一种稳定的培哚普利吲达帕胺片及制备工艺的制作方法
本发明属于医药技术领域,具体涉及一种稳定的培哚普利吲达帕胺片及其制备工艺。
背景技术:
高血压是指以体循环动脉压升高、周围小动脉阻力增高同时伴有不同程度的心排血量和血容量增加为主要表现的临床综合症。高血压是最常见的慢性病,也是心脑血管病最主要的危险因素,其脑卒中、心肌梗死、心力衰竭及慢性肾脏病等主要并发症。目前,我国人群高血压病患病率呈增长态势,据统计,我国高血压患者人数已达到2.5亿,成人高血压患病率为25%-30%,每年心脑血管病死亡350万人,其中一半以上死亡与高血压有关。我国高血压病不仅患病人数多,而且知晓率、治疗率和控制率显著低于发达国家,呈现“三低”(分别为30.2%,24.7%,6.1%)特点,特别是经济文化发展水平较低的农村或边远地区情况尤为严重,治疗患者的血压达标率仅为25%。
原发性高血压是病因不明的高血压病,占总高血压患者的95%以上;目前认为,原发性高血压是一个终身性疾病,医学界目前没有发现确切的能够彻底治愈原发性高血压的治疗方法,国内外的实践证明,高血压是可以预防和控制的疾病,降低高血压患者的血压水平,可明显减少脑卒中及心脏病事件,显著改善患者的生存质量,有效降低疾病负担。
高血压的治疗主要分为非药物治疗和药物治疗,目前治疗高血压的药物产品主要由以下六大类构成:钙通道阻滞剂(CCB)、血管紧张素转化酶抑制剂(ACEI)、血管紧张素Ⅱ受体阻滞剂(ARB)、α受体阻滞剂、β受体阻滞剂及利尿药,以及由上述药物组成的固定配比复方制剂。目前全球上市的高血压药已经有上百种,而且新的药物仍在不断研发中。当前抗高血压药的研发主要从以下几点出发:(1)提高治疗效率,包括从降压、稳压、降低副作用等方面的考虑使血压尽快达标,增加血压达标率;(2)提高患者的依从性,特别是如何解决联合用药的适应性;(3)提高降压药的患者可及性,经济问题是患者和社会对抗高血压治疗认识提高的关键因素之一。
固定配比复方降压药是临床上常用的一组高血压联合治疗药物,通常由不同作用机制的两种小剂量降压药组成,也称为单片固定复方制剂,而对复方降压制剂的研发与临床效果仍是国内外研发热点。中国高血压防治指南(2010修订版第三版)中指出“与分别处方的降压联合治疗相比,固定剂量复方制剂其优点是使用方便,可改善治疗的依从性,是联合治疗的新趋势。对2或3级高血压或某些高危患者可作为初始治疗的药物选择之一。高血压复方药物中,血管紧张素转换酶抑制剂和利尿剂组成的复方是最经典的复方之一,血管紧张素转换酶抑制剂可能带来血钾升高的不良反应,而噻嗪类利尿剂可以明显地减少这种不良反应的发生,同时两类药物具有协同降压的效果,这种复方的组合在临床上受到广泛的欢迎。
培哚普利活性代谢物培哚普利拉都能够与血管紧张素转化酶(ACE)竞争性结合,降低ACE活性,阻断ACE将血管紧张素Ⅰ转化为血管紧张素Ⅱ,降低血管阻力,从而降低血压。培哚普利拉对血管紧张素转化酶(ACE)具有强有力的特异竞争性抑制作用。体外及临床研究结果表明:对血浆ACE抑制的强度,培哚普利拉与雷米普利拉(Ramiprilat)相似,而依次大于色拉普利拉(Cilazaprilat),依那普利拉(Enalaprilat),赖诺普利拉(Lisinopril)和卡托普利拉(Captopril)。研究还发现,培哚普利抗高血压作用持续时间较对血浆ACE活性抑制持续时间长,与依那普利比较,同等程度抑制血浆ACE活性的培哚普利,对心衰病人扩张血管影响血压的程度并不相同,提示组织ACE参与了对RAAS的调节,并且不同的ACEI对组织ACE的抑制程度也有差异。与雷米普利和依那普利相比,培哚普利对脑组织ACE抑制作用最强。
吲达帕胺为利尿药,可排钠,减少血容量,降低外周血管阻力,降压疗效肯定,在防治心血管事件中能发挥一定作用。吲达帕胺无噻嗪环,化学结构不属于噻嗪类,但与后者相似,都含有磺酰胺基,利尿作用和噻嗪类相似,作用于肾远曲小管的近端部分,抑制氯化钠和水的重吸收而发挥利尿作用。同时具有扩张血管作用,降压温和,疗效确切,对心脏有保护作用。吲达帕胺低血钾症的发生率比噻嗪类利尿药低,且不影响糖、脂代谢。
复方制剂质量稳定性产品开发的重点内容之一,与单药制剂相比其成分相对较多,不同药物本身之间的相互作用及辅料的作用均不可测,均极大影响产品的杂质谱和稳定性。采用液相色谱法进行培哚普利吲达帕胺片的有关物质研究,结果表明降解杂质主要为I、II、III、IV、V、VI,其中III为培哚普利拉,为培哚普利叔丁胺体内主要代谢产物;IV为培哚普利结构中的羧基和近位氨基,发生酰化反应环合生成具有内酰胺结构的培哚普利二酮哌嗪,除上述降解杂质外,V和异构体VI均为杂质IV的进一步酯基水解产物;杂质I和II均为吲达帕胺的降解杂质。某些情况下,药物的晶型不仅与产品的临床疗效有关,甚至可影响药物的稳定性及安全性,培哚普利叔丁胺除存在上述稳定性问题之外,也存在多晶型的特性,目前公开报道有α,β,γ,δ,ε等晶型,各晶型均有相应的药用组合物专利,国内原研上市品种及采用α晶型;而吲达帕胺的晶型及稳定性均良好。如前述,由于培哚普利存在结构上的不稳定性,因此其着湿热稳定性问题,但研究表明上市产品在40℃条件下放置10和30天时,不仅有关物质大幅增加甚至超出标准规定的限度,更严重的是其晶型也可发生改变,推测其稳定性可能与晶型有关,而目前的研究资料均未明确不同的进行由此产生的稳定性问题。固体制剂口服给药后,药物的吸收取决于药物从制剂中的溶出或释放、药物在生理条件下的溶解以及在胃肠道的渗透等,因此,药物的体内溶出和溶解对吸收具有重要影响,体外溶出试验常用于指导药物制剂的研发、评价制剂批内批间质量的一致性、评价药品处方工艺变更前后质量和疗效的一致性等。
对上市培哚普利吲达帕胺片进行研究分析,其采用铝塑泡罩包装,置加干燥剂的铝箔袋内存放,并且要求储存在30℃以下,虽然通过隔绝氧气和水分极大增加了产品的稳定性,然而研究结果表明制剂在此条件并不足以保证培哚普利叔丁胺和吲达帕胺的稳定,即其也存在高温降解,同时该包装也极大了增加了成本。另一方面,其更严重的问题上市产品中的吲达帕胺溶出曲线表明是存在明显的批间差异和批内差异,这将对临床疗效产生影响。
综上所述,培哚普利吲达帕胺片的稳定性中杂质的降解速率、晶型转变、批内及披间的溶出变异过大是产品开发的难点,发明安全、有效、稳定、质量可控且简单、经济的培哚普利吲达帕胺片的配方工艺,实现产品的产业申报时符合仿制药一致性评价的原则,将变的很有意义。
技术实现要素:
本发明针对现有不足,提供了一种稳定的培哚普利吲达帕胺片及其制备工艺,该制备工艺能使培哚普利吲达帕胺片药学稳定性各方面与上市产品一致或更优。
为实现发明目的,本发明采取如下的技术手段:
一种稳定的培哚普利吲达帕胺片,由下列组份按照重量份数组成:
α晶型培哚普利叔丁胺:1-5份;
吲达帕胺:1-5份;
乳糖:60-80份;
羟丙基纤维素:1-10份;
羧甲基淀粉钠:1-5份;
二氧化硅:0.1-1.0;
硬脂酸镁:0.8-1.5份。
作为优选,所述的培哚普利吲达帕胺片由下列组份按照重量份数组成:
α晶型培哚普利叔丁胺:4份;
吲达帕胺:1.25份;
乳糖:79.65份;
羟丙基纤维素:3.6份;
羧甲基淀粉钠:1.5份;
二氧化硅:0.5份;
硬脂酸镁:1份;
或
α晶型培哚普利叔丁胺:2份;
吲达帕胺:0.625份;
乳糖:82.275份;
羟丙基纤维素3.6份;
羧甲基淀粉钠1.5份;
胶态二氧化硅0.5份;
硬脂酸镁:1份。
作为优选,所述的培哚普利叔丁胺的粒径分布为MEAN D(4,3)为30-50um,D90为70~90um;吲达帕胺的粒径为:D(0.5)<48(10μm),D(0.9)<147(10μm)。
作为优选,所述的吲达帕胺的晶型为文献Phamazie 2006,61(12):99-1004报道的晶型相同。
作为优选,所述的乳糖为一水乳糖,所述的羟丙基纤维素为SSL羟丙基纤维素,所述的二氧化硅为无水胶态二氧化硅。
本发明采用常用的乳糖为填充剂,在此基础上加入羟丙基纤维素为粘合剂保证产品可压性,并结合适量的羧甲基淀粉钠协同控制产品的溶出度,考虑到培哚普利的难流动性,加入适量的无水胶态二氧化硅调节整个处方优良的粉体流动性,润滑剂硬脂酸镁的适量加入是为了制备压片过程中的顺利进行。
本发明的另一个目的是提供上述稳定的培哚普利吲达帕胺片的制备工艺,采取如下技术方案:
一种稳定的培哚普利吲达帕胺片的制备工艺,采用直接压片工艺,包括以下步骤:
(1)取乳糖干燥至水分质量含量低于5.5%后,与胶态二氧化硅共同过筛;
(2)加入过筛的羟丙基纤维素和羧甲基淀粉钠混合;
(3)加入过筛的吲达帕胺混合;
(4)加入过筛的培哚普利叔丁胺混合均匀;
(5)加入硬脂酸镁混合,取样检测中间体含量,采用4mm*8mm压片。
作为优选,所述的制备工艺还包括步骤(6)采用铝塑泡罩包装,内置干燥剂,外覆复合膜袋。
作为优选,所述步骤(1)中过筛的筛目为30目-60目,所述步骤(2)中过筛的筛目为50目-80目,所述步骤(3)中过筛的筛目为80目-100目,所述步骤(4)中过筛的筛目为40目-80目,
作为优选,所述步骤(5)压片的硬度为:50~70N。
作为优选,所述的制备片剂的工艺环境湿度低于40%RH。
作为优选,所述步骤(4)的混合时间为10分钟-30分钟;步骤(5)的混合时间为1分钟-10分钟。
作为优选,所述步骤(6)中的铝塑泡罩包装材料为PVC铝箔材料。
作为优选,所述步骤(6)中干燥剂为硅胶或分子筛。
本发明提供的培哚普利吲达帕胺片与已上市产品相比,在外观、溶出度、水分、含量上无明显变化,且稳定性实验过程中其杂质降解速率小于上市产品,其晶型稳定性与上市产品无明显差异,其溶出曲线的批内差异与批件差异均明显由于上市产品。
本发明在制备培哚普利叔丁胺片方法中的具体优势如下:
本发明具有工艺简单、生产周期短、成本低的优势。培哚普利吲达帕胺片规格和片重均较小,培哚普利属易溶性药物,而吲达帕胺属于难溶性药物,本发明采用常用的乳糖为填充剂,在此基础上加入羟丙基纤维素为粘合剂保证产品可压性,并结合适量的羧甲基淀粉钠协同控制产品的溶出度,考虑到培哚普利的难流动性,加入适量的无水胶态二氧化硅调节整个处方优良的粉体流动性,润滑剂硬脂酸镁的适量加入是为了压片过程中的顺利进行。
本发明的培哚普利吲达帕胺片其稳定性中杂质降解速率与上市产品相比明显降低,且晶型稳定性和变化同上市产品相同。主要方法是对工艺环境的湿度和水分进行了规定,即产品水分质量含量低于5.5%,工艺制备环境低于RH40%,避免湿法制粒等过程中溶于水后的转晶问题和湿热引起的稳定降低问题。
本发明采用粉末直压工艺,其产品批内差异和披间差异与上市产品更优,另一方面且采用本发明可有效解决培哚普利吲达帕胺片的含量均匀性问题;从而表现出其整体的质量优于上市产品。
附图说明
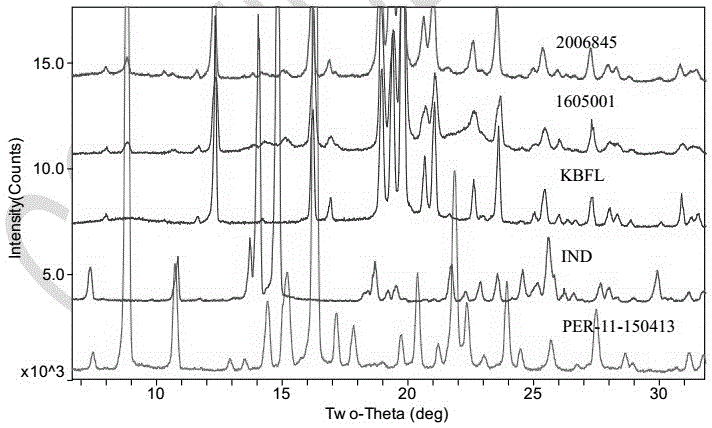
图1为上市产品、自制产品、原料药与空白辅料的晶型图,结果显示扣除空白辅料峰,自制产品、原料药与上市产品晶型一致,其中2006848:上市产品;1605001:本发明产品;KBFL:空白辅料;IND:吲达帕胺;PER-11-150413:培哚普利。
图2为上市产品、自制产品、原料药分别在影响因素光照、高温、高湿条件下30天的晶型图,结果显示在光照、高湿条件下自制产品、原料药与上市产品晶型一致,而光照条件下上市产品、自制产品、原料药均存在转晶,其中2006848:上市产品;1605001:本发明产品;KBFL:空白辅料;IND:吲达帕胺;PER-11-150413:培哚普利;L30d:4500Lux光照30天;H30d:75%RH放置30天;T10d:40℃放置30天。
具体实施方式
下面结合具体实施例对本发明作进一步说明。
实施例1:
一种培哚普利吲达帕胺片,所述的培哚普利吲达帕胺片由下列组份按照重量份数组成:
α晶型培哚普利叔丁胺:4份;
吲达帕胺:1.25份;
一水乳糖:79.65份;
羟丙基纤维素3.6份;
羧甲基淀粉钠1.5份;
胶态二氧化硅0.5份;
硬脂酸镁:1份;吲达帕胺晶型与文献Phamazie 2006,61(12):99-1004
报道的晶型相同;
最后制的片重约90mg的片剂,其制备工艺包括如下步骤:
(1)取乳糖干燥至水分质量含量低于5.5%后,与胶态二氧化硅共同过50目筛,混合;
(2)加入经过筛的羟丙基纤维素(过50目)、羧甲基淀粉钠(过50目)混合;
(3)继续加入入经过80目筛的吲达帕胺混合,混合;吲达帕胺的粒径为D(0.5)<48(10μm),D(0.9)<147(10μm);
(4)继续加入经过40目筛的培哚普利叔丁胺,混合均匀30分钟,制剂工艺环境湿度低于40%RH;培哚普利叔丁胺的粒径为MEAN D(4,3)为30-50um,D90为70~90um;
(5)最后加入最后加入硬脂酸镁混合5分钟,取样检测中间体含量,采用4mm*8mm压片,压片硬度为50~70N;
(6)采用铝塑泡罩包装,内置干燥剂为硅胶或分子筛;外覆复合膜袋;
通过对上述实施例的原辅料进行相容性实验。用量较大的填充剂乳糖,按主药:辅料=1:5的比例混合,用量中等的羟丙基纤维素和羧甲基淀粉钠按主药:辅料=1:10的比例混合,,用量较小的二氧化硅和润滑剂硬脂酸镁,按主药:辅料=20:1的比例混合,按稳定性影响因素试验方法,分别在强光、高温、高湿的条件下放置10天,考察放置前后有关物质变化,同时观察外观性状等的变化。实验结果显示,降解杂质I、III、IV在高温高湿条件下随时间增加,此现象与理论上培哚普利叔丁胺湿热不稳定相符合,因存在辅料下易吸湿导致高温和高湿条件产生降解。而各辅料混合前后,杂质并无明显增加,特别是无明显的未知杂质增加,因此可认为本发明中所用的原辅料相容性良好,在制剂生产和贮藏过程应该避免高温高湿的影响。
表1原辅料相容性实验
表中,PER表示培哚普利叔丁胺,MS表示硬脂酸镁,HPC表示羟丙基纤维素,CMS-Na表示羧甲基淀粉钠,SiO2表示二氧化硅,IND表示吲达帕胺。
实施例2
一种培哚普利吲达帕胺片,所述的培哚普利吲达帕胺片由下列组份按照重量份数组成:
α晶型培哚普利叔丁胺:2份;
吲达帕胺:0.625份;
一水乳糖:82.275份;
SSL羟丙基纤维素3.6份;
羧甲基淀粉钠1.5份;
胶态二氧化硅0.5份;
硬脂酸镁:1份;
吲达帕胺晶型与文献Phamazie 2006,61(12):99-1004报道的晶型相同。
最后制的片重约90mg的片剂,其制备工艺包括如下步骤:
(1)取乳糖干燥至水分低于5.5%后,与胶态二氧化硅共同过50目筛,混合;
(2)别加入经过筛的羟丙基纤维素(过50目)、羧甲基淀粉钠(过50目)混合;
(3)继续加入入经过80目筛的吲达帕胺混合,混合;吲达帕胺的粒径为D(0.5)<48(10μm),D(0.9)<147(10μm);
(4)继续加入经过40目筛的培哚普利叔丁胺,混合均匀30分钟,制剂工艺环境湿度低于40%RH;培哚普利叔丁胺的粒径为MEAN D(4,3)为30-50um,D90为70~90um;
(5)最后加入最后加入硬脂酸镁混合5分钟,取样检测中间体含量,采用4mm*8mm压片,压片硬度为50~70N;
(6)采用铝塑泡罩包装,内置干燥剂为硅胶或分子筛;外覆复合膜袋;
采用上述实施例的配方平行制备3批,进行质量对比。结果,本发明所制备的产品与上市产品晶型一致,性状、有关物质、溶出度、含量均匀度、水分、含量等关键质量属性均一致。见表2。
表2自制产品与上市产品的质量对比
注:ND为未检出
采用上述实施例的配方制备样品,进行溶出曲线对比。结果,本发明所制备的产品与本发明与上市产品的溶出曲线均相互匹配,但产品中的吲达帕胺在溶出曲线5分钟和10分钟累积溶出量的相对标准偏差(RSD)明显优于上市产品,见表3。
表3自制产品与上市产品的溶出曲线对比
注:ND为未检出
对上述实施例产品与上市产品同时进行影响因素(高温40℃、光照4500lux/h、高湿RH75%)放置10天、30天研究,性状、有关物质、溶出度、含量、晶型的对比。结果自制产品各时间点的性状、溶出度、含量均与上市产品一致,有关物质变化趋势明显优于上市产品,晶型变化与上市产品一致,见下表4与图1和图2。
表4自制产品与上市产品的影响因素实验质量对比
对上述实施例产品与上市产品分别进行影响因素条件下和加速条件(40℃/RH75%)进行放置1月、3月下进行性状、有关物质、溶出度、含量、晶型的对比。结果自制产品各时间点的性状、溶出度、含量均与上市产品一致,有关物质的降解速率明天低于上市产品,晶型的转变规律和上市产品一致。见下表5。
表5自制产品与上市产品的加速实验质量对比
实施例3
一种培哚普利吲达帕胺片,所述的培哚普利吲达帕胺片由下列组份按照重量份数组成:
α晶型培哚普利叔丁胺:4份;
吲达帕胺:1.25份;
一水乳糖:76.25份;
SSL羟丙基纤维素1.5份;
羧甲基淀粉钠4.5份;
胶态二氧化硅1份;
硬脂酸镁:1.5份;吲达帕胺晶型与文献Phamazie 2006,61(12):99-1004报道的晶型相同最后制的片重约90mg的片剂,其制备工艺如实施例1所述,实施例所得的产品与上市产品晶型一致,性状、有关物质、溶出度、含量均匀度、水分、含量等关键质量属性均一致。
实施例4
一种培哚普利吲达帕胺片,所述的培哚普利吲达帕胺片由下列组份按照重量份数组成:
α晶型培哚普利叔丁胺:5份;
吲达帕胺:1份;
一水乳糖:73.2份;
SSL羟丙基纤维素6份;
羧甲基淀粉钠3份;
胶态二氧化硅1份;
硬脂酸镁:0.8份;
吲达帕胺晶型与文献Phamazie 2006,61(12):99-1004报道的晶型相同,最后制的片重约90mg的片剂,其制备工艺如实施例1所述,实施例所得的产品与上市产品晶型一致,性状、有关物质、溶出度、含量均匀度、水分、含量等关键质量属性均一致。
实施例5
一种培哚普利吲达帕胺片,所述的培哚普利吲达帕胺片由下列组份按照重量份数组成:
α晶型培哚普利叔丁胺:1份;
吲达帕胺:1份;
一水乳糖:80份;
羟丙基纤维素2.5份;
羧甲基淀粉钠3份;
胶态二氧化硅1份;
硬脂酸镁:1.5份吲达帕胺晶型与文献Phamazie 2006,61(12):99-1004报道的晶型相同,最后制的片重约90mg的片剂,其制备工艺如实施例1所述,实施例所得的产品与上市产品晶型一致,性状、有关物质、溶出度、含量均匀度、水分、含量等关键质量属性均一致。
- 还没有人留言评论。精彩留言会获得点赞!