纳米乳液的制作方法
本发明涉及纳米乳液和纳米乳液组合物的组分、制造方法和用途。纳米乳液特别用于口腔药物递送。
背景技术:
药物递送是相当活跃和具有挑战的领域。递送已知的活性成分的新方法可以带来相当大的益处,例如最小化治疗特定病症所需的药物的量、保证药物达到期望位置、提高便利性、控制释放分布和改善患者依从性。特别地,口腔递送经常是期望的,但是由于大部分活性成分的疏水性质而可能是困难的。
许多研究者已经寻找了处理该问题的方式以及特别是处理药物的不良水溶性的方式。
可以将包封用于分离和递送活性材料。例如,wo2013/024307公开了使用两亲性支链聚合物形成包含活性材料的水包油乳液,随后交联聚合物形成胶囊。
weaveretal.,angew.chem.2009,121,2165-2168公开了将大乳液液滴组装到更大规模的工程化结构中。
技术实现要素:
本发明是基于已经确定了在水性体系内携带疏水材料的有效方式的研究。
本发明的第一方面提供了水包油乳液,其包含乳化剂,乳化剂是非凝胶的支链聚合物,其中所述聚合物的至少一些链的端部以5个或更多个碳原子的烷基链封端,并且其中水包油乳液采取具有不大于约1000nm的z均直径的颗粒的形式。
因为它们的尺寸小,所以在本文中还将本发明的乳液称为纳米乳液。它们具有其中至少一个尺寸或其中z均直径(还称为平均流体力学直径)不大于约1000nm的平均大小。它们可以具有小于800nm、或小于500nm、或小于300nm、例如约50至500nm或200至300nm的z均直径。
相反,weaveretal.,angew.chem.2009,121,2165-2168没有考虑到纳米级结构而是涉及较大的结构。从具有9.2微米的平均直径的水包油液滴开始,其将那些液滴凝聚为mm尺度的更大的球体,然后将那些球体凝聚为cm尺度的管或其他结构。weaver等人没有涉及药物递送应用。weaver等人的较大尺度的结构感兴趣的是墨喷式印刷和农用化学品领域以及使用时关系到稀释的其他领域。
烷基链是疏水的并因此与油相关联,且将油相稳定在水相中。
我们发现有效的稳定在c5或更大的烷基、例如c6(己基)或更大的烷基发生,以及在某些情况下更大的稳定发生在c8或更大的烷基、例如c10或更大的烷基、例如c12(十二烷基)或更大的烷基、例如c5至c20。
烷基链可以是支链或非支链的,或是支链和非支链的混合。
烷基链可以是饱和的或不饱和的,或饱和或不饱和的混合。
烷基链可以可选地是取代的。
合适的化合物的一个子集包括具有饱和的、未取代的c5-c20烷基链的那些;这些制备起来方便且划算。
支链聚合物是非凝胶的和可加工的,即表现出合适的溶解度/相容性以允许溶解和用作乳化剂。其可以与不可溶和/或表现出高粘度的聚合物结构,如大量交联的不可溶聚合物网络、高分子量直链聚合物或微凝胶相反。
支链聚合物可以是例如加成聚合物。支链聚合物可以是例如由不饱和的,例如乙烯基或烯丙基单体,如例如丙烯酸酯或甲基丙烯酸酯单体制成的聚合物。
可以通过已知的方法由单官能的乙烯基单体和二官能的乙烯基单体(支化剂)制备支链乙烯基聚合物。它们可以通过以下制备但不限于通过以下制备:活性聚合、受控聚合或常规的链生长聚合技术,如自由基聚合。几种类型的活性和受控聚合是本领域已知的并适合用于本发明。优选类型的受控的自由基聚合是原子转移自由基聚合(atrp);然而通过有意添加链转移剂控制的其他技术如可逆加成-断裂链转移(raft)和氮氧调控聚合(nitroxidemediatedpolymerization)(nmp)或常规的自由基聚合也是合适的合成反应。
技术人员知道提供支链但非凝胶化的乙烯基聚合物的技术。例如,以下描述了合适的流程:n.o’brien,a.mckee,d.c.sherrington,a.t.slarkanda.titterton,polymer2000,41,6027-6031;t.he,d.j.adams,m.f.butler,c.t.yeoh,a.i.cooperands.p.rannard,angew.chem.int.ed.2007,46,9243-9247;v.bütün,i.bannister,n.c.billingham,d.c.sherringtonands.p.armes,macromolecules2005,38,4977-4982;i.bannister,n.c.billingham,s.p.armes,s.p.rannardandp.findlay,macromolecules2006,39,7483-7492;和r.a.slater,t.omcdonald,d.j.adams,e.r.draper,j.v.m.weaverands.p.rannard,softmatter2012,8,9816-9827。本发明的非凝胶且可溶的产物不同于l.a.connal,r.vestberg,cj.hawkerandg.g.qiao,macromolecules2007,40,7855-7863中公开的,已知在凝胶化网络中包含多重交联的材料。
每个乙烯基聚合物链的聚合起始于引发剂。单官能乙烯基单体的聚合产生直聚合物链。
与二官能乙烯基单体的共聚产生链之间的支化。为了控制支化和防止凝胶化,每条链应存在少于一个有效的支化剂(二官能乙烯基单体)。在某些条件下,这可以通过使用小于一的支化剂与引发剂的摩尔比实现:这假设单体(即单官能乙烯基单体)和支化剂(即二官能乙烯基单体)具有相同的反应性,不存在分子内反应,支化剂的两个官能团具有相同或类似的反应性,以及反应性即使在部分反应之后也保持相同。当然,体系和条件可以不同,但是技术人员理解如何控制反应和在没有过度实验的情况下确定如何实现非凝胶结构。例如,在稀释条件下,一些支化剂形成限制在链之间支化的支化剂的数目的分子内环,即使反应中的支化剂与引发剂(即聚合物链)的摩尔比高于1:1。
用于聚合过程的引发剂和其他试剂是本领域已知的。例如,在atrp中,方便的和有效的引发剂包括烷基卤化物(例如烷基溴化物),以及在常规的自由基聚合中,有效的引发剂包括偶氮化合物。
其他合适类型的支链聚合物包括支链聚酯。这些可以通过例如单官能的内酯单体和二官能的内酯单体(支化剂)的开环聚合制备。开环聚合方法和材料是本领域例如由nguyenetal.,polymchem2014,5,2997-3008已知的。
合适的支链聚合物的一个子集包括含有醚或聚醚部分的那些,例如含有聚乙二醇(peg)或聚氧化乙烯(peo)的那些,例如由包含醚基团的乙烯基单体制成的那些。我们发现这些便于制备且表现出良好的性能。不希望受到理论限制,看起来虽然烷基链充当油颗粒中的锚定物,但是醚部分促进水中的稳定性。用于在制备具有peg基团的支链聚合物的方法中使用的合适的单体的实例是低聚(乙二醇)甲基丙烯酸酯(oegma),也称为peg-甲基丙烯酸酯。
这允许在两个水平上并入多个醚部分:首先该单体已经包含若干(例如5至15)个氧化乙烯部分;以及其次该单体可以经由其乙烯基部分聚合,使得在经由支链连接乙烯基聚合物链之前,其可以包含例如30至300个、例如50至200个、例如60至100个、例如约80个oegma单元(并因此高一个数量级的环氧乙烷部分)。
其他合适的单官能单体包括但不限于例如n-甲基丙烯酸丁酯、甲基丙烯酸羟丙基酯、n,n-二乙基氨基甲基丙烯酸乙酯、丙三醇甲基丙烯酸酯和2-甲基丙烯酰氧基乙基磷酸胆碱。可以使用不同单体的混合物来形成共聚物。
合适类型的二官能单体(即支化剂)包括例如包含两个或更多个可聚合官能团,例如丙烯酸酯或甲基丙烯酸酯单体的那些。
合适的支化剂的一个实例是二甲基丙烯酸乙二醇酯(egdma)。其是方便且有效的。
可以经由引发剂或链转移剂,例如经由溴化物引发剂(如溴代异丁酸酯)或硫醇链转移剂将疏水的烷基链并入到支链聚合物中。因此,引发剂(或链转移剂)可以包含5个或更多个碳原子的烷基链(如以上所定义的)。这是赋予要求的疏水特征以稳定或“锚定”油滴的方便且有效的方式。而且,其是灵活的:其使得可以简单地通过改变引发剂来容易地改变生成的聚合物的烷基链端部,并从而提供定制组合物的重要方式。
可以将本发明的支链聚合物理解为用链之间的支链保持在一起的许多直链聚合物链(优选地每条链一条支链或更少),使得一些链以疏水的烷基部分封端。这些疏水的“锚定物”不需要存在于每个聚合物链上。我们出乎意料地发现当90%或更少、或75%或更少、或50%或更少、或甚至25%或更少的聚合物链端部携带要求的烷基链时,可以实现有效的乳化和有效的稳定性。这意味着经由引发剂并入疏水部分时,仅一些引发剂可以携带这些部分,且其他引发剂可以具有不同的结构,例如可以具有更简单的结构,或可用于并入其他化学性质或功能,例如靶向能力或其他用于治疗、诊断或其他生物用途的能力。
我们还发现用于本发明的支链聚合物出乎意料地比包含类似的基团的对应的直链聚合物,例如由除支化剂外的对应的组分制成的那些更有效。
有利地,本发明的水包油乳液可以进一步包含由油相携带或溶解在油相中的材料。这可以是有机化合物、疏水材料或可溶于油或有机溶剂的材料。这种材料可以例如是生物学上有用的材料,例如治疗或诊断上有用的材料,例如药物或前药。
本发明的产物因此可以优选地是药物组合物。
因此,本发明的进一步方面提供了如上所述的水包油乳液,用作药剂。
本发明还提供了对应的医疗方法,包括将有效量的以上定义的水包油乳液给予至需要其的受试者。
该组合物在口腔药物递送中是特别有效的。我们发现该乳液不仅在保持优异的稳定性上而且在穿过模型内脏系统上出乎意料地有效。
本发明的进一步方面提供了制备水包油乳液的方法,包括在乳化剂的存在下混合油相与水相,其中所述乳化剂是非凝胶的支链聚合物,其中所述聚合物的至少一些链的端部以5个或更多个碳原子的烷基链封端,并且其中水包油乳液采取具有不大于约1000nm的z均直径的颗粒的形式。
乳液的乳化剂和其他组分的特征可以如以上所描述的。
在制备纳米乳液的初始步骤中,可以将药物和/或其他药物组分溶解在油中。对于药物用途和治疗给予,油当然必须选自适用于并安全用于那些应用的油。技术人员熟知满足这种标准的油。有利地,油将是用于携带的材料的良好的溶剂。
一些合适的油包括篦麻油、椰子油、十二烷酸、角鲨烯、花生油、芝麻油和大豆油。篦麻油对于某些药物是特别优选的。
用药物来饱和油将给出药物在最终的乳液中的最大可能的浓度。如果需要,可以容易地稀释纳米乳液。相反,一旦形成则可能难以浓缩乳液:一种浓缩方法包括冻干,但是这可以降低稳定性和改变直径。
我们制备了具有各种不同的药物的许多乳液。理论上,本发明适用于具有不良的水溶性的任何亲脂性的/疏水性的药物。
一些合适的药物的非限制性实例包括例如抗逆转录病毒药物洛匹那韦(lpv)和依法韦仑(efv)及抗生素利福平和红霉素。实际上,可以将约50mg/ml的依法韦仑或25mg/ml的洛匹那韦溶解在篦麻油中。
我们还制备了其中携带各种其他疏水材料例如姜黄素、荧光素和尼罗红的制剂。
可选地,在制备流程中,除了油之外,可以使用另外的溶剂。其与油是可互溶的且还溶解药物。另外的溶剂存在于最终的乳液中并因此是可以通过蒸发或其他方法除去的溶剂。
合适的挥发溶剂包括例如乙酸乙酯、己烷、丙酮或thf。乙酸乙酯是优选的溶剂:其与水不互溶,溶解油,容易和迅速地蒸发,并具有低毒性。
将油(其中溶解药物等)与挥发溶剂混合。可以选择油与溶剂的比率来定制纳米乳液液滴的大小。典型地,溶剂与油比率越高,最终的乳液中的液滴越小。
按体积计溶剂相对于油的量可以是例如50:50或更大、例如60:40或更大、例如70:30或更大、例如80:20或更大、例如90:10或更大、例如95:5或更大、例如99:1或更大、例如95:5至99.9:0.1、例如约99:1。
通过混合油相(可选地包含挥发溶剂)与支链聚合物的水溶液方便地形成乳液。
可以选择水相相对于油相的量和水相内的支链聚合物的浓度来定制乳液的性质和性能。应该使用足够的聚合物来稳定纳米乳液液滴。聚合物的浓度可以影响液滴的大小。不希望受到理论限制,相信较低量的聚合物导致较大的液滴,因为没有足够的聚合物来完全包封液滴并因此导致聚集。相反,通常存在需要的聚合物的上限,使得在该量以上,将观察不到进一步的稳定益处并产生溶液中游离的聚合物。
在一些情况下,水相中的聚合物的优选的浓度(w/v)选自约0.1-99.9%、0.5-99%、1-90%、1-50%、1-20%、2-10%、3-7%或约5%。
在一些情况下,油(+溶剂)相的量相对于水相的量(v/v)是约90:10至10:90、或75:25至25:75、或60:40至40:60、或约50:50。
可以使用任何合适的方法或装置混合和均匀化油相和水相来产生水包油乳液。通常,乳液将包含纳米尺寸的液滴并将通过乳白色和黏稠度识别。
可以通过任何合适的方法,例如通过使得其蒸发、和/或通过稀释和搅拌和/或通过使气体(例如惰性气体,例如氮气)流过材料,除去挥发性溶剂。我们发现的一个简单和有效的方法是简单地将材料置于未密封的容器中并事情蒸发(例如在通风橱中)12-48小时、通常约24小时的时间。
蒸发或除去溶剂导致形成处于其最终形式的乳液。其具有存在于油相中的药物和/或其他疏水的组分,由于纳米液滴上的聚合物的“涂覆”,油相稳定在水中。通过动态光散射(dls)确定的z均直径通常是100-500nm、例如200-300nm。
可以在25℃下通过dls测量z均直径。
纳米乳液在存储和稀释时是稳定的。
如以下所描述的,我们还观察到纳米乳液制剂的抗凝血剂效果,并因此本发明的进一步方面提供了所述乳液,用作抗凝血剂。
水包油乳液可以具有与上述的那些不同尺寸的颗粒或液滴,即不一定具有不大于约1000nm的z均直径。换句话说,在进一步的方面,本发明提供了包含乳化剂的水包油乳液,乳化剂是非凝胶的支链聚合物,其中所述聚合物的至少一些链的端部以5个或更多个碳原子的烷基链封端。这种乳液和对应的组合物的其他特征、用途和方法可以如上所述。
附图说明
现在将参考以下实施例和附图描述本发明的进一步的非限制性的细节,其中:
图1示出了通过在制备过程中改变溶剂与油的比率所导致的本发明的纳米乳液的直径的变化;
图2示出了存储时纳米乳液样品的z均直径;
图3示出了聚合物的量对纳米乳液直径的影响;
图4示出了连续稀释之后纳米乳液的z均直径;
图5至7示出了与对照相比关于本发明的纳米乳液的免疫结果;
图8示出了纳米乳液对血浆凝结的影响-用依法韦仑、空白纳米乳液和包含依法韦仑的纳米乳液处理健康志愿者的血浆,之后分析凝血酶原时间(a)、凝血酶时间(b)和活化部分凝血活酶时间(c);
图9示出了本发明的纳米乳液-药物制剂与对照药物制剂相比的疗效;
图10和11示出了与对应的直链聚合物乳化剂(比较例)相比,由根据本发明的支链聚合物乳化剂提供的稳定性;
图12示出了与其他引发剂相比,用根据本发明的混合引发剂制备的某些乳液的平均液滴尺寸是如何作为疏水引发剂的量的函数变化的;
图13示出了与lpv的水溶液的跨细胞渗透相比,负载lpv的纳米乳液的跨细胞渗透;以及
图14示出了与efv的水溶液的跨细胞渗透相比,负载efv的纳米乳液的跨细胞渗透。
具体实施方式
实施例
第1部分:支链聚合物(包含十二烷基部分、使用egdma链接的polyoegmadp80链)的制备、及其制备并入药物的纳米乳液的用途以及这些纳米乳液的安全性和疗效研究
聚合物的制备
可以使用原子转移自由基聚合(atrp)制造用于稳定纳米乳液的聚合物,原子转移自由基聚合是受控的聚合方法,允许限定具体的聚合度(dp)。根据本发明的可溶于水的聚合物的一个实例基于polyoegmadp80。
使用dp80,是指每聚合物链存在约80个单体单元(oegma)。通过将egdma用作支化剂一起支化这些直链。
首先,在圆底烧瓶中称取单体的量(oegma,mw300)。去除天平皮重,然后添加恰当量的egdma支链使得与单体相比摩尔比是0.95。然后添加十二烷基引发剂(2-溴代异丁酸十二烷基酯),随后添加少量的苯甲醚,其用作之后的nmr分析的内标。最后,添加反应溶剂(ipa:h2o;92.5:7.5)。将烧瓶用橡胶塞盖住,并固定在通风橱中的夹具上。将氮气通过塞泵入烧瓶中以通过塞中在氮气入口针头旁边插入的小规格针头除去烧瓶中的任何氧气(氧气通过淬灭自由基物质灭活反应)。
除气10分钟之后,迅速称重氯化铜和bpy并在秤盘中组合,然后通过稍微移开塞子小心添加到反应混合物中来滴入cucl/bpy混合物。其添加启动反应,并再密封烧瓶并使其进一步除气10分钟,该点之后移除出口针头和氮气入口。消耗约8小时来完成反应,并通过每小时的nmr样品测定聚合%确认反应完成。通过分析凝胶渗透色谱确定聚合物的分子量。
该流程的精确反应量如下:
oegma9.44g
十二烷基引发剂0.118g
egdma62.5ul
苯甲醚400ul
cucl0.034g
bpy0.137g
溶剂12mls
纳米乳液的制备
生产溶解在篦麻油中的药物的储备溶液(50mg/ml依法韦仑在篦麻油中或25mg/ml的洛匹那韦在相同的油中)。
将负载药物的油转移到玻璃小瓶中,并添加溶剂以达到99:1的溶剂与油比率。对于该比率,将30ul的油添加到小瓶中,然后与2970ul的挥发溶剂(乙酸乙酯)混合。
改变溶剂与油的比率对改变最终的纳米乳液的直径有影响(图1)。图1示出了基于使用本文所描述的支链聚合物与篦麻油、乙酸乙酯和依法韦仑,通过dls确定的最终的纳米乳液的z平均值。
以5%(w/v)的浓度下向3ml的油/溶剂中添加3ml可溶于水的支链聚合物。均匀化之前最终的溶液组成6ml。
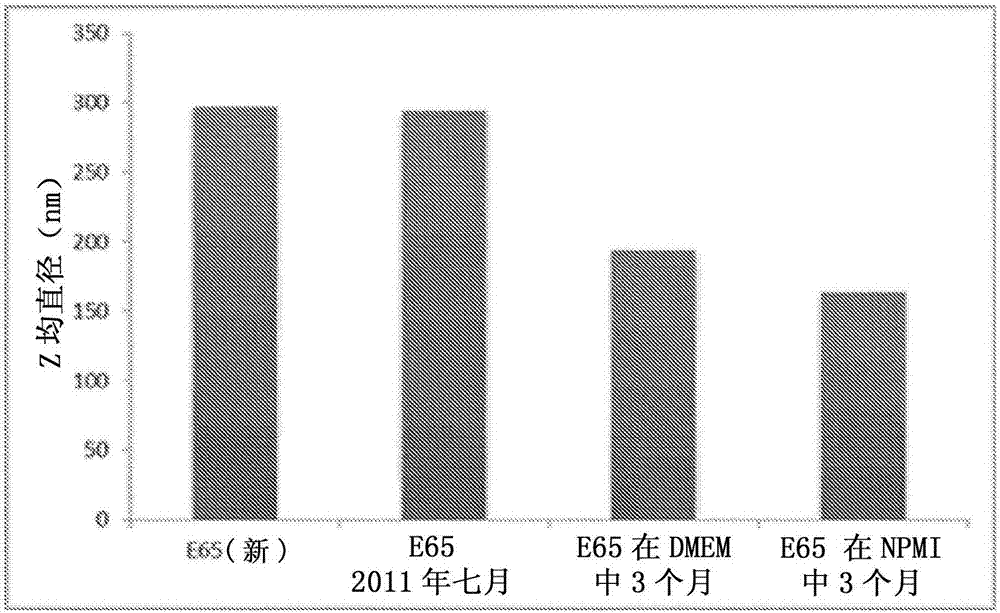
使用5%(w/v)的浓度,因为其完全稳定纳米乳液液滴,给予它们最小的直径,并允许在均匀化之后在该直径下稳定大于2年(图2)。在图2中,“e652011年7月”是在2011年7月制造的样品,存储然后在2013年7月改变尺寸。随着时间观察到优异的稳定性。dmem和rmpi是其中分散了e65的细胞培养物介质,然后存储在冰箱中并在三个月后改变尺寸(e65是基于使用本文所描述的支链聚合物与篦麻油、乙酸乙酯和依法韦仑的样品)。
即使当溶剂与油的比率保持不变时,改变聚合物的浓度也增加液滴的直径。这表明在较低的浓度下不存在足够的聚合物来完全包封油滴,导致它们聚集(图3)。图3示出了作为水相中的支链聚合物的浓度的函数的对于最终的乳液的z均直径。在5%(w/v)以上的浓度下,没有观察到直径的进一步降低,因此使用该值以上的浓度仅导致纳米乳液中残留的游离聚合物。
使用安装有s25n-10g分散元件并设置为25,000rpm的最大速度的ultrathuraxxt-25数字匀浆器使两相溶液均匀化2分钟。在2分钟期间,顺时针方向旋转小瓶30秒,逆时针旋转30秒,然后上下旋转1分钟。
可以使用任何合适的匀浆器,条件是其能够具有充分混合物溶液并将油“切碎”为纳米尺寸的液滴所必需的力。可以改变旋转小瓶的过程,但是在本实验中,将其保持相同以实现纳米乳液的批次之间的连续性。
均匀化之后,可以通过它们现在的乳白色和黏稠度看出样品被乳化。
在不封盖的情况下将小瓶置于通风橱中24小时,这使得挥发溶剂在环境温度下蒸发并使油滴瓦解,导致形成高度稳定的水包油乳液。用于除去挥发溶剂的其他方法包括稀释到水中和搅拌,并且稀释到水中并使氮气流过样品。这些方法中生产的液滴的直径与简单地将小瓶放置整夜相同。
药物存在于油相中,通过聚合物的覆盖而稳定,并具有通过dls确定的约200-300nm的z均直径(图2)。在该方法中制备的样品的最终体积是3ml。
当稀释以得到对于各种细胞分析适当的浓度时,最终的纳米乳液也保持稳定。通过稀释纳米乳液直到不再能通过dls看到而确认(图4)。
纳米乳液安全性
使用三个单独的试验流程进行免疫学评估。对于所有实验,使用了利用ficoll离心梯度法从全血中提取的外周血单核细胞。
最初,使用通过磁分离使用macs系统(miltenyibiotech)从pbmc分离的cd4+和cd8+细胞两者确定细胞表面活化标记物(cd25、cd44、cd69和cd95)的表达。数据示出不存在由于暴露于纳米乳液制剂导致的活化标记物的表达的增加,指示至少在体外非免疫原性的化合物。实际上,在对细胞表面标记物的表达的影响方面,纳米乳液与标准水溶液是相当的(图5)。
图5示出了暴露于洛匹那韦(lpv)和依法韦仑(efv)的水溶液或纳米乳液(n)制剂24小时之后细胞表面激活标记物的表达水平。如上所述,使用99:1乙酸乙酯:篦麻油,以及如上所述的支链聚合物(具有c12烷基部分并使用egdma支化的polyoegma)制备纳米乳液,且纳米乳液包含lpv或efv(“nlpv”或“nefv”-“n”表示纳米乳液)或完全不包含药物(“空白”)。从上到下的图例在每种情况下对应于从左到右的条。
分离表达试验的细胞外介质并使用bioplex200系统和试验试剂盒(bio-rad)分析细胞因子il-2、il-10和ifnγ的分泌水平。用t细胞活化试剂盒(miltenyibiotech)预刺激的细胞的数据示出了由暴露于纳米乳液的那些细胞分泌的细胞因子的水平与在暴露于相同药物的水溶液之后观察到的那些类似。在ifnγ的情况下,lpv纳米乳液在其分泌分布方面相比lpv的水溶液与对照更加相当(图6)。图6示出了用含水或纳米乳液arv制剂温育24小时之后细胞因子il-2(1)、il-10(2)和ifng(3)的分泌水平。在未刺激的细胞上进行相同的实验,结果示出在所有条件下不可检测的细胞因子水平(数据未示出)。
还使用3h-胸苷并入试验研究了纳米乳液对pbmc的细胞增殖的影响。简要地,在水溶液或纳米乳液等价物存在下,使pbmc生长72小时,其中在温育的最后16小时期间添加3h-胸苷。使用放射分析确定这些温育对增殖的任何影响。在添加或不添加用于刺激细胞增殖的植物血球凝集素(pha)的情况下进行实验。数据示出在所有制剂和添加下,在处理和未处理的细胞之间没有观察到差异(图7)。
抗凝血剂疗效
还评估了纳米乳液制剂对血浆凝结的影响。通过静脉穿刺术将来自三个供体的人血液收集到用柠檬酸钠抗凝结的管中;在收集的一个小时内使用血液。通过在2500xg下、在21℃下离心血液10分钟来制备测试血浆,并且收集并聚集得到的血浆。将聚集的血浆稳定在室温下8小时。以需要的最终浓度的10x制备纳米乳液样品以在添加到测试血浆中时提供稀释。浓度和随后的稀释是基于包含在样品内的依法韦仑的浓度,并且以相同的方式稀释空白纳米乳液。测试的最终浓度是40μg/ml、4μg/ml、0.8μg/ml和0.16μg/ml。将纳米乳液样品与测试血浆混合并在37℃下温育30分钟。制备三份每种纳米乳液制剂。将代表正常和不正常血浆(凝结延迟的血浆)的冻干的对照用蒸馏水(2ml)重组并在使用之前置于室温下平衡30分钟。
试验设计为捕获纳米乳液与三种主要凝结途径的组分的相互相用;内源性途径(也称为接触活化途径,因为其由损伤的表面激活)、外源性途径(也称为组织因子途径)和最终共同途径(finalcommonpathway)。将活化部分凝血活酶时间(aptt)试验用于评估内源性途径,而凝血酶原时间(pt)试验是外源性途径的量度。凝血酶时间(tt)是最终共同途径的作用的指示。
将试管放置到抗凝计上的a、b、c和d测试行,并将一个金属球添加到每个试管(在使用之前温热至少3分钟)中。当测试pt或凝血酶时间时,将100μl的对照或测试血浆的任一种添加到试管中,并且当测试aptt时,添加50μl,其中对于每个血浆样品用三个重复的试管。另外,对于aptt测定,还添加50μl的ptt-a。对于每个测试行开始计时器,并且在响铃提示之前10秒将试管转移到pip行中。一旦温育时间完成,将凝结试剂添加到每个试管中并记录凝结时间。根据下式计算每个对照和测试样品的百分比变化系数:%cv=sd/平均值x100%。如果对于研究样品%cv大于5%,则重新分析该样品。将数据表达为对于不存在纳米材料的血浆(仅血浆的对照)记录的凝结时间的百分比。
观察到依法韦仑溶液、空白纳米乳液或包含依法韦仑的纳米乳液对pt(图8a)或tt(图8b)没有影响。对于40μg/ml(大159%的凝结时间)和4μg/ml(大52%的凝结时间)的空白纳米乳液观察到aptt试验中(图8c)凝结时间显著延长。40μg/ml(大147%的凝结时间)和4μg/ml(大88%的延长时间)的包含依法韦仑的纳米乳液对延长时间具有类似的影响。然而,依法韦仑溶液对凝结时间完全没有影响,表明aptt的延长受纳米乳液或组成材料本身的影响。
此处,我们证明了新型纳米乳液的抗血凝性质。另外,已证明这种纳米乳液经由一种特殊的凝结途径延长凝结时间,从而潜在地减轻不希望的副作用。
抗病毒药物的疗效
针对适应实验室的菌株hiv-1iiib评估纳米乳液制剂的抗病毒活性,在暴露于病毒(有和没有水溶液或纳米乳液)之后使用mtt试验确定人淋巴细胞cd4+细胞系(mt4)的存活率。将细胞温育5天的时间。数据示出纳米乳液制剂的抗病毒活性与水溶液的那些类似,分别是对于lpv和efv的0.46μm和0.18μm的ic50值。
图9示出基于用lpv(顶部)和efv(底部)的含水或纳米乳液制剂温育的mt4细胞的存活率的hiv-1iiib的病毒杀伤曲线。
纳米乳液efv的剂量响应曲线与含水efv的那些相同,而纳米乳液lpv与水溶液相比在较低的浓度下具有更大的病毒杀伤。
因此,本发明提供了不仅表现出良好的稳定性而且还没有显示细胞毒性或免疫应答问题且此外有效的组合物。
第2部分:改变疏水基团的类型和量,包括使用混合引发剂的影响以及与类似的直链聚合物相比支链聚合物的性能的进一步研究
引发剂
通过用伯醇酯化α-溴代异丁酰溴制备具有不同的疏水性的若干atrp引发剂。这些引发剂以疏水性降序包括以下:
-溴代异丁酸十二烷基酯(dbib),c12引发剂
-溴代异丁酸己基酯(hbib),c6引发剂
-异丁酸乙酯(ebib),c2引发剂
-peg750溴代异丁酸酯(pbib),聚氧化乙烯大分子引发剂
根据在以下反应方案中概括的反应制备这些引发剂(ebib也购自sigmaaldrich)。从sigmaaldrich购买试剂。由具有750gmol-1平均分子量的单乙氧基peg合成peg引发剂。
聚合物的制备
将引发剂用于聚合oegma来得到直链聚合物以及用于制备oegma和egdma以得到统计学的支链聚合物。使用0.80:1的支化剂(egdma)与引发剂的比率以避免凝胶化。
使用单种引发剂进行聚合以产生相同官能化的端基,以及使用混合的引发剂来产生具有混合的端基官能团的统计学聚合物。
以每条直链80单元的单体oegma的目标聚合度(dp)进行聚合;将得到的直链聚合物表示为poegma80。这用每种引发剂进行;在每种情况下,聚合达到85-90%的转化率来产生具有类似的重量性质(根据gpc)的直链聚合物,如下表所示。
在egdma存在下通过聚合制备支链类似物。优化聚合以得到超过
99%的单体转化率,并且在引发剂范围内再次观察到类似的分子量性质,
如下表所示。
因此,通过改变引发剂可以定制疏水性而不显著改变聚合物的其他性质。
使用混合的引发剂制备另外的支链类似物。测试了各种体系。通过举例的方式,一种代表性体系使用不同比率的dbib和ebib。再次,反应进行为超过99%的单体转化率并产生类似的分子量特征的聚合物,如下表所示。因此,本发明允许细调疏水性,以及允许灵活性,因为可以出于其他原因改变引发剂。
聚合物作为乳化剂的用途
在模型体系中,使用按体积计1:1的十二烷油和聚合物水溶液制备乳液。使用高剪切混合器均匀化乳液两分钟。在不存在聚合物乳化剂的情况下,相分离在5分钟内产生。
由直链聚合物和支链聚合物制备的乳液的比较
将dbib、hbib和ebib各自单独用于直链poegma80的聚合和支链poegma80-egdma0.8的聚合。然后将这些用于制备乳液。在29天时间周期内研究乳液的平均液滴尺寸和分布。
直链聚合物产生具有开始的类似液滴尺寸的乳液,但是之后平均液滴尺寸取决于使用的引发剂增加不同的程度(ebib产生最不稳定的乳液,随后是hbib,随后是pbib)。相反,所有支链聚合物在整个时间周期内保持稳定;没有观察到凝聚且没有看到平均液滴尺寸随时间的显著变化,这对于所有支链聚合物是类似的。与图10(示出使用对应的直链聚合物制备的乳液作为时间的函数的平均液滴尺寸)相比,在图11(示出使用支链聚合物制备的乳液作为时间的函数的平均液滴尺寸)中看到差异。
不希望受到理论限制,支链构造似乎由于多端基效应,多个锚定物组装在同一结构中而提供了增加的锚定能力。
由具有混合引发剂的支链聚合物制备的乳液
使用混合的引发剂体系制备若干支链poegma80-egdma0.8聚合物,并将其用于制备乳液。一个非限制性的实例组使用不同比率的dbib和ebib,如下表所示。
与疏水的dbib相比,ebib是相对亲水的。即使当存在一些ebib时,dbib也提供良好的锚定效果。事实上,可以看出即使少至25mol%的dbib也足以在平均液滴尺寸的任何显著变化之前产生稳定的乳液,如图12所示。
跨细胞渗透实验
图13示出了与lpv的水溶液的跨细胞渗透(左手侧的柱)相比负载lpv的纳米乳液(右手侧的柱)在顶端至基底外侧方向(血液至内脏)(a)和基底外侧至顶端方向(内脏至血液)(b)中的跨细胞渗透。星形表示数据组之间的统计显著性。
图14示出了与efv的水溶液的跨细胞渗透(左手侧的柱)相比负载lpv的纳米乳液(右手侧的柱)在顶端至基底外侧方向(血液至内脏)(a)和基底外侧至顶端方向(内脏至血液)(b)中的跨细胞渗透。星形表示数据组之间的统计显著性。
跨细胞渗透试验
以35,000细胞每孔的密度将caco-2细胞接种在24孔
在试验期间,将培养介质从孔中除去并用具有10μm最终浓度的lpv或efv的水溶液或纳米乳液等同物的补充有15%fbs的dmem的替换。在不同的孔中,顶端或底外侧腔室(供体腔室)负载有包含药物或纳米乳液的介质,并且那些孔的相对的腔室(受体腔室)填充有新鲜的不含药物的介质。以1、2和24小时的时间周期从所有孔中从顶端和基底外侧腔室两者中取出样品。然后立即将样品冷冻在-40℃下用于随后经由高效液相色谱的批次分析,来确定样品中的药物的浓度。
使用以下等式计算表观渗透性(papp):papp=((a/t)*(v))/(s*0.3)。
其中a是样品的浓度,t是收集样品时的以秒计的时间,v是包含在采样的腔室中的样品的总体积,s是药物初始的起始浓度,以及0.3是以cm2计算的caco-2在其上生长的迁移室嵌入物的表面积。
负载lpv的纳米乳液的跨细胞渗透
在1小时和2小时时间点对于lpv的纳米乳液制剂看到统计上显著的(通过使用spss软件的独立样品t检验确定的)更大的渗透,其中在1小时的papp值是1x10-4对比8.4x10-6(p=<0.05)以及在2小时的papp值是6.4x10-5对比2.7x10-6(p=<0.05)。24小时之后,水溶液和纳米乳液体系之间看不到差异。
水溶液和纳米乳液lpv的papp数据最初示出在1小时时间点处顶端至底外侧的渗透对于纳米乳液改善(papp值是1x10-4对比8.4x10-6(p=<0.05))。然而,还观察到在相反方向上的渗透,即基底外侧至顶端也比水溶液值高得多(papp值9.3x10-6相比6.1x10-5)。lpv是已知的p-gp(在某些细胞表面发现的药物转运蛋白质底物,其使得可能在b-a方向上经历从caco-2细胞的流出(模拟从需要药物的体循环返回至肠道系统的移动)。然而,迁移室试验体系的条件没有理想地匹配在动物模型或实际上人体中发现的那些,因为药物不会在基底外侧隔室中聚集和累积,反而是被体循环运走(caco-2迁移室是静态试验)。另外,与转运(移动到细胞中,然后在另一侧移出)相反,caco-2细胞没有形成与在体内发现的那些一样的致密的单层,其被认为是低估经由细胞外路途径(即细胞之间,而不是穿过它们)渗透的化合物的原因。因此,非常有希望的是在1小时时在a-b方向渗透的大幅增加将可能导致体内体系中更多的药物越过肠屏障。
2小时时间点的数据再次示出与lpv的水溶液相比,对于纳米乳液存在从顶端至基底外侧的渗透的增加(6.4x10-5对比2.7x10-6(p=<0.05))。然而,在这种情况下,没有观察到基底外侧至顶端方向的相同增加(6.0x10-6对比1.3x10-5(p=0.11)),表明lpv纳米乳液的渗透优于lpv水溶液。
水溶液和纳米乳液制剂之间的渗透的差异可以表明药物通过不同的机制越过单层的转运。由于纳米乳液将不会经由转运蛋白越过单层转运,所以其意味着纳米乳液液滴经由细胞外转运越过单层渗透,这在之前的纳米颗粒渗透的研究中已经看出。这还可以解释为什么纳米乳液超过水溶液的在b-a渗透中的大幅增加,因为该制剂允许药物在其陷入乳液液滴内时在单层的小间隙之间移动。对于纳米乳液2小时时与1小时时相比b-a渗透的降低的可能解释是纳米乳液开始聚集且不再能经由细胞外方式穿过单层。
负载efv的纳米乳液的跨细胞渗透
1小时之后,在乳液制剂和含水efv溶液之间没有观察到表观渗透性的增加。然而,在2和24小时之后,efv纳米乳液具有显著升高的渗透性(2小时时与efv纳米乳液的1.1x10-5相比,水溶液为8.2x10-6(p=<0.05)和24小时时与efv纳米乳液的6.4x10-6相比,水溶液为7.0x10-7(p=<0.05))。
在1和2小时时,与efv纳米乳液相比,efv水溶液的b-a渗透性增加,但是再次应注意在静态体系如迁移室试验中,b-a渗透性相对a-b是较不有用的量度。仍然有希望的是看到b-a渗透性降低。caco-2迁移室体系的另一个问题是caco-2细胞不表达细胞色素p4502b6,并且因为这是efv的新陈代谢的主要途径,所以如果受新陈代谢保护,将低估在乳液液滴内包含药物可以具有的保护作用。还应注意efv已经示出了良好的肠道渗透性,因为其具有fda的2类状况(不良的溶解度、高肠道渗透性),所以看到进一步的改善表明纳米乳液制剂具有固有的良好的渗透性特征。
另外,可以将纳米乳液制剂直接稀释到含水培养介质中并用于测定而不需要提前溶解到溶剂中(其是制造lpv和efv的水溶液所必需的)。这对于制造容易调节剂量的更耐受的和安全的口服制剂是高度有吸引力的,尤其作为其中考虑使用溶剂如乙醇的儿科设置。
- 还没有人留言评论。精彩留言会获得点赞!