至少20%的响应者中观察到这些生物标志物基线水平的下降。
实施例5–针对ALS进展的基线LPS水平的检测
通过比较使用1mg/kg或2mg/kg的亚氯酸盐/NP001治疗6个月之前和之后的ALSFRS-R值来确定ALS进展率。低于0.5pg/ml的基线LPS水平被认为是阴性的。
图22显示LPS基线为阴性的患者比LPS基线水平为阳性的患者疾病进展更慢。血浆LPS作为试验项标记为+或-。基于所有64位患者(2mg/kg和安慰剂组)的已知的参加试验日期和症状发作日期计算中值进展率。基于症状发作日期计算进展率。LPS基线为阴性的患者(41.2)也比LPS基线为阳性的患者(37.7)的基线ALSFRS-R评分高(见图23)。此外,如图24所示,当将在6个月的LPS血浆水平与基线比较时,没有采用亚氯酸盐/NP001治疗的LPS基线为阴性的患者(安慰剂组)在6个月内转变为LPS阳性。这些患者表现出ALSFRS-R功能评分的下降(图25)。简而言之,16/19的LPS阴性患者转变为LPS阳性;而4/19的患者在试验期间病情没有进展,而是发展成LPS阳性血液。
实施例6–ALS的治疗
虽然亚氯酸盐治疗的正响应被发现与生物标志物的初始浓度或基线水平有关,但生物标志物的高于截止值的不同浓度可能引起对治疗的不同程度的响应。
生物标志物的初始浓度高于截止值的ALS患者参加试验。研究包括IL-18、IL-6、INF-g或CRP的不同生物标志物。对于每种生物标志物,选择至少5个基线水平,并使用每对相邻水平(用于确定每组包含的范围)创建4个组。各组患者在至少6个月的过程中使用1-2mg/kg亚氯酸钠输注进行治疗。在治疗期间的不同时间点,例如,基线(第1周期、第1周给药前)、第1个月、2个月、3个月、4个月、5个月和6个月测定并记录ALSFRS-R和LPS水平。通过将在每个时间点获得的每组的所有ALSFRS-R水平值平均来计算每组的ALSFRS-R水平的平均值。通过获得每组患者的所有生物标志物基线水平的平均值来确定该组基线水平的平均值。然后将ALSFRS-R水平的平均值与生物标志物的平均基线值作图。对于所有研究的生物标志物,可以发现ALSFRS-R水平的平均值与生物标志物的平均基线水平之间具有正斜率的线性关系,这表明患者的正响应的程度与生物标志物的基线水平成比例。
在治疗过程中LPS水平的下降也表明对治疗的正响应。治疗后LPS水平下降越多意味着治疗效果越好。对于每种生物标志物,测量并记录所有组在基线和第25周的LPS水平。计算在第25周和基线的水平之间差值的绝对值并将其平均。通过获得每组内患者的所有生物标志物基线水平的平均值来确定该组基线水平的平均值。然后将治疗后生物标志物水平的差值的平均绝对值与生物标志物的平均基线水平作图。对于所有的生物标志物,患者的生物标志物基线水平越低,LPS水平下降越少,因此对治疗的响应越差。
实施例7–阿尔茨海默病(AD)的治疗
生物标志物的初始浓度高于或低于截止值的AD患者参加试验。研究包括IL-18、IL-6、INF-g或CRP的不同生物标志物。对于每种生物标志物,选择至少5个基线水平,并使用每对相邻水平(用于确定每组包含的范围)创建4个组。
各组患者在6个月的过程中使用1-2mg/kg亚氯酸钠输注进行治疗。在治疗期间的不同时间点,例如,基线(第1周期、第1周给药前)、第1个月、2个月、3个月、4个月、5个月和6个月确定并记录疾病进展评价。
预期亚氯酸盐能够治疗AD患者,并且具有不同生物标志物水平的AD患者对亚氯酸盐治疗会有不同响应。
实施例8–PD的治疗
生物标志物的初始浓度高于截止值的PD患者参加试验。研究包括IL-18、IL-6、INF-g或CRP的不同生物标志物。对于每种生物标志物,选择至少5个基线水平,并使用每对相邻水平(用于确定每组包含的范围)创建4个组。各组患者在6个月的过程中使用1-2mg/kg亚氯酸钠输注进行治疗。在治疗期间的不同时间点,例如,基线(第1周期、第1周给药前)、第1个月、2个月、3个月、4个月、5个月和6个月评价并记录帕金森病的进展。
预期亚氯酸盐治疗能够治疗帕金森病患者,并且具有不同生物标志物水平的帕金森病患者对亚氯酸盐治疗会有不同响应。
实施例9–ALS中巨噬细胞活化标志物的NP001调节
为了评估施用NP001对单核细胞活化标志物的作用,Neuraltus制药公司(Neuraltus Pharmaceuticals,Inc.)(Palo Alto,CA)和西方ALS研究组(Western ALS Study Group)(Clinicaltrials.org NCT01091142)在ALS患者中进行了NP001的I期、双盲、以安慰剂为对照的、单一递增剂量安全性和耐受性临床研究。
根据修改的E1Escorial标准(Brooks等人,(2000)Research Group on Motor Neuron Diseases,1(5):293-299)可能或确定患有ALS的32位男性和女性被分配为5组:1个安慰剂组(8位),或4个组之一(每种剂量6位)单一递增静脉内给药(0.2、0.8、1.6和3.2mg/kg NP001)。包括年龄小于75岁、稳定给药利鲁唑30天且能够提供知情同意书的患者。采用气管造口术、除了ALS还患有其他活动性疾病或接受免疫抑制剂治疗的患者被排除在外。患者的临床特征在表4中列出。监测接受安慰剂或NP001递增剂量的患者的主要终点(安全性以及临床状态的变化)以及次要终点(在给药前至少24小时和给药后至少24小时在血液单核细胞中血液单核细胞免疫活化标志物CD16和HLA-DR对NP001的响应)。统计计划中包括每种单核细胞标志物相对于基线的变化,并且这些值由位于UCSF的独立流式细胞仪实验室使用经验证的程序获得。由独立的统计人员进行CD16值的统计分析,并由Neuraltus科学家进行HLA-DR值的统计分析。
表4基线ALS特征(安全性分析群体)
知情同意和伦理学声明
本研究在美国的以下三个临床地点进行:加州太平洋医疗中心(California Pacific Medical Center),旧金山(San Francisco),CA;堪萨斯大学医学中心研究所(University of Kansas Medical Center Research Institute),堪萨斯城(Kansas City),KS;肯塔基大学ALS中心(University of Kentucky ALS Center),列克星敦市(Lexington),KY。ALS患者提供符合由加州太平洋医疗中心和加州大学旧金山分校(UCSF)的人类研究委员会制定,由艾滋病和癌症样本资源(AIDS and Cancer Specimen Resource)(ACSR)协调确定的指导原则的知情同意书。在其他两个临床地点获得了相似的批准。所有研究均根据赫尔辛基宣言原则(Declaration of Helsinki principles)进行。每位参与者通过数字而不是名字来识别。患者和评估员均对治疗的分配不知情。
血液单核细胞活化/炎症测定
为了探索单一剂量的NP001对可能与ALS的发病机理与进展有关的巨噬细胞炎症活化标志物的作用,使用修改后的ALS功能评定量表(ALSFRS-R)(评分0-48),来评估所有患者的功能状态(Cedarbaum等人,(1999)Journal of the neurological sciences,169(1-2):13-21)。如下计算估计的疾病进展率:
平均每月下降率=(48-基线ALSFRS-R评分)/疾病持续时间
测量的单核细胞活化标志物为CD16和HLA-DR在染色的全血中CD14+单核细胞中表达的水平。CD16和HLA-DR在CD14+单核细胞中的表达是单核细胞/巨噬细胞炎症活化在细胞水平的量度(Zhange等人,(2005)I Neuroimmunol.159(1-2):215-224;Belge等人,(2002)J Immunol168(7):3536-3542;Scherberich和Nockher,(1999)Clin Chem Lab Med.37(3):209-213;Ziegler-Heitbrock,(2007)Journal of leukocyte biology81(3):584-592;Merino等人,(2011)J Immunol 186(3):1809-1815)。在给药前和给药后24小时从患者中收集用于探索性单核细胞活化标志物分析的血液样本。将样本从临床地点运送到指定的实验室以在同一天制备样品。随后将染色并固定的样品运送到UCSF核心免疫学实验室(UCSF Core immunology laboratory)(UCSF,San Francisco,CA)以使用FACSDiva软件(BD Biosciences,San Jose,CA)通过LSR II型流式细胞仪(Becton Dickinson)进行流式细胞仪测量。
通过FlowJo软件(TreeStar Inc.,Ashland,OR)来补偿并分析数据。流式细胞术分析的结果表示为单核细胞活化标志物的几何平均荧光强度(Geo MFI)。通过流式细胞术鉴定HLA-DR和CD16在CD14+单核细胞中的表达的通常门控策略包括:使用CD3和CD16来排除在单核细胞门中污染的CD3+淋巴细胞和CD16+粒细胞。然后从HLA-DR与CD14点图中门控CD14+HLA-DR+细胞以排除残余淋巴细胞(包括B细胞和NK细胞)。然后从CD14与侧向散点图(CD14+)中门控所有单核细胞。从CD14+门中测量HLA-DR的几何平均荧光强度(MFI)。在CD14与CD16点图中,还从CD14+细胞中门控CD16+与CD16明亮细胞的比例。CD16亮门(通常比标准CD16强度亮10倍)捕获所有暗淡的CD14+CD16+明亮细胞。
安全性和临床状态变量
在NP001治疗后,在输注期间和输注后8小时内以及在给药后第1、4和7天,监测患者的多种安全性和临床状态变量。这包括包含检查反应的输注部位的体检,以及包含血细胞计数、多通道化学组(multi-channel chemistry panel)、尿分析、心电图和肺活量的临床实验室检测。在上升至下一个更高剂量之前,由安全监测委员会来检查每个剂量水平的所有8位患者的安全性数据。在给药前和给药后24小时进行血液单核细胞NP001治疗的流式细胞仪评估。
统计分析
通过GraphPad Prism 6.0程序(GraphPad软件,San Diego,California,USA)进行统计分析。除非另有说明,流式细胞仪的结果表示为平均值±SED。如表和图例中所示,使用单因素方差分析(One-way ANOVA)和线性回归来评估统计显著性。对于所有分析,认为p<0.05是统计学显著的。
安全性结果
这一针对ALS受试者的NP001的I期安全性和耐受性研究由西方ALS研究组(Western ALS Study Group)和Neuraltus制药公司(Neuraltus Pharmaceuticals,Inc.)在2010年完成。在该试验中,有32位患者(临床特征列于表4中)参加,并且4组患者在第1天接受了单次剂量的NP001(0.2、0.8、1.6或3.2mg/kg NP001亚氯酸盐,每组N=6,总共24位NP001患者)或安慰剂(盐水,每组N=2,总共8位安慰剂患者)的30分钟输注。所有剂量的NP001通常都是安全且耐受良好的,并且没有治疗相关的严重不良事件(表5)或安全性相关实验参数的临床相关变化。此外,在基线以及在单次剂量的药物或安慰剂输注之后24小时,定量血液单核细胞活化标志物CD16和HLA-DR。
表5在所有NP001剂量或安慰剂组(安全性分析群体)中在≥2位受试者中发生的治疗突发不良事件的总结
ALS患者中基线单核细胞/巨噬细胞活化相关炎症细胞表面标志物上升与ALS疾病进展率相关。
在先前的报道中,发现全身单核细胞/巨噬细胞活化的程度(HLA-DR和CD16的单核细胞超表达)与ALS疾病进展率相关(Zhang等人,(2005)J Neuroimmunol 159(1-2):215-224);活化的水平越高,ALS疾病进展越快。在NP001I期研究中在基线处获得的ALS患者血液单核细胞显示了如通过CD14细胞的HLA-DR共表达确定的单核细胞活化的证据,其水平与估计的ALS疾病进展率(基于对患者症状持续时间的评价的每月ALSFRS-R评分减少)相关(r=0.4310,p=0.0138;n=32)(图26A)。还观察到ALS疾病进展率与CD16明亮单核细胞中CD16的水平之间的正相关性,CD16明亮单核细胞为充当分化的单核细胞或组织巨噬细胞的最活跃的促炎单核细胞亚群(Belge等人,(2002)J Immunol 168(7):3536-3542;Ziegler-Heitbrock,(2007)Journal of leukocyte biology 81(3):584-592;Sadeghi等人,(1999)Experimental gerontology34(8):959-970;Takeyama等人,(2007)Annals of hematology 86(11):787-792;Thieblemont等人,(1995)European journal of immunology 25(12):3418-3424)(404244-46)(r=0.4499,p=0.0098,n=32)(图26B)。此外,多重回归分析显示这两种单核细胞活化标志物在与ALS疾病进展率的关系中彼此独立,并且当它们结合时显示与ALS疾病进展率的相关性增强(多重R=0.5734,p=0.0031)。没有发现基线ALSFRS-R评分与单核细胞HLA-DR或单核细胞CD16明亚组共表达水平之间的关系。NP001降低了基线HLA-DR值升高的患者的单核细胞HLA-DR的水平
在NP001治疗之后,单核细胞HLA-DR水平的变化没有显示出剂量依赖性效应;然而在单核细胞HLA-DR基线水平较高的患者中所有剂量的NP001组中的HLA-DR表达均下调。图27示出了在NP001I期研究中32位给药受试者的作为单核细胞HLA-DR基线水平的函数的NP001诱导的单核细胞HLA-DR表达水平变化的散点图。x轴代表单核细胞HLA-DR表达的几何平均荧光强度的基线值(Geo MFI CD14/HLA-DR)。y轴代表总单核细胞HLA-DR表达相对于基线的变化百分比。红线代表8位安慰剂患者中单核细胞HLA-DR表达的平均百分比变化;黑色方块和线代表安慰剂组的实际个体变化(r=-0.07721,p=0.8558;N=8)。蓝色三角和线代表在非剂量依赖的NP001施用后单核细胞HLA-DR表达的变化(r=-0.4967,p=0.0135;N=24)。安慰剂组显示在治疗后相对稳定的单核细胞HLA-DR(r=-0.07721,p=0.8558;N=8)。在NP001治疗响应中单核细胞中HLA-DR表达的变化与治疗后24小时的基线单核细胞HLA-DR表达的程度线性相关(r=-0.4967,p=0.0135;N=24)。基线初始单核细胞HLA-DR水平越高,HLA-DR对NP001的响应越大。图28示出了基于基线初始单核细胞HLA-DR水平的数据的图示。基于参加I期临床研究的整个组的全部32位患者的基线单核细胞HLA-DR的中值(Geo MFI CD14/HLA-DR=1200),将使用NP001治疗的患者分为两组。如x轴所示,将基线Geo MFI CD14/HLA-DR分为两组(Geo MFI CD14/HLA-DR>1200,N=12;Geo MFI CD14/HLA-DR<1200,N=12)。y轴代表在24小时中与基线相比HLA-DR的单核细胞几何平均水平的变化百分比。正值表示HLA-DR表达的增加,而负值表示HLA-DR表达的相对下降。在基线单核细胞HLA-DR升高的12位患者组中,施用NP001后24小时相对于基线的平均变化百分比超过10%,而单核细胞HLA-DR较低的患者显示相对于基线没有变化(p=0.0153)。
单核细胞HLA-DR表达的NP001相关变化与估计的ALS疾病进展率相关
进行用于评估估计的ALS疾病进展率对这些结果的影响的事后分析。图29示出了作为每位患者自从症状发作开始的历史ALS疾病进展率(基于参与机构临床图表的总结)的函数的NP001治疗后单核细胞HLA-DR表达变化的结果(合并数据)。将I期试验的患者基于其历史ALS疾病进展率分为亚组,通过ALSFRS-R的每月平均变化进行评估(DP率=疾病进展率),并与安慰剂组(N=8)进行比较。如x轴所示,DP率分为三组(DP率<0.5,N=8;DP率在0.5和1之间,N=11;DP率≥1,N=5)。y轴代表在24小时中与基线相比HLA-DR的单核细胞几何平均水平的变化百分比。正值表示HLA-DR表达的增加,而负值表示HLA-DR表达的相对下降。R2=0.2310,p=0.0058,在单因素方差分析(One-way ANOVA)后进行针对线性趋势的后续测试。在使用ALSFRS-R评分量表的情况下,ALS患者平均以大约1单位/月的速率下降。进展缓慢的患者(定义为估计的进展率<0.5单位/月)的HLA-DR均没有显示出变化,不论该患者接受NP001还是安慰剂。相比之下,估计的进展率≥1单位/月的患者在NP001给药后显示出HLA-DR表达的最大变化(对于线性趋势比较:R2=0.2310,p=0.0058)。
NP001诱导体内CD14单核细胞中明CD16亚组的CD16表达水平的剂量依赖性下降
与安慰剂组相比,在NP001治疗的患者中观察到单核细胞中CD16明亚组的CD16表达水平的剂量依赖性变化。单核细胞CD16调节的程度与基线CD16表达或估计的下降率(如通过自从疾病发作开始ALSFRS-R的变化评估的)无关。图30示出了单核细胞CD16表达相对于基线的变化与施用的NP001剂量之间的剂量依赖关系趋势(对于线性趋势比较:R2=0.1958,p=0.0085;安慰剂,N=8;NP001,每种剂量N=6)。在给药后24小时,用获得的同样的测量方式比较使用单剂量NP001或安慰剂治疗的ALS患者的单核细胞CD16表达的基线值。将给药后24小时单核细胞中CD16明亚组的CD16表达水平的变化百分比绘制在y轴上。将安慰剂(N=8)和剂量水平(每种剂量N=6)绘制在x轴上。R2=0.1958,p=0.0085,在单因素方差分析后进行针对线性趋势的后续测试。注意到在安慰剂组中单核细胞CD16表达的水平没有显著变化。
图31示出了如通过定量流式细胞术确定的接受1.6mg/kg剂量的NP001的患者的单核细胞CD16明亚组的CD16绝对水平。左边和中间的条代表ALS患者的CD16明亮单核细胞中CD16表达在基线(左边)和NP001输注后24小时(中间)的平均水平(N=6)。右边的条代表在健康的对照组中通常观察到的CD16表达的平均水平(N=7)。在NP001一次给药后24小时,与ALS患者的基线预处理水平相比,单核细胞CD16表达在ALS和正常对照水平之间的差异相对于正常值减少了大约50%。
ALS患者的NP001 1期研究与对基线炎症标志物升高的患者的单核细胞/巨噬细胞活化的以下两种可确定的作用有关:1)全身性抗炎作用,以及2)能够从血液迁移到组织的单核细胞亚群的CD16水平的明显下降。没有发现安全性或耐受性问题。不受任何理论约束,NP001治疗可以减少全身性炎症和血液单核细胞迁移至脊髓——被认为是对ALS进展很重要的关键过程,且有可能减慢该疾病的进展。
尽管本文中已经示出并描述了本发明的优选实施方案,但对于本领域技术人员显而易见的是这些实施方案仅以示例的方式提供。本领域技术人员在不脱离本发明的情况下现将想到多种变化、改变和替代。应当理解本文中所述的本发明实施方案的各种替代方案可用于实施本发明。目的在于以下述权利要求限定本发明的范围,并由此涵盖这些权利要求范围内的方法和结构及其等同项。
- 还没有人留言评论。精彩留言会获得点赞!