治疗或预防雌激素相关疾病的方法与流程
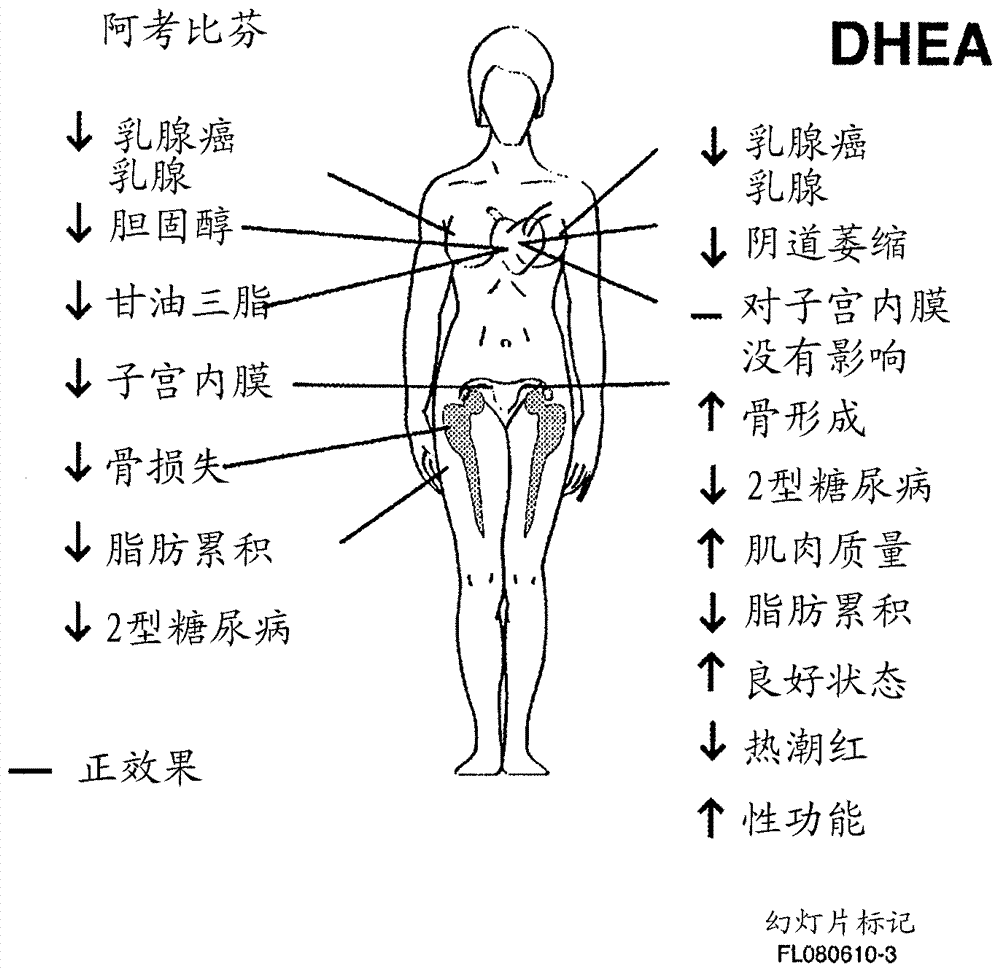
本申请是申请日为2011年6月16日、申请号为201180039155.0、发明名称为“治疗或预防雌激素相关疾病的方法”的发明专利申请的分案申请。相关申请的交叉引用本申请要求2010年6月16日申请的美国临时申请no.61/355,465的优先权,其全部内容以引证的方式包括在本文中。发明领域本发明涉及在易感的恒温动物包括人中使用新的组合疗法治疗雌激素相关(例如雌激素加重)疾病,包括子宫内膜异位或减少患上所述疾病可能性的方法。尤其是,一种组合方案包括给予选择性雌激素受体调节剂(serm),和例如通过给予lhrh激动剂或拮抗剂来抑制卵巢分泌。在一些实施方案中,也给予性甾体前体物,所述前体物选自脱氢表雄甾酮(dhea),硫酸脱氢表雄甾酮(dhea-s),雄-5-烯-3β,17β-二醇(5-二醇),和雄烯二酮或可转变为这些之一的化合物。相关领域的背景子宫内膜异位是常见的妇科疾病,导致显著的不孕比例,和痛经以及骨盆、腹部或阴道疼痛的高发病率。在发病率方面,子宫内膜异位是平滑肌瘤(jonesandjones1981)后第二大常见的妇科病症,其可见于20%年龄大于35岁的女性(conleyandlacey1984)。子宫内膜异位定义为“存在具有子宫内膜组织学结构和功能的异位组织”(sampson1921)。其是可能从初潮至绝经期都影响女性的虚弱性疾病。具(kistner1979)估计30至40%患有子宫内膜异位症的女性是不育的。因为为了生长和增殖,子宫内膜组织需要雌激素,在女性以及实验动物自然或手术经绝之后观察到,雌激素过少的状况会导致子宫内膜异位症的萎缩和衰退(dizerega,barber等人,1980)。在后面可能讨论到,除去卵巢雌激素不会除去子宫内膜异位组织的全部雌激素,而在缺乏制备雌二醇的酶尤其是芳香酶的正常人子宫内膜中,则会除去全部雌激素。据报道假孕和孕酮疗法可在超过80%的患者中改善子宫内膜异位症,减轻骨盆疼痛和痛经,但通常是短暂地(kistner1959;riva,wilson等人,1961;kistner1962;moghissiandboyce1976)。对雄激素也有类似的观察报道:甲基睾丸素在减轻痛经和腹痛中部分有效,但如果使用的剂量较高,则显现显著的雄性化迹象。此外,不能不断地抑制排卵,用雄激素治疗会提高雌性胎儿男性化的可能性或导致泌尿生殖器的致畸性(kistner1979)。对gnrh-a(也称为lhrh激动剂)疗法的初步研究显示在患有子宫内膜异位症患者中的主观和客观的改善(lemay,maheux等人,1984;ericksonandory1989),且对照比较研究证明了类似的效果和与达那唑相比对于gnrh-a有较好的耐受性(henzl,corson等人,1988;rock,truglia等人,1993)。由此较好地确立了由lhrh激动剂诱导的雌激素缺陷显示出对子宫内膜异位症的临床益处(meldrum,chang等人,1982;lemay,maheux等人,1984)。尽管有上述益处,gnrh-a疗法在子宫内膜异位症中的广泛应用已经受到雌激素过少的副作用的限制,例如血管舒缩的症状,阴道干燥,情绪不稳定,失眠,和骨矿物密度的损失(bmd)。担心这些副作用的长期效应,特别是bmd损失,限制了用gnrh-a疗法治疗对于大多数妇科病症的治疗持续时间为6个月(surrey1995)。根据我们的原始观测,在雄性大鼠中慢性给予lhrh激动剂导致以卵巢lh受体损失为特征的卵巢功能的抑制(auclair,kelly等人,1977a;auclair,kelly等人,1977b;auclair,ferland等人,1978)和类固醇生成的阻断(bélanger,auclair等人,1979;rivier,rivier等人,1979;bélanger,labrie等人,1980),因此有兴趣研究在母畜中卵巢促性腺激素受体类似损失的可能性。事实上,在发情间期i单一注射8nglhrh激动剂会导致卵巢lh受体的明显减少(30%)(kledzik,cusan等人,1978)。卵巢lh受体的接近最大抑制(60%)可在40ng的剂量处观察到,该抑制效果保持类似大小,直到至多25µg的剂量。卵巢lh受体减少伴随有子宫湿重、子宫内液体和血浆黄体酮浓度的显著的减少(在预期动情前期的早上测定);血清lh和fsh水平保持没有变化(kledzik,cusan等人,1978)。事实上,已知雌激素缺陷可导致骨损失和骨质疏松症,由此限制用于治疗子宫内膜异位症和子宫肌瘤的lhrh激动剂的另外有效使用。这导致建议使用lhrh激动剂单一6个月疗程来治疗子宫内膜异位症(fogelman1992)。当停止lhrh激动剂治疗时,这样的治疗程序通常伴有由于子宫内膜异位的细胞不充分细胞程序死亡而导致的子宫内膜异位症的反复。“反向加入”激素代替治疗(hrt),即各种药剂与gnrh-a联合治疗,是推荐的一种维持治疗响应和减少gnrh疗法潜在的不利事件的方法。该方法的基本原理得自雌激素阈限假设,其规定在某一浓度范围内的雌激素可以部分预防骨损失,而非刺激子宫内膜病变的生长(barbieri1992)。为了预防与lhrh-a疗法相关的骨损失,对加入小剂量雌激素(反向加入疗法)进行了研究。然而,反向加入雌激素疗法不太可能实现对子宫内膜异位症的最大作用。当将孕激素醋酸甲羟孕酮(mpa)和lhrh激动剂一起使用6个月,在前臂近端和远端没有发现显著的bmd变化(friedman1989)。已经在一些小规模的研究中评价了以醋酸甲羟孕酮(mpa)(lemay,dodin等人,1989;cedars,lu等人,1990)形式的孕激素疗法或炔诺酮(riis,christiansen等人,1990;eldred,haynes等人,1992;surreyandjudd1992)与gnrh-a组合疗法。两种疗法看来似乎可消除与gnrh-a疗法相关的血管舒缩症状和bmd损失,但持续的给予mpa看来似乎可逆转gnrh-a的有益效果,而炔诺酮在脂质特性上具有副作用(surrey1995)。然而,mpa显示了增加的乳腺癌危险(women'shealthinitiative2002)。雌激素受体拮抗剂还可以有效用于阻断对子宫内膜异位的组织增生起刺激作用的雌激素。可以使用的雌激素受体拮抗剂是:氟维司群,雷诺昔酚,三苯氧胺,枸橼酸托瑞米芬,阿佐昔芬,ly335563(去甲阿佐昔芬),ly335124,ly326315,chf-4227,萘氧啶,拉索昔芬,ly-2066948,ly-2120310,奥培米芬,司韦芬(a-007),tas-108,醋酸巴多昔芬(1-(4-[2-(氮杂环庚烷-1-基)乙氧基]苄基(-2-(4-羟基苯基)-3-甲基-1h-吲哚-5-醇乙酸酯),era-923,阿非昔芬,(z)-4-羟基它莫西芬,顺式氯米芬,非培米芬,阿考比芬,em-652,em-800,屈洛昔芬,艾多昔芬,gw5638,tat-59,gw-7603,森可曼(centchroman),左美洛昔芬,ici-164384,bl-3040,ch-4893237,sr16158,sr16137,rad-1901,serm3471(psk-3471),hmr3339,hmr3656,cc8490,11β-氟-7α-[5-(甲基-3-[(4,4,5,5,5-五氟戊基)硫烷基]丙氨基)戊基]雌甾-1,3,5(10)-三烯-3,17β-二醇(sh646,见wo1998/007740),11β-氟-17α-甲基-7α-5-[甲基(8,8,9,9,9-五氟壬基)氨基]戊基雌甾-1,3,5(10)-三烯-3,17β-二醇(见wo2003/045972)或(+)-3-(4-羟基苯基)-2-[4-(2-哌啶-1-基乙氧基)苯基]-4-(三氟甲基)-2h-苯并吡喃-7-醇(见wo2001/68634)。在us5,395,842;us5,393,785和us5,204,337中已经公开了甾体和非甾体抗雌激素药(选择性雌激素受体调节剂)治疗雌激素-相关疾病,包括子宫内膜异位症。其它用于治疗子宫内膜异位症的选择性雌激素受体调节剂公开在wo97/04763;uk2303628a;wo96/09040;wo96/09041;ep0652005a1;ep0731093a1;us5,484,797;ep0761669a2;us5,567,828;ep0729951a1;ep0703228a1;wo2004/009086;wo2004/259915;wo2005/073190;wo2005/073205;wo2005/073206;和wo2005/073244中。治疗雌激素敏感的疾病的组合治疗公开在us5,550,107中。lhrh激动剂或拮抗剂与选择性雌激素受体调节剂(serm)或抗雌激素药的组合治疗雌激素相关疾病包括子宫内膜异位症公开在ep1424080a1,us7,309,691b2,us2001/0041672a1和wo02/056903a2中。已经提出用氯米芬在lhrh激动剂治疗子宫内膜异位症期间用于保护骨骼(gouldingandfisher1991)。dhea,dhea-s,5-二醇和雄烯二酮可以以细胞和组织特异的方式通过内分泌学的方法转变为雌激素和/或雄激素(labrie,bélanger等人,1988;labrie1991;labrie,luu-the等人,2005和labrie2007)。本发明描述了治疗雌激素相关疾病包括子宫内膜异位症的新方法。本发明概述相应地,本发明的一个目标是提供治疗雌激素相关疾病包括子宫内膜异位症、同时将不希望的副作用减到最小的有效方法。另一个目标是提供减少获得上述疾病的危险的方法。另一个目标是提供适合上面方法使用的试剂盒。在一个实施方案中,本发明涉及治疗雌激素-相关疾病包括子宫内膜异位症或减少患上所述疾病的危险的方法,该方法包括给予需要所述治疗或所述减少的患者治疗有效量的lhrh激动剂或拮抗剂,和进一步包括给予所述患者作为组合治疗的部分的治疗有效量的选择性雌激素受体调节剂(serm)。在另一个实施方案中,本发明涉及治疗雌激素-相关疾病包括子宫内膜异位症或减少患上所述疾病的危险的方法,该方法包括给予需要所述治疗或所述减少的患者治疗有效量的lhrh激动剂或拮抗剂,和进一步包括给予所述患者治疗有效量的选择性雌激素受体调节剂(serm)和治疗有效量的选自下列的性甾体前体物:脱氢表雄甾酮(dhea),硫酸脱氢表雄甾酮(dhea-s),4-雄甾烯-3,17-二酮,雄甾-5-烯-3β,17β-二醇(5-二醇),体内转染入二者之一的化合物(前体药物),和其盐。本文使用的抗雌激素药是直接或通过其活性代谢物阻断雌激素受体的化合物,由此使受体不能被雌激素化合物利用,否则雌激素化合物可以活化受体。选择性雌激素受体调节剂(serm)是一种化合物,其可在子宫内膜和乳房组织中直接地或通过其活性代谢物起雌激素受体拮抗剂(“抗雌激素药”)的作用,但可在骨组织和血清胆固醇水平上提供类雌激素的作用(即通过减少血清胆固醇)。在体外,在人乳腺癌细胞系中,或在人乳腺癌模型中体内起雌激素受体拮抗剂作用的非甾体化合物(特别是如果化合物在人乳腺癌细胞(作为在裸鼠中的异种移植物生长)中起抗雌激素药的作用)同样起serm的作用。反之,甾体抗雌激素药则趋于不起serms的作用,因为它们趋于对骨不显示任何有益的作用。我们所发现的非甾体抗雌激素药或文献中报道的起serms的作用的非甾体抗雌激素药包括:em-800,em-652·hcl(阿考比芬),雷诺昔酚,ly335563,ly353381(阿佐昔芬),艾多昔芬,gw5638,它莫西芬,(z)-4-羟基它莫西芬,托瑞米芬,奥培米芬,屈洛昔芬,拉索昔芬,巴多昔芬(tse-424),和哌喷昔芬(era-923),但不局限于这些化合物。当在乳腺癌治疗中或为了减少乳腺癌或骨质疏松症形成的危险而使用这些化合物时,可以按本领域已知的相同剂量给予根据本发明的serms。本文使用的术语子宫内膜异位症包括但不局限于腹膜疾病,卵巢子宫内膜异位,和直肠阴道的疾病。它包括子宫内膜组织在任何位置的生长,包括子宫的内层或子宫内膜。在另一个实施方案中,本发明涉及一种治疗其它雌激素-相关疾病或减少患上所述疾病的危险的方法,雌激素-相关疾病例如子宫肌瘤,子宫平滑肌瘤,子宫内膜癌,子宫癌,子宫平滑肌肉瘤,卵巢癌,乳腺癌,多囊卵巢综合征,功能性月经失调,阴道出血,月经过多,经前期综合征,偏头痛,宫颈上皮内瘤样病变,内在性子宫内膜异位症,和阿尔海默氏疾病,包括给予需要所述治疗或所述减少的患者治疗有效量的lhrh激动剂或拮抗剂,和进一步包括给予所述患者治疗有效量的选择性雌激素受体调节剂(serm)和任选选自下列的治疗有效量的性甾体前体物:脱氢表雄甾酮(dhea),硫酸脱氢表雄甾酮(dhea-s),4-雄甾烯-3,17-二酮,雄甾-5-烯-3β,17β-二醇(5-二醇),体内转变为二者之一的化合物(前体药物),和其盐。本发明的其它特征和优点将由下列非限制性的、引用附图的优选实施方案的说明变得更清楚。附图的简要说明图1是血清dhea随年龄的渐进性减少的略图。在经绝时,血清dhea已经减少了60%,且此后继续降低。对于全部雄激素接触观察到相似的降低。图2举例说明了年龄30至40岁和50至75岁的正常女性中血清dhea水平的广泛改变。数据是独立地以及以平均值,且为5%-95%百分数给出的(labrie等人,未发表资料)。图3是柱状图,显示了完整的绝经后女性(与卵巢切除的绝经后女性相比)(年龄在42至74岁)血清dhea高出22.3%,取样器的编号显示于x轴。图4是绝经后女性的性甾体的独特来源的略图,即dhea。经绝之后,全部雌激素和雄激素是在周围靶向胞分泌组织从肾上腺(80%)或卵巢(20%)dhea局部制备的。在整个一生期间,雄激素仅仅来源于肾上腺和卵巢dhea。周围靶组织制备的性甾体的量取决于在每个组织中特异性表达的甾体形成酶(steroid-formingenzymes)的水平。图5是大鼠中用dhea(10mg,经皮,每日一次)或em-800(em-652的前体物,其也得自于em-652·hcl(阿考比芬))(75µg,口服,每日一次)单独治疗与组合治疗9个月的血清甘油三酯(a)和胆固醇(b)水平的效果的对比柱状图。数据以平均值±sem来表示。**:p<0.01实验组对相应的对照组。图6是在切除卵巢的大鼠中用脱氢表雄甾酮(dhea)单独治疗或用氟他米特(flu)或em-800组合治疗12个月对小梁骨体积的效果的对比柱状图。添加了完整动物另外的对照组。数据以平均值±sem给出。**p<0.01对卵巢切除的(ovx)对照组。图7是在切除卵巢的大鼠中用脱氢表雄甾酮(dhea)单独治疗或用氟他米特(flu)或em-800组合治疗12个月对小梁骨数目的效果的对比柱状图。添加了完整动物另外的对照组。数据以平均值±sem给出。**p<0.01对卵巢切除的(ovx)对照组。图8显示了完整对照组(a)、切除卵巢的对照组(b)和用dhea单独治疗的切除卵巢的大鼠(c)或用氟他米特(flu)(d)或em-800(e)组合治疗的近端胫骨干骺端。注意到在切除卵巢的对照动物(b)中小梁骨(t)数量减少,而给予dhea(c)后诱导的小梁骨体积(t)显著的增加。向dhea加入氟他米特(flu)部分地阻断了dhea在小梁骨体积上的效果(d),而dhea和em-800组合提供了对卵巢切除术相关的骨损失的彻底保护。改性的三色masson-goldner,magn.x80。t:小梁,gp:生长面。图9显示了标准hrt(雌激素)和选择性雌激素受体调节剂(serm)在绝经参数上的效果比较。图10显示了标准hrt(雌激素)和脱氢表雄甾酮在绝经参数上的效果比较。图11显示serm(阿考比芬)和dhea在绝经参数上的联合效果。没有预期的副作用。详细说明本文引用的参考文献的全部引用列表在以下以简表形式列出:ailawadi,r.k.,s.jobanputra,m.kataria,b.gurates和s.e.bulun(2004)。“treatmentofendometriosisandchronicpelvicpainwithletrozoleandnorethindroneacetate:apilotstudy.”fertilsteril81(2):290-6。allen,l.v.withthecontributionsbyd.b.worthen和b.mink(2008)。suppositorybasesandtheircharacteristics(chapter3)。suppositories.pharmaceuticalpress,london,uk:27-49。auclair,c.,p.a.kelly,d.h.coy,a.v.schally和f.labrie(1977a)。“potentinhibitoryactivityof[d-leu6,des-gly-nh210]ethylamideonlh/hcgandprltesticularreceptorlevelsintherat.”endocrinology101:1890-1893。auclair,c.,p.a.kelly,f.labrie,d.h.coy和a.v.schally(1977b)。“inhibitionoftesticularluteinizingreceptorlevelbytreatmentwithapotentluteinizinghormone-releasinghormoneagonistofhumanchorionicgonadotropin.”biochem.biophys.res.commun.76:855-862。auclair,c.,l.ferland,l.cusan,p.a.kelly,f.labrie,g.azadian-boulanger和j.p.raynaud(1978)。“effetinhibiteurdelalhrhsurlesrécepteursdelalhdansletesticulechezlerat.”c.r.acad.sci.paris,séried286:1305-1307。barbieri,r.(1992)。“hormonetreatmentofendometriosis:theestrogenthresholdhypothesis.”am.j.obstet.gynecol.166:740-745。bardon,s.,f.vignon,d.chalbos和h.rochefort(1985)。“ru486,aprogestinandglucocorticoidantagonist,inhibitsthegrowthofbreastcancercellsviatheprogesteronereceptor.”j.clin.endocrinol.metab.60:692-697。baxendale,p.m.,m.j.reed和v.h.james(1981)。“inabilityofhumanendometriumormyometriumtoaromatizeandrostenedione.”jsteroidbiochem14(3):305-6。bélanger,a.,c.auclair,c.séguin,p.a.kelly和f.labrie(1979)。“down-regulationoftesticularandrogenbiosynthesisandlhreceptorlevelsbyanlhrhagonist,roleofprolactin.”mol.cell.endocrinol.13:47-53。bélanger,a.,f.labrie,a.lemay,s.caron和j.p.raynaud(1980)。“inhibitoryeffectsofasingleintranasaladministrationof[d-ser(tbu)6,des-gly-nh210]lhrhagonist,apotentlhrhagonist,onserumsteroidlevelsinnormaladultmen.”j.steroidbiochem.13:123-126。black,l.j.,m.sato,e.r.bowley,d.e.magee,a.bekele,d.c.williams,g.j.cullinan,r.bendele,r.f.kaufman,w.r.bensch,c.a.frolik,j.d.termine和h.u.bryant(1994)。“raloxifene(ly139481hcl)preventsbonelossandreducesserumcholesterolwithoutcausinguterinehypertrophyinovariectomizedrats.”j.clin.invest.93:63-69。bulun,s.e.,s.yang,z.fang,b.gurates,m.tamura,j.zhou和s.sebastian(2001)。“roleofaromataseinendometrialdisease.”jsteroidbiochemmolbiol79(1-5):19-25。bulun,s.e.,z.lin,g.imir,s.amin,m.demura,b.yilmaz,r.martin,h.utsunomiya,s.thung,b.gurates,m.tamura,d.langoi和s.deb(2005)。“regulationofaromataseexpressioninestrogen-responsivebreastanduterinedisease:frombenchtotreatment.”pharmacolrev57(3):359-83。burger,h.g.,j.hailes,m.menelaus,j.nelson,b.hudson和n.balazs(1984)。“themanagementofpersistentmenopausalsymptomswithoestradiol-testosteroneimplants:clinical,lipidandhormonalresults.”maturitas6:351-358。casson,p.r.,r.n.andersen,h.g.herrod,f.b.stentz,a.b.straughn,g.e.abraham和j.e.buster(1993)。“oraldehydroepiandrosteroneinphysiologicdosesmodulatesimmunefunctioninpostmenopausalwomen”,am.j.obstet.gynecol.169:1536-1539。cedars,m.,j.lu,d.meldrum和h.judd(1990)。“treatmentofendometriosiswithalong-actinggonadotropin-releasinghormoneagonistplusmedroxyprogesteroneacetate.”obstet.gynecol.5:641-645。colditz,g.a.,s.e.hankinson,d.j.hunter,w.c.willett,j.e.manson,m.j.stampfer,c.hennekens,b.rosner和f.e.speizer(1995)。“theuseofestrogensandprogestinsandtheriskofbreastcancerinpostmenopausalwomen.”n.engl.j.med.332:1589-1593。coleman,d.l.,e.h.leiter和r.w.schwizer(1982)。“therapeuticeffectsofdehydroepiandrosterone(dhea)indiabeticmice.”diabetes31:830-833。conley,g.和m.d.lacey(1984)。currentobstetricandgynecologicdiagnosisandtreatment.r.c.benten.lange:258-263。corbin,a.,f.j.bex和r.c.jones(1984)。“comparisonoflhrhagonist(ag)andantagonist(ant):antifertilityandtherapeuticdevelopments.”j.steroidbiochem.20(6b)(1369):a9。couillard,s.,m.gutman,c.labrie,a.bélanger,b.candas和f.labrie(1998)。“comparisonoftheeffectsoftheantiestrogensem-800andtamoxifenonthegrowthofhumanbreastzr-75-1cancerxenograftsinnudemice.”cancerres.58:60-64。couillard,s.,c.labrie,a.bélanger,b.candas,f.pouliot和f.labrie(1998)。“effectofdehydroepiandrosteroneandtheantiestrogenem-800onthegrowthofhumanzr-75-1breastcancerxenografts.”j.natl.cancerinst.90:772-778。coy,d.h.,a.horvath,m.v.nekola,e.j.coy,j.erchegyi和a.v.schally(1982)。“peptideantagonistsoflhrh:largeincreasesinantiovulatoryactivitiesproducedbybasicd-aminoacidsinthesixposition.”endocrinology110:1445-1447。diamond,p.,l.cusan,j.l.gomez,a.bélanger和f.labrie(1996)。“metaboliceffectsof12-monthpercutaneousdheareplacementtherapyinpostmenopausalwomen.”j.endocrinol.150:s43-s50。dizerega,s.g.,d.l.barber和g.d.hodgen(1980)。“endometriosis:roleofovariansteroidsininitiation,maintenance,andsuppression.”fertil.steril.33:649-653。draper,m.w.,d.e.flowers,j.a.neild,w.j.huster和r.l.zerbe(1995)。“antiestrogenicpropertiesofraloxifene.”pharmacology50(4):209-217。draper,m.w.,d.e.flowers,w.j.huster,j.a.neild,k.d.harper和c.arnaud(1996)。“acontrolledtrialofraloxifene(ly139481)hcl:impactonboneturnoverandserumlipidprofileinhealthypostmenopausalwomen.”j.boneminer.res.11(6):835-842。dutta,a.s.,b.j.a.furr,m.b.giles和b.valcaccia(1978)。“synthesisandbiologicalactivityofhighlyactivea-azaanaloguesofluliberin.”j.med.chem.21(10):1018-1024。eldred,j.,p.haynes和c.thomas(1992)。“arandomizeddouble-blindplacebocontrolledtrialoftheeffectsofbonemetabolismofthecombinationofnafarelinacetateandnorethisterone.”clin.endocrinol.37:354-359。erchegyi,j.,d.h.coy,m.v.nekola,e.j.coy,a.v.schally,i.mezo和i.teplan(1981)。“luteinizinghormone-releasinghormoneanalogswithincreasedactivity.”biochem.biophys.res.commun.100:915-920。erickson,l.d.和s.j.ory(1989)。“gnrhanaloguesinthetreatmentofendometriosis.”obstet.gynecol.clin.northam.16:23-45。fang,z.,s.yang,b.gurates,m.tamura,e.simpson,d.evans和s.e.bulun(2002)。“geneticorenzymaticdisruptionofaromataseinhibitsthegrowthofectopicuterinetissue.”jclinendocrinolmetab87(7):3460-6。fogelman,i.(1992)。“gonadotropin-releasinghormoneagonistsandtheskeleton.”fertil.steril.57:715-724。friedman,a.j.(1989)。“treatmentofleiomyomatauteriwithshort-termleuprolidefollowedbyleuprolideplusestrogen-progestinhormonereplacementtherapyfor2years:apilotstudy.”fertil.steril.51:526-528。gauthier,s.,b.caron,j.cloutier,y.l.dory,a.favre,d.larouche,j.mailhot,c.ouellet,a.schwerdtfeger,g.leblanc,c.martel,j.simard,y.mérand,a.bélanger,c.labrie和f.labrie(1997)。“(s)-(+)-4-[7-(2,2-dimethyl-1-oxopropoxy)-4-methyl-2-[4-[2-(1-piperidinyl)-ethoxy]phenyl]-2h-1-benzopyran-3-yl]-phenyl2,2-dimethylpropanoate(em-800):ahighlypotent,specific,andorallyactivenonsteroidalantiestrogen.”j.med.chem.40:2117-2122。gordon,g.b.,l.m.shantz和p.talalay(1987)。“modulationofgrowth,differentiationandcarcinogenesisbydehydroepiandrosterone.”adv.enzymeregul.26:355-382。goulding,a.和l.fisher(1991)。“preventiveeffectsofclomiphenecitrateonestrogen-deficiencyosteopeniaelicitedbylhrhagonistadministrationintherat.”j.boneminer.res.6(11):1177-81。gurates,b.,s.sebastian,s.yang,j.zhou,m.tamura,z.fang,t.suzuki,h.sasano和s.e.bulun(2002)。“wt1anddax-1inhibitaromatasep450expressioninhumanendometrialandendometrioticstromalcells.”jclinendocrinolmetab87(9):4369-77。henderson,e.,j.y.yang和a.schwartz(1992)。“dehydroepiandrosterone(dhea)andsysntheticdheaanalogsaremodestinhibitorsofhiv-1iiibreplication.”aidsres.hum.retroviruses8:625-631。hennernan,p.m.和s.wallach(1957)。“theroleofandrogensandestrogensandtheirmetaboliceffects.areviewoftheprolongeduseofestrogensandandrogensinpostmenopausalandsenileosteoporosis.”ama:arch.int.med.100:715-723。henzl,m.r.,s.l.corson,k.moghissi,v.c.buttram,c.bergvist和j.jacobson(1988)。“administrationofnasalnafarelinascomparedwithoraldanazolforendometriosis:amulticentredouble-blindcomparativetrial.”n.engl.j.med.318:485-489。jankowski,c.m.,w.s.gozansky,r.s.schwartz,d.j.dahl,j.m.kittelson,s.m.scott,r.e.vanpelt和w.m.kohrt(2006)。“effectsofdehydroepiandrosteronereplacementtherapyonbonemineraldensityinolderadults:arandomized,controlledtrial.”jclinendocrinolmetab91(8):2986-93。johnstonjr,c.c.和s.epstein(1981)。“clinical,biochemical,radiographic,epidemiologic,andeconomicfeaturesofosteoporosis.”orthop.clin.north.am.12:559-569。jones,h.w.和g.s.jones(1981)。novak's,textbookofgynecology.baltimore,williams和whilkins。kauffman,r.f.和h.u.bryant(1995)。“effectivetherapeuticmanagementofthepostmenopausalstatewillbeacornerstoneinstrategiesforpreservingorimprovingwomen'shealthinthe21stcentury.”drugnewsandperspectives8:531-539。kistner,r.w.(1959)。“thetreatmentofendometriosisbyinducingpseudopregnancywithovarianhormones:areportoffifty-eightcases.”fertil.steril.10:539-556。kistner,r.w.(1962)。“infertilitywithendometriosis:aplanoftherapy.”fertil.steril.13:237-245。kistner,r.w.(1979)。“endometriosisandinfertility.”clin.obstet.gynecol.22:101-119。kitawaki,j.,t.noguchi,t.amatsu,k.maeda,k.tsukamoto,t.yamamoto,s.fushiki,y.osawa和h.honjo(1997)。“expressionofaromatasecytochromep450proteinandmessengerribonucleicacidinhumanendometrioticandadenomyotictissuesbutnotinnormalendometrium.”biolreprod57(3):514-9。kledzik,g.s.,l.cusan,c.auclair,p.a.kelly和f.labrie(1978)。“inhibitionofovarianlhandfshreceptorlevelswithanlh-releasinghormoneagonistduringtheestrouscycleintherat.”fertil.steril.30:348-353。labrie,c.,a.bélanger和f.labrie(1988)。“androgenicactivityofdehydroepiandrosteroneandandrostenedioneintheratventralprostate.”endocrinology123:1412-1417。labrie,f.(1991)。“intracrinology.”mol.cell.endocrinol.78:c113-c118。labrie,f.,j.simard,v.luu-the,a.bélanger和g.pelletier(1992a)。“structure,functionandtissue-specificgeneexpressionof3b-hydroxysteroiddehydrogenase/5-ene-4-eneisomeraseenzymesinclassicalandperipheralintracrinesteroidogenictissues.”j.steroidbiochem.mol.biol.43:805-826。labrie,f.,j.simard,v.luu-the,g.pelletier,a.bélanger,y.lachance,h.f.zhao,c.labrie,n.breton,y.delaunoit,m.dumont,e.dupont,e.rhéaume,c.martel,j.couet和c.trudel(1992b)。“structureandtissue-specificexpressionof3b-hydroxysteroiddehydrogenase/5-ene-4-eneisomerasegenesinhumanandratclassicalandperipheralsteroidogenictissues.”j.steroidbiochem.mol.biol.41:421-435。labrie,f.,j.simard,v.luu-the,a.bélanger,g.pelletier,y.morel,f.mebarki,r.sanchez,f.durocher,c.turgeon,y.labrie,é.rhéaume,c.labrie和y.lachance(1996)。the3b-hydroxysteroiddehydrogenase/isomerasegenefamily:lessonsfromtypeii3b-hsdcongenitaldeficiency.signaltransductionintesticularcells,ernstscheringresearchfoundationworkshop.v.hansson,f.o.levy和k.taskén.berlin,heidelberg,springer-verlag.suppl.2:185-218。labrie,f.,a.belanger,l.cusan和b.candas(1997a)。“physiologicalchangesindehydroepiandrosteronearenotreflectedbyserumlevelsofactiveandrogensandestrogensbutoftheirmetabolites:intracrinology.”jclinendocrinolmetab82(8):2403-2409。labrie,f.,a.belanger,l.cusan,j.l.gomez和b.candas(1997b)。“markeddeclineinserumconcentrationsofadrenalc19sexsteroidprecursorsandconjugatedandrogenmetabolitesduringaging.”jclinendocrinolmetab82:2396-2402。labrie,f.,p.diamond,l.cusan,j.l.gomez,a.belanger和b.candas(1997)。“effectof12-monthdehydroepiandrosteronereplacementtherapyonbone,vagina,andendometriuminpostmenopausalwomen.”jclinendocrinolmetab82(10):3498-505。labrie,f.,v.luu-the,s.x.lin,c.labrie,j.simard,r.breton和a.bélanger(1997)。“thekeyroleof17b-hsdsinsexsteroidbiology.”steroids62:148-158。labrie,f.,c.labrie,a.bélanger,j.simard,v.giguère,a.tremblay和g.tremblay(2001)。“em-652(sch57068),apuresermhavingcompleteantiestrogenicactivityinthemammaryglandandendometrium.”j.steroidbiochem.mol.biol.79:213-225。labrie,f.,v.luu-the,c.labrie,a.bélanger,j.simard,s.-x.lin和g.pelletier(2003)。“endocrineandintracrinesourcesofandrogensinwomen:inhibitionofbreastcancerandotherrolesofandrogensandtheirprecursordehydroepiandrosterone.”endocrinereviews24(2):152-182。labrie,f.,v.luu-the,a.bélanger,s.-x.lin,j.simard和c.labrie(2005)。“isdheaahormonestarlingreview.”jendocrinol187:169-196。labrie,f.(2006)。“futureperspectivesofsermsusedaloneandincombinationwithdhea.”endocrrelatcancer13(2):335-355。labrie,f.,a.bélanger,p.bélanger,r.bérubé,c.martel,l.cusan,j.l.gomez,b.candas,i.castiel,v.chaussade,c.deloche和j.leclaire(2006)。“androgenglucuronides,insteadoftestosterone,asthenewmarkersofandrogenicactivityinwomen.”journalsterbiochem&molbiol99:182-188。labrie,f.(2007)。“druginsight:breastcancerpreventionandtissue-targetedhormonereplacementtherapy.”natureclinicalpractice,endocrinology&metabolism3(8):584-593。labrie,f.,a.belanger,p.belanger,r.berube,c.martel,l.cusan,j.gomez,b.candas,v.chaussade,i.castiel,c.deloche和j.leclaire(2007)。“metabolismofdheainpostmenopausalwomenfollowingpercutaneousadministration.”jsteroidbiochemmolbiol103(2):178-88。labrie,f.,l.cusan,j.l.gomez,i.côté,r.bérubé,p.bélanger,c.martel和c.labrie(2008)。“effectofintravaginaldheaonserumdheaandelevenofitsmetabolitesinpostmenopausalwomen.”journalsterbiochem&molbiol111:178-94。labrie,f.,d.archer,c.bouchard,m.fortier,l.cusan,j.l.gomez,g.girard,m.baron,n.ayotte,m.moreau,r.dubé,i.côté,c.labrie,l.lavoie,l.berger,l.gilbert,c.martel和j.balser(2009a)。“effectonintravaginaldehydroepiandrosterone(prasterone)onlibidoandsexualdysfunctioninpostmenopausalwomen.”menopause16:923-931。labrie,f.,d.archer,c.bouchard,m.fortier,l.cusan,j.l.gomez,g.girard,m.baron,n.ayotte,m.moreau,r.dubé,i.côté,c.labrie,l.lavoie,l.berger,l.gilbert,c.martel和j.balser(2009b)。“intravaginaldehydroepiandrosterone(prasterone),aphysiologicalandhighlyefficienttreatmentofvaginalatrophy.”menopause16:907-922。labrie,f.,d.archer,c.bouchard,m.fortier,l.cusan,j.l.gomez,g.girard,m.baron,n.ayotte,m.moreau,r.dubé,i.côté,c.labrie,l.lavoie,l.berger,l.gilbert,c.martel和j.balser(2009c)。“serumsteroidlevelsduring12-weekintravaginaldehydroepiandrosteroneadministration.”menopause16:897-906。labrie,f.,l.cusan,j.l.gomez,c.martel,r.berube,p.belanger,a.belanger,l.vandenput,d.mellström和c.ohlsson(2009)。“comparableamountsofsexsteroidsaremadeoutsidethegonadsinmenandwomen:stronglessonforhormonetherapyofprostateandbreastcancer.”jsteroidbiochemmolbiol113:52-56。labrie,f.(2010)。dhea,importantsourceofsexsteroidsinmenandevenmoreinwomen.neuroendocrinology,thenormalneuroendocrinesystem,progressinbrainresearch.l.martini,chrousosgp,labrief,pacakk和d.pfaff,eds.,elsevier.182:chapter4,97-148。labrie,f.,c.martel,s.gauthier,g.pelletier和j.y.sancéau(2010)。“effectoftoremifeneandospemifene,comparedtoacolbifene,onestrogen-sensitiveparametersinratandhumanuterinetissues.”hormmolbiolclininvest1:139-146。labrie,f.,c.martel和j.balser(2011)。“widedistributionoftheserumdehydroepiandrosteroneandsexsteroidlevelsinpostmenopausalwomen:roleoftheovary”menopause18:30-43。labrie,y.,f.durocher,y.lachance,c.turgeon,j.simard,c.labrie和f.labrie(1995)。“utiliserl'autreréf.thehumantypeii17beta-hydroxysteroiddehydrogenasegeneencodestwoalternativelysplicedmrnaspecies.”dnacellbiol14(10):849-61。leiblum,s.,g.bachmann,e.kemmann,d.colburn和l.swartzman(1983)。“vaginalatrophyinthepostmenopausalwomen.theimportanceofsexualactivityandhormones.”jama249:2195-2198。lemay,a.,r.maheux,n.faure,c.jean和a.t.a.fazekas(1984)。“reversiblehypogonadisminducedbyaluteinizinghormone-releasinghormone(lhrh)agonist(buserelin)asanewtherapeuticapproachforendometriosis.”fertil.steril.41:863-871。lemay,a.,s.dodin和s.dewailly(1989)。“long-termuseofthelowdoselhrhanaloguecombinedwithmonthlymedroxyprogesteroneadministration.”horm.res.32(suppl.1):141-145。li,s.,x.yan,a.bélanger和f.labrie(1993)。“preventionbydehydroepiandrosteroneofthedevelopmentofmammarycarcinomainducedby7,12-dimethylbenz(a)anthracene(dmba)intherat.”breastcancerres.treat.29:203-217。luo,s.,c.martel,s.gauthier,y.mérand,a.bélanger,c.labrie和f.labrie(1997a)。“longterminhibitoryeffectsofanovelantiestrogenonthegrowthofzr-75-1andmcf-7humanbreastcancertumorsinnudemice.”int.j.cancer73:735-739。luo,s.,c.martel,a.sourla,s.gauthier,y.mérand,a.bélanger,c.labrie和f.labrie(1997b)。“comparativeeffectsof28-daytreatmentwiththenewantiestrogenem-800andtamoxifenonestrogen-sensitiveparametersintheintactmouse.”int.j.cancer73:381-391。luo,s.,a.sourla,c.labrie,a.bélanger和f.labrie(1997)。“combinedeffectsofdehydroepiandrosteroneandem-800onbonemass,serumlipids,andthedevelopmentofdimethylbenz(a)anthracene(dmba)-inducedmammarycarcinomaintherat.”endocrinology138:4435-4444。luo,s.,c.labrie和f.labrie(1998)。“preventionofdevelopmentofdimenthylbenz(a)anthracene(dmba)-inducedmammarycarcinomaintheratbythenewnonsteroidalantiestrogenem-800(sch57050)。”breastcancerres.treat.49:1-11。luo,s.,m.stojanovic,c.labrie和f.labrie(1998)。“inhibitoryeffectofthenovelantiestrogenem-800andmedroxyprogesteroneacetate(mpa)onestrone-stimulatedgrowthofdimethylbenz(a)anthracene(dmba)-inducedmammarycarcinomaintherat.”int.j.cancer73:580-586。luu-the,v.,i.dufort,n.paquet,g.reimnitz和f.labrie(1995)。“structuralcharacterizationandexpressionofthehumandehydroepiandrosteronesulfotransferasegene.”dnacellbiol.14:511-518。macewen,e.g.和i.d.kurzman(1991)。“obesityinthedog:roleoftheadrenalsteroiddehydroepiandrosterone(dhea)。”j.nutr.121:s51-s55。martel,c.,a.sourla,g.pelletier,c.labrie,m.fournier,s.picard,s.li,m.stojanovic和f.labrie(1998)。“predominantandrogeniccomponentinthestimulatoryeffectofdehydroepiandrosteroneonbonemineraldensityintherat.”j.endocrinol.157:433-442。meldrum,d.r.,r.j.chang,j.lu,w.vale,j.rivier和h.l.judd(1982)。"medicaloophorectomy"usingalong-actinggnrhagonist--apossiblenewapproachtothetreatmentofendometriosis.”j.clin.endocrinol.metab.54:1081-1083。michalska,d.,j.j.stepan,b.r.basson和i.pavo(2006)。“theeffectofraloxifeneafterdiscontinuationoflong-termalendronatetreatmentofpostmenopausalosteoporosis.”jclinendocrinolmetab91(3):870-7。moghissi,k.s.和c.r.boyce(1976)。“managementofendometriosiswithoralmedroxyprogesteroneacetate.”obstet.gynecol.47:265-267。morales,a.j.,j.j.nolan,j.c.nelson和s.s.yen(1994)。“effectsofreplacementdoseofdehydroepiandrosteroneinmenandwomenofadvancingage.”j.clin.endocrinol.metab.78:1360-1367。morales,a.j.,r.h.haubrich,j.y.hwang,h.asakura和s.s.yen(1998)。“theeffectofsixmonthstreatmentwitha100mgdailydoseofdehydroepiandrosterone(dhea)oncirculatingsexsteroids,bodycompositionandmusclestrengthinage-advancedmenandwomen.”clinendocrinol(oxf)49(4):421-32。nair,k.s.,r.a.rizza,p.o'brien,k.dhatariya,k.r.short,a.nehra,j.l.vittone,g.g.klee,a.basu,r.basu,c.cobelli,g.toffolo,c.dallaman,d.j.tindall,l.j.melton,3rd,g.e.smith,s.khosla和m.d.jensen(2006)。“dheainelderlywomenanddheaortestosteroneinelderlymen.”nengljmed355(16):1647-59。need,a.g.,m.horowitz,a.bridges,h.a.morris和b.e.nordin(1989)。“effectsofnandrolonedecanoateandantiresorptivetherapyonvertebraldensityinosteoporoticpostmenopausalwomen.”arch.intern.med.149:57-60。nestler,j.e.,c.o.barlascini,j.n.clore和w.g.blackard(1988)。“dehydroepiandrosteronereducesserumlowdensitylipoproteinlevelsandbodyfatbutdoesnotalterinsulinsensitivityinnormalmen.”j.clin.endocrinol.metab.66:57-61。nestor,j.j.j.,t.l.ho,r.tahilramani,b.l.horner,r.a.simpson,g.h.jones,g.i.mcrae和b.h.vickery(1984)。lhrhagonistsandantagonistscontainingveryhydrophobicaminoacids.lhrhanditsanalogs.b.h.vickery,j.j.nestor和e.s.e.hafez.lancaster,england,mtppress:22-33。noble,l.s.,e.r.simpson,a.johns和s.e.bulun(1996)。“aromataseexpressioninendometriosis.”jclinendocrinolmetab81(1):174-9。noble,l.s.,k.takayama,k.m.zeitoun,j.m.putman,d.a.johns,m.m.hinshelwood,v.r.agarwal,y.zhao,b.r.carr和s.e.bulun(1997)。“prostaglandine2stimulatesaromataseexpressioninendometriosis-derivedstromalcells.”jclinendocrinolmetab82(2):600-6。notelovitz,m.,n.watts,c.timmons,a.addison,b.wiita和l.downey(1991)。effectsofestrogenpluslowdoseandrogenvsestrogenaloneonmenopausalsymptomsinoophorectomized/hysterectomizedwomen.northam.menopausesoc.,montreal。pye,j.k.,r.e.mansel和l.e.hughes(1985)。“clinicalexperienceofdrugtreatmentsformastalgia.”lancet2:373-377。rasmussen,k.r.,m.j.arrowood和m.c.healey(1992)。“effectivenessofdehydroepiandrosteroneinreductionofcryptosporidialactivityinimmunosuppressedrats.”antimicrob.agentschemother.36:220-222。riis,b.,c.christiansen,j.johansen和j.jacobson(1990)。“isitpossibletopreventbonelossinyoungwomentreatedwithluteinizinghormone-releasinghormoneagonists”j.clin.endocrinol.metab.70:920-924。riva,h.l.,j.h.wilson和d.m.kowasaki(1961)。“effectofnorethynodrelonendometriosis.”am.j.obstet.gynecol.82:109-118。rivier,c.,j.rivier和w.vale(1979)。“chroniceffectsof[d-trp6,pro9-net]luteinizinghormone-releasingfactoronreproductiveprocessesinthemalerat.”endocrinology105:1191-1201。rivier,j.,c.rivier,m.perrin,j.porter和w.vale(1984)。lhrhanalogsasantiovulatoryagents.lhrhanditsanalogs.b.h.vickery,j.j.nestorjr.和e.s.e.hafez.lancaster,mtppress:11-22。rock,j.a.,j.a.truglia,r.j.caplan和z.e.s.group(1993)。“zoladex(goserelinacetateimplant)inthetreatmentofendometriosis:arandomizedcomparisonwithdanazol.”obstet.gynecol.82:198-205。ruttimann,j.(2008)。“themenopausebraineffect:canhormonetherapyhelp”endocrinenews.:15-16。sampson,j.a.(1921)。“perforatinghemorrhagic(chocolate)cystsoftheovary.”archivesofsurgery3:245-250。schriock,e.d.,c.k.buffington,g.d.hubert,b.r.kurtz,a.e.kitabchi,j.e.buster和j.r.givens(1988)。“divergentcorrelationsofcirculatingdehydroepiandrosteronesulfateandtestosteronewithinsulinlevelsandinsulinreceptorbinding.”j.clin.endocrinol.metab.66:1329-1331。schwartz,a.g.,l.pashko和j.m.whitcomb(1986)。“inhibitionoftumordevelopmentbydehydroepiandrosteroneandrelatedsteroids.”toxicol.pathol.14:357-362。sherwin,b.b.和m.m.gelfand(1984)。“effectsofparenteraladministrationofestrogenandandrogenonplasmahormonelevelsandhotflushesinthesurgicalmenopause.”am.j.obstet.gynecol.148:552-557。sherwin,b.b.和m.m.gelfand(1985)。“differentialsymptomresponsetoparenteralestrogenand/orandrogenadministrationinthesurgicalmenopause.”am.j.obstet.gynecol.151:153-160。sherwin,b.b.和m.m.gelfand(1987)。“theroleofandrogeninthemaintenanceofsexualfunctioninginoophorectomizedwomen.”psychosommed.49:397-409。sherwin,b.b.(1988)。“affectivechangeswithestrogenandandrogenreplacementtherapyinsurgicallymenopausalwomen.”j.affect.disord.14:177-187。simard,j.,c.labrie,a.bélanger,s.gauthier,s.m.singh,y.mérand和f.labrie(1997)。“characterizationoftheeffectsofthenovelnon-steroidalantiestrogenem-800onbasalandestrogen-inducedproliferationoft-47d,zr-75-1andmcf-7humanbreastcancercellsinvitro.”int.j.cancer73:104-112。simard,j.,r.sanchez,d.poirier,s.gauthier,s.m.singh,y.mérand,a.bélanger,c.labrie和f.labrie(1997)。“blockadeofthestimulatoryeffectofestrogens,oh-tamoxifen,oh-toremifene,droloxifeneandraloxifeneonalkalinephosphataseactivitybytheantiestrogenem-800inhumanendometrialadenocarcinomaishikawacells.”cancerres.57:3494-3497。simon,j.a.(2009)。“vulvovaginalatrophy:newandupcomingapproaches.”menopause16(1):5-7。sourla,a.,s.luo,c.labrie,a.bélanger和f.labrie(1997)。“morphologicalchangesinducedbysix-monthtreatmentofintactandovariectomizedmicewithtamoxifenandthepureantiestrogenem-800.”endocrinology138:5605-5617。studd,j.w.w.,w.p.collins,s.chakravarti,j.r.newton,d.oram和a.parsons(1977)。“oestradiolandtestosteroneimplantsinthetreatmentofpsychosexualproblemsinthepost-menopausalwomen.”britishjournalofobstetricsandgynaecology.84:314-315。sun,h.s.,k.y.hsiao,c.c.hsu,m.h.wu和s.j.tsai(2003)。“transactivationofsteroidogenicacuteregulatoryproteininhumanendometrioticstromalcellsismediatedbytheprostaglandinep2receptor.”endocrinology144(9):3934-42。surrey,e.和h.judd(1992)。“reductionofvasomotorsymptomsandbonemineraldensitylosswithcombinednorethindroneandlong-actinggonadotropin-releasinghormoneagonisttherapyofsymptomaticendometriosis:aprospectiverandomizedtrial.”j.clin.endocrinol.metab.75:558-563。surrey,e.(1995)。“steroidalandnonsteroidal"add-back"therapy:extendingsafetyandefficacyofgonadotropin-releasinghormoneagonistsinthegynecologypatients.”fertil.steril.64:673-685。suzuki,t.,n.suzuki,r.a.daynes和e.g.engleman(1991)。“dehydroepiandrosteroneenhancesil2productionandcytotoxiceffectorfunctionofhumantcells.”clin.immunol.immunopathol.61:202-211。takayama,k.,k.zeitoun,r.t.gunby,h.sasano,b.r.carr和s.e.bulun(1998)。“treatmentofseverepostmenopausalendometriosiswithanaromataseinhibitor.”fertilsteril69(4):709-13。tchernof,a.,j.p.després,a.bélanger,a.dupont,d.prud'homme,s.moorjani,p.j.lupien和f.labrie(1995)。“reducedtestosteroneandadrenalc19steroidlevelsinobesemen.”metabolism44:513-519。tremblay,a.,g.b.tremblay,c.labrie,f.labrie和v.giguère(1998a)。“em-800,anovelantiestrogen,actsasapureantagonistofthetranscriptionalfunctionsofestrogenreceptorsaandb.”endocrinology139:111-118。tremblay,g.b.,a.tremblay,n.g.copeland,d.j.gilbert,n.a.jenkins,f.labrie和v.giguere(1997)。“cloning,chromosomallocalizationandfunctionalanalysisofthemurineestrogenreceptorb.”mol.endocrinol.11:353-365。tremblay,g.b.,a.tremblay,f.labrie和v.giguere(1998b)。“ligand-independentactivationoftheestrogenreceptoraandbbymutationsofaconservedtyrosinecanbeabolishedbyantiestrogens.”cancerres.58:877-881。tremblay,g.b.,a.tremblay,f.labrie和v.giguère(1999)。“dominantactivityofactivationfunction-1(af-1)anddifferentialstoichiometricrequirementsforaf-1and-2intheestrogenreceptora-bheterodimericcomplex.”mol.cell.biol.19(3):1919-1927。tsai,s.j.,m.h.wu,c.c.lin,h.s.sun和h.m.chen(2001)。“regulationofsteroidogenicacuteregulatoryproteinexpressionandprogesteroneproductioninendometrioticstromalcells.”jclinendocrinolmetab86(12):5765-73。villareal,d.t.和j.o.holloszy(2004)。“effectofdheaonabdominalfatandinsulinactioninelderlywomenandmen:arandomizedcontrolledtrial.”jama292(18):2243-8。willson,t.m.,j.d.norris,b.l.wagner,i.asplin,p.baer,h.r.brown,s.a.jones,b.henke,h.sauls,s.wolfe,d.c.morris和d.p.mcdonnell(1997)。"dissectionofthemolecularmechanismofactionofgw5638,anovelestrogenreceptorligand,providesinsightsintotheroleofestrogenreceptorinbone."endocrinology138(9):3901-3911。women'shealthinitiative(2002)。“risksandbenefitsofestrogenplusprogestininhealthypostmenopausalwomen.”jama288:321-333。yang,s.,z.fang,t.suzuki,h.sasano,j.zhou,b.gurates,m.tamura,k.ferrer和s.bulun(2002)。“regulationofaromatasep450expressioninendometrioticandendometrialstromalcellsbyccaat/enhancerbindingproteins(c/ebps):decreasedc/ebpbetainendometriosisisassociatedwithoverexpressionofaromatase.”jclinendocrinolmetab87(5):2336-45。zeitoun,k.,k.takayama,m.d.michael和s.e.bulun(1999)。“stimulationofaromatasep450promoter(ii)activityinendometriosisanditsinhibitioninendometriumareregulatedbycompetitivebindingofsteroidogenicfactor-1andchickenovalbuminupstreampromotertranscriptionfactortothesamecis-actingelement.”molendocrinol13(2):239-53。zumoff,b.,j.levin,r.s.rosenfeld,m.markham,g.w.strain和d.k.fukushima(1981)。“abnormal24-hrmeanplasmaconcentrationsofdehydroepiandrosteroneanddehydroisoandrosteronesulfateinwomenwithprimaryoperablebreastcancer.”cancerres.41:3360-3363。综述尽管在正常子宫内膜中没有芳香酶允许将dhea转化为雌二醇,芳香酶在子宫内膜异位症中被表达。见kitawaki,j.,t.noguchi等人,(1997)“expressionofaromatasecytochromep450proteinandmessengerribonucleicacidinhumanendometrioticandadenomyotictissuesbutnotinnormalendometrium”biol.reprod.57(3):514-19.;balun,s.e,s.yang等人,(2001)roleofaromataseinendometrialdisease”j.steroidbiochem.mol.biol.79(1-5):19-25;fang,z.,s.yang等人,(2002)“geneticorenzymaticdisruptionofaromataseinhibitsgrowthofectopicuterinetissue,”j.clin.endocrin.metab.87(7):3460-6。在这里申请人提出了以与抑制卵巢激素分泌联合治疗的方式提供选择性雌激素受体调节剂(serm),以阻断在子宫内膜异位组织中局部产生的雌激素的作用。在一些实施方案中,serm的存在允许进一步给予外源性的性甾体前体物例如:获得下文记录的这种前体物的益处的dhea。serms的有益效果阿考比芬,即本发明的serm,是最初开发为用于治疗乳腺癌的纯抗雌激素药的苯并吡喃衍生物。(gauthier,caron等人,1997;luo,martel等人,1997a;luo,martel等人,1997b;luo,sourla等人,1997;simard,labrie等人,1997;simard,sanchez等人,1997;tremblay,tremblay等人,1997;couillard,gutman等人,1998;couillard,labrie等人,1998;luo,labrie等人,1998;luo,stojanovic等人,1998;tremblay,tremblay等人,1998a;tremblay,tremblay等人,1998b;tremblay,tremblay等人,1999)。em-800是可在完整细胞以及体内定量转变为活性化合物em-652的非活性前体物。阿考比芬(em-1538)是em-652的盐酸盐。这些口服活性抗雌激素药在大鼠、猴子和小鼠中对乳腺和子宫内膜上皮组织(luo,martel等人,1997b;sourla,luo等人,1997)、以及在人乳房和子宫内膜体外人乳腺癌癌瘤细胞中(gauthier,caron等人,1997;simard,labrie等人,1997)和在裸鼠体内异种移植物中(couillard,gutman等人,1998)显示了纯的抗雌激素活性。em-652,即em-800和阿考比芬的活性代谢物,是对人雌激素受体具有最高的已知亲合性的化合物。由此,em-652比替换来源于人乳腺癌和正常子宫组织雌激素受体的[3h]雌二醇的17β-雌二醇和己烯雌酚更有效1.5至3.0倍。在结合试验中,em-652比它莫西芬的活性代谢物羟基它莫西芬更有效5倍,比它莫西芬本身更有效200倍。根据结合试验的条件,em-652在对人乳腺癌雌激素受体竞争中比甾体抗雌激素药ici182780更有效10至140倍,比ici164384更有效20至85倍,比枸橼酸托瑞米芬(toremifene)更有效100至1500倍。em-652对于人雌激素受体的亲合性ki值为非常低的值,0.05nm,由此显示出迄今为止已知的化合物对于雌激素受体的最高亲合性。如上所述,em-800(em-652)在人子宫内膜石川(ishikawa)癌细胞中显示纯的抗雌激素活性(simard,sanchez等人,1997)。应当提及的是雷诺昔酚,阿佐昔芬,屈洛昔芬,艾多昔芬,枸橼酸托瑞米芬,奥培米芬和它莫西芬在人子宫内膜石川癌细胞中以各种程度刺激了雌激素-敏感的参数碱性磷酸酶(gauthier,caron等人,1997;simard,sanchez等人,1997;labrie,martel等人,2010)。雷诺昔酚,来源于苯并噻吩系列抗雌激素药的化合物(black,sato等人,1994),据报道可对骨损失产生保护作用和对血清脂质具有有益作用(black,sato等人,1994;draper,flowers等人,1996)。em-652在serms中是独特的,在人乳房和子宫细胞中都具有单纯的抗雌激素活性(gauthier,caron等人,1997;simard,labrie等人,1997;simard,sanchez等人,1997;couillard,gutman等人,1998)。如上所述,em-652在大鼠中似乎是防止骨矿物密度损失最有效的serm和血清胆固醇抑制剂。尽管如上面所综述,em-652是在乳腺和子宫内膜中具有单纯的抗雌激素活性的唯一化合物,但由于其对骨和血脂的有益作用,可以将其归类为选择性雌激素受体调节剂(serm),如最初对于雷诺昔酚的提议(draper,flowers等人,1995;kauffman和bryant1995)。事实上,已经显示em-800在大鼠中在卵巢切除术之后可抑制骨吸收,与0.1mg/kg的雷诺昔酚相比,用0.01mg/kg即可实现最大效果(martel,sourla等人,1998)。已经在大鼠中发现了em-800(em-652)对血清胆固醇类似的高效能(martel,sourla等人,1998)。此外,在绝经后女性中以每日口服5,10,20和40mg的剂量口服em-800(em-652)2周,导致全部血清胆固醇10%的减少,而在1周时已经观察到血清甘油三酯水平15%的减少(labrie等人,未发表的资料)。性甾体前体物的有益效果人以及其它灵长类在动物种类之中是独特的,其具有分泌大量非活性前体物甾体dhea特别是dhea-s的肾上腺,其可以在周围组织中转变为有效的雄激素和/或雌激素。血浆dhea-s水平比成年男子的睾酮水平高200-1000倍,比在成年女性中的雌二醇水平高5000至25000倍,由此提供了形成雄激素和/或雌激素的大批底物。如上所述,在周围靶组织中性甾体的局部合成和作用被称为内分泌学;所选择的例子包括dhea和雄烯二酮(labrie,bélanger等人,1988;labrie1991)。血清dhea随年龄改变和高度可变化性从30岁开始,dhea分泌显著地降低,并已经观察到在绝经时有60%的平均损失(labrie,bélanger等人,2006;labrie,luu-the等人,2005;labrie,luu-the等人,2003;labrie,belanger等人,1997b)。随衰老而由肾上腺导致的dhea分泌的显著减少可导致在周围靶组织中雄激素和雌激素形成的类似的降低(labrie,belanger等人,1997b),这种情况被认为是与一系列下列医学问题有关:绝经(胰岛素抗性(coleman,leiter等人,1982),脂肪累积(tchernof,després等人,1995),骨损失,肌肉损失,ii型糖尿病,阴道萎缩和皮肤萎缩(labrie,luu-the等人,2005;simon2009;diamond,cusan等人,1996;labrie,diamond等人,1997;labrie2007),记忆和认知能力丧失(ruttimann2008)等等。绝经后被充分认识的这样的一些问题也可能在绝经前变得明显,例如骨损失、阴道干燥和热潮红。图1显示了dhea水平随年龄变化的减少。除dhea的血清水平随年龄显著地降低之外,dhea的血清水平变化是剧烈的,一些女性具有低的dhea水平,甚至在生殖期。参见图⒉。人卵巢分泌大量的dhea当前很重要的观察结果是卵巢分泌大量的dhea。相应地,作为本发明的一部分,通过利用lhrh激动剂或拮抗剂实现的停止卵巢雌激素分泌的治疗应该也能降低在体循环中卵巢的dhea分泌,由此在年龄大于30岁的女性中添加了性甾体活性的缺乏(图1)。此外,我们当前的数据表明女性的全部雄激素来源于dhea,而且人卵巢不会直接分泌雄激素,其对于女性的正常内分泌生理学是很重要的(labrie,martel等人,2011)。事实上,女性的雄激素为男子的大约40%(labrie2007)。因为当甾体血清浓度低时,没有增加dhea分泌的反馈机制,因此在没有用外源性dhea替代治疗的情况下,具有低dhea分泌率的女性在他们余生中一直缺乏性甾体。从442个年龄在46至74岁的绝经后完好的女性(平均值:59.9岁;中间值:60.5岁),71个年龄在42至72岁先前做过双向卵巢切除术的绝经后女性(平均值:60.6岁;中间值:62.0岁)和47个年龄在30至39岁绝经前正常循环女性(平均值和中间值:33岁)中获得数据。在血液样品中测定甾体水平,该样品在筛选时收集或在女性参与各种临床试验的第1天时收集。所有样品是在给予任何调查研究的药物之前收集。在irb批准并且给与了书面的通知许可之后使女性参与临床试验。将血样加工成血清制剂,并在-20℃或以下温度冷藏直到测定甾体为止。通过质谱测定dhea,dhea-s,雄甾-5-烯-3β,17β-二醇(5-二醇),雄烯二酮(4-二酮),睾酮(testo),二氢睾丸酮(dht),雌二醇(e2),雌酮(e1),雌酮硫酸盐(e1-s),雄酮葡糖苷酸(adt-g),雄甾烷-3α,17β-二醇-3-葡糖苷酸(3α-二醇-3g)和3α-二醇-17g的血清甾体水平,如先前所描述(labrie,bélanger等人,2006;labrie,belanger等人,2007;labrie,cusan等人,2009)。试验的操作,以及精确性和敏感度的详情可以在(labrie,bélanger等人,2006)和(labrie,cusan等人,2008)中得到。表1给出的描述统计学使用sas软件进行。使用t检验、用sas软件测定基于完好的和切除卵巢的绝经后女性的平均甾体水平比较值的统计显著性(表1)。如表1所示,所给出的数据表明在该年龄组绝经后卵巢分泌大约全部dhea的18%。与在本研究中完好的绝经后女性中所观察到的dhea相比,在卵巢切除的(ovx)绝经后女性中的dhea血清水平较低,这最好地被解释为卵巢分泌相应数量的dhea到循环中。然后卵巢起源的dhea与肾上腺起源的dhea具有相同的胞分泌机理。因为当血清dhea低时没有增加dhea分泌的调节机制,修正这种缺陷的唯一方法似乎是提供外源性的dhea,以补偿不存在dhea分泌反馈控制的缺陷。在卵巢切除的(ovx)女性中发现18%的、但相似较低的dhea水平和其全部代谢物的血清水平(图3),包括e2和睾酮,说明在42-至74-岁年龄组中绝经后卵巢分泌全部dhea的18%,没有直接被卵巢分泌的大量e2或睾酮(图4)。没有理由相信绝经后卵巢对全部循环dhea数量(pool)的显著贡献(~18%)在绝经前会是较低的。我们认为,增加的对称为内分泌学的、雄激素和雌激素在周围靶组织中的形成和作用的了解(labrie1991;labrie,simard等人,1992a;labrie,simard等人,1992b;labrie,durocher等人,1995;luu-the,dufort等人,1995;labrie,simard等人,1996;labrie,belanger等人,1997a;labrie,belanger等人,1997b;labrie,diamond等人,1997;labrie,luu-the等人,1997),以及我们当前的观察结果表明雄激素对在大鼠中卵巢切除术之后骨损失的预防相对于雌激素占主要作用(martel,sourla等人,1998),并在绝经后女性中观察到相似情况(labrie,diamond等人,1997),这为在性甾体代替治疗领域及时和潜在的高度显著的进程铺平了道路。这种可能性很好地受到了我们的观察结果和在绝经后女性中观察到的dhea的一系列其它有益效果的支持(morales,nolan等人,1994;diamond,cusan等人,1996;labrie,diamond等人,1997;labrie2007,2010;labrie,archer等人,2009a,2009b,2009c),该状况类似于用lhrh激动剂或拮抗剂治疗子宫内膜异位症所引起的医学阉割。近来在安慰剂对照、随机化的关键的iii期临床试验中获得了dhea的效果和安全性的引人注目的证明,其中患有阴道萎缩的绝经后女性每日阴道内接受dhea或安慰剂3个月。观察到阴道萎缩的所有病况和征侯有快速和非常明显的改进,而循环雌二醇或睾酮则没有变化。发现了用雌激素没有看出的另外的益处,即发现性功能障碍在所有领域即欲望、觉醒、高潮和快乐都有显著的改善(labrie,archer等人,2009a,2009b)。在子宫内膜异位症中雌激素的形成尽管在正常人子宫内膜中不存在雌激素形成所需要的酶特别是芳香酶(bulun,lin等人,2005;baxendale,reed等人,1981),但芳香酶是高度表达的,且局部雌激素的生产存在于子宫内膜异位的组织中((kitawaki,noguchi等人,1997;zeitoun,takayama等人,1999;bulun,yang等人,2001;fang,yang等人,2002;gurates,sebastian等人,2002;yang,fang等人,2002)。随后在子宫内膜异位症治疗中引入芳香酶抑制剂成功地强调了在子宫内膜异位的组织中存在芳香酶(takayama,zeitoun等人,1998;ailawadi,jobanputra等人,2004)。在子宫内膜异位症中,原型异常是在子宫内膜异位症的基质细胞组分中存在显著性水平的star和芳香酶活性以及蛋白和mrna的表达,而在健康女性的正位的子宫内膜中则不存在star或芳香酶表达,或有几乎不可检测的star或芳香酶表达(noble,simpson等人,1996;noble,takayama等人,1997;tsai,wu等人,2001;gurates,sebastian等人,2002;sun,hsiao等人,2003)。患有子宫内膜异位症的女性的正位子宫内膜含有低的但显著性水平的芳香酶mrna和酶活性,代表了该疾病的中间状态。似乎当经血倒流(retrogrademenstruation)和在骨盆腹膜表面上植入该固有的异常组织时,芳香酶表达和酶活性增强至多400倍(noble,simpson等人,1996;noble,takayama等人,1997)。然而,体内缺乏star和芳香酶是区分正常子宫内膜和子宫内膜异位症的因素。在正常子宫内膜组织或pge2-刺激的子宫内膜基质细胞中没有检测到生理学显著性水平的这些基因产物(bulun,lin等人,2005)。芳香酶活性或mrna不能由健康女性的基质细胞中的pge2或camp类似物诱导(noble,takayama等人,1997)。serm+性甾体前体物的组合的有益效果已知dhea在大鼠中预防二甲基苯并(a)蒽-诱导的乳腺肿瘤形成和抑制其生长(labrie,luu-the等人,2003)。此外dhea可在裸鼠中抑制人乳腺癌异种移植物的生长(labrie,luu-the等人,2003)。由此,与产生刺激效应的雌激素和孕酮不同,可预期dhea能够抑制女性乳腺癌的形成和生长两者,如在大部分人乳腺癌细胞系中所证明(labrie,luu-the等人,2003;labrie2010,2006;labrie,bélanger等人,2006)。为了避免使用常规hrt而由whi研究所说明的问题(women'shealthinitiative,jama288:321-333,2002),在更年期和绝经后使用组织特异的抗雌激素/雌激素(取决于组织)化合物(serm)与组织靶向的雄激素性和/或雌激素代替治疗相结合似乎是合理的。这一策略可能是维持每个组织的每个细胞中雄激素和雌激素之间的生理平衡、同时预防乳房和子宫癌的最好的或可能是唯一的途径。这样的目标可潜在地通过serm与dhea联用得到(labrie,luu-the等人,2005;labrie,luu-the等人,2003;labrie2007)。尽管serms在骨中具有局限于抑制骨吸收的效果,但dhea通过其雄激素性或合成代谢组分刺激骨形成(michalska,stepan等人,2006;martel,sourla等人,1998)。这样的合成代谢或造骨效果不能用serms、双磷酸盐类、雌激素或降钙素实现,其只会降低骨吸收的速率。事实上,这些抗吸收疗法不会改善正常骨损失的全部特性,特别是微体系结构。尽管在临床前水平已经证明了阿考比芬在骨上的高效能(比雷诺昔酚高10倍)(labrie,labrie等人,2001),已经观察到用dhea治疗通过合成代谢作用可增加绝经后女性中骨形成(labrie,diamond等人,1997)。除了增加骨形成之外,dhea还在绝经后女性中显示了刺激阴道成熟、降低肥胖倾向以及血清葡萄糖和胰岛素水平。描述在某些研究中的dhea在脂肪和葡萄糖代谢上的效果(diamond,cusan等人,1996;villareal和holloszy2004;morales,haubrich等人,1998)还没有在其它研究中被发现(jankowski,gozansky等人,2006;nair,rizza等人,2006)。还有可能的是serms可能在绝经后女性中产生另外的有益效果。事实上用阿考比芬获得的临床前数据包括下列有益效果:降低了胆固醇和甘油三酯水平,减少了脂肪累积和改善了胰岛素敏感度(labrie,labrie等人,2001;labrie2007)。serm加dhea的联用(图7)还可以通过dhea的雄激素性效果帮助控制热潮红,同时预防乳腺癌、子宫癌、卵巢癌、骨和肌肉损失以及降低脂肪累积、ii型糖尿病和血清胆固醇(表2)。在这方面,重要的是表明在正常人子宫内膜上不存在dhea的刺激效应(labrie,diamond等人,1997),其排除了对给予孕酮以中和雌激素对子宫内膜的潜在效果的需要。述及乳房,已知dhea可预防大鼠中二甲基苯并(a)蒽乳腺肿瘤的形成(luo,sourla等人,1997)和抑制其生长(li,yan等人,1993)。此外,dhea可在裸鼠中抑制人乳腺癌异种移植物的生长(couillard,labrie等人,1998)。由此,与产生刺激效应的雌激素和孕酮相反,可预料到dhea可抑制女性乳腺癌的形成和生长两者。雄激素在骨生理学中的作用在骨质疏松症形成中,据报道蛋白同化甾类(anabolicsteroids)可帮助预防骨损失(hennernan和wallach1957)。如用癸酸诺龙治疗所观察到的,已经发现雄激素疗法可以在绝经后女性中提高脊椎骨矿物密度(need,horowitz等人,1989)。尽管由于雄激素在绝经后女性中独特的作用,使雄激素赢得越来越多的支持,但借助于睾酮观察到了雄性化的效果(burger,hailes等人,1984;studd,collins等人,1977)。雄激素在女性中的其它作用越来越多地认识到由dhea产生的雄激素在绝经后女性中具有许多有益效果。已经描述了向ert或hrt加入雄激素在综合健康、活力、情绪、和综合的生活质量方面的详细益处(sherwin和gelfand1985;sherwin1988)。已经观察到在向雌激素替代治疗(ert)中加入雄激素之后可改善主要的心理学和心理数学症状,即烦躁、紧张、记忆和失眠(notelovitz,watts等人,1991)。性欲和/或性满足的丧失是绝经后初期常见的。已知向激素代替治疗(hrt)中加入雄激素对这些难题具有有益的作用(leiblum,bachmann等人,1983;sherwin和gelfand1987;sherwin1988)。此外,发现在绝经后女性中性行为和雄激素的循环水平之间存在正相关性。此外,已经发现雄激素性化合物对治疗通常由hrt引起的乳腺痛有好处(pye,mansel等人,1985)。事实上,雌激素替代治疗可以导致严重的乳房疼痛,其导致治疗的中止。已经发现加入雄激素在用雌激素单独治疗的结果不能令人满意的女性中可有效减轻热潮红(sherwin和gelfand1984)。dhea的其它益处在衰老期间由肾上腺导致的dhea和dhea-s形成减少70至95%,可在周围靶组织中导致雄激素和雌激素形成的显著减少,其很可能与年龄相关疾病例如胰岛素抗性(coleman,leiter等人,1982;schriock,buffington等人,1988)和肥胖症(nestler,barlascini等人,1988;macewen和kurzman1991;tchernof,després等人,1995)的发病原理有关。事实上,在患有乳腺癌的患者中发现了dhea-s和dhea的低循环水平(zumoff,levin等人,1981),且在一系列动物模型中发现dhea可产生抗致癌的活性(schwartz,pashko等人,1986;gordon,shantz等人,1987;li,yan等人,1993)。还显示dhea具有体外免疫调节效果(suzuki,suzuki等人,1991)和体内对真菌和病毒疾病(rasmussen,arrowood等人,1992)的免疫调节效果,包括hiv(henderson,yang等人,1992)。另一方面,已经描述了dhea在绝经后女性中对免疫系统的刺激效应(casson,andersen等人,1993)。用dhea在女性中获得的初步数据如上所述,骨质疏松症是老年女性的主要难题,其主要通过增加骨折比例导致发病和致死(johnstonjr和epstein1981)。使用雌激素替代治疗需要加入孕酮以抑制由雌激素诱导的子宫内膜增殖,然而雌激素和孕酮两者都可能增加乳腺癌的危险(bardon,vignon等人,1985;colditz,hankinson等人,1995)。为了避免标准雌激素(ert)或激素代替治疗(hrt)的限制,我们研究了对60-至70-岁女性给予dhea12个月对骨矿物密度,骨形成和循环参数,血清脂质,葡萄糖和胰岛素,脂肪组织质量,肌肉质量,活力,健康以及阴道和子宫内膜组织学的效果(diamond,cusan等人,1996;labrie,diamond等人,1997)。经皮给予dhea,以避免甾体前体物通过肝的首过(firstpassage)效应。我们由此评价了用10%dhea乳膏剂慢性代替治疗的效果,在60-至70-岁女性(n=15)中每日应用一次,应用12个月。人体测量显示在12个月时体重无变化,但皮下皮肤褶襞厚度减少9.8%(p<0.05)(diamond,cusan等人,1996)。骨质量密度在臀部增加2.3%,在臀部ward's三角增加3.75%,和在腰脊椎增加2.2%(全部p<0.05)(labrie,diamond等人,1997)。这些骨矿物密度的变化在12个月时伴随有尿羟脯氨酸和血浆骨碱性磷酸酶分别明显降低38%和22%(全部p<0.05)。伴随观察到血浆骨钙蛋白与对照物相比增加135%(p<0.05)。通过计算断层分析,在腿中部(midthigh)脂肪和肌肉部位的测量显示在12个月时股骨脂肪(femoralfat)降低3.8%(p<0.05),股骨肌肉面积(femoralmusculararea)增加3.5%(p<0.05)(diamond,cusan等人,1996)。在腹部脂肪测量中没有显著的变化。这些在体脂肪和肌肉表面积方面的变化与空腹血糖降低12%(p<0.05)和空腹血浆胰岛素水平降低17%(p<0.05)有关。用dhea治疗对脂质或脂蛋白特性没有不希望的作用。事实上,存在总胆固醇和其脂蛋白分数3%至10%减少的总体趋势。血浆甘油三酯不受影响。用dhea治疗12个月后皮脂分泌指数增加79%,治疗停止后3个月时恢复到预处理时的值。给予dhea在10个女性中有8个可刺激阴道上皮组织成熟,这些女性在开始治疗时成熟值为零,而还可以在治疗前具有中等阴道成熟的三个女性中观察到刺激作用。最重要的是,在子宫内膜中没有发现在阴道中观察到的雌激素的刺激效应,子宫内膜在用dhea治疗12个月后的所有女性中均保持完全的萎缩(labrie,diamond等人,1997)。通过其在特定的胞分泌靶组织中转化为雄激素和/或雌激素,给出的数据清楚地表明dhea治疗在绝经后女性中的有益效果,而没有显著的副作用。dhea没有对子宫内膜的刺激作用,消除了对孕酮代替治疗的需求,由此避免对于孕酮诱导的乳腺癌的担忧。观察到的dhea对骨矿物密度的刺激效应和血清骨钙蛋白的增加(其是骨形成的标志),对于预防和治疗骨质疏松症是特别有益的,表明了dhea在骨生理学即骨形成上的独特活性,而ert和hrt只能减少骨损失的速率。serm和dhea联用在用lhrh激动剂或拮抗剂治疗的女性中的益处我们已经证明,dhea对雌性大鼠(luo等人,endocrinology138:4435-4444,1997)和绝经后女性(labrie等人,j.clin.endocrinol.metab.82:3498-3505,1997)两者的骨骼具有有益作用。由此,在完好的雌性大鼠中用dhea治疗提高了全部骨骼、腰脊椎和股骨的骨矿物密度(bmd)(luo等人,endocrinology138:4435-4444,1997)。此外,如图5至图8所举例说明,我们发现性甾体前体物(dhea)和serm(em-800)的联用不但可维持dhea对骨形成的刺激效应,而且当两种化合物联合使用时,可增强单独用serm(em-800)对骨循环和再吸收的抑制效果,其由尿羟脯氨酸和钙排出的进一步降低所证明(luo,sourla等人,1997)。简言之,上述数据清楚地证明serm(em-800)和性甾体前体物(dhea)联用对由dmba诱导的乳腺癌发展的有益效果,以及这种联用对骨质量和血清脂质的保护作用。这种数据清楚地表明这种联用对于治疗和预防骨质疏松症,同时改善脂质特性和预防乳腺癌和子宫内膜癌症的另外的有益效果。特别重要的是表明了dhea和em-800联用对骨代谢的重要生化参数产生了意想不到的有益效果。事实上,单独的dhea不会影响尿羟脯氨酸/肌酐比例,其是骨吸收标志。此外,单独的dhea在每日尿钙或磷排出上可能检测不到效果(luo,sourla等人,1997)。另一方面,em-800可使尿羟脯氨酸/肌酐比例降低48%,同时,类似地对于dhea,em-800在尿钙或磷排出上没有看出效果。此外,em-800对血清碱性磷酸酶活性没有影响,其是骨形成的标志,而dhea使该参数值增加大约75%(luo,sourla等人,1997)(表2)。因此,dhea和em-800联用的意想不到的效果之一涉及尿羟脯氨酸/肌酐比例,其是骨再吸收的标志,当dhea和em-800两者组合时其减少了69%,该值是统计上不同于(p<0.01)由单独的em-800实现的48%抑制,而单独的dhea不显示任何效果。因此,向em-800中加入dhea使em-800对骨再吸收的抑制效果提高了50%。最重要地,向em-800中加入dhea的另一个意想不到的效果是分别大约减少84%的尿钙(从23.17±1.55至3.71±0.75μmol/24h/100g(p<0.01)和减少尿磷55%(从132.72±6.08至59.06±4.76μmol/24h/100g(p<0.01))(luo,sourla等人,1997)(表2)。在大鼠中获得的本结果清楚地证明了dhea可提供单独使用选择性雌激素受体调节剂(serm)例如em-800,raloxifene等等所缺乏的有益效果。尽管serm具有局限于骨再吸收抑制的效果,据信加入dhea可刺激骨形成(是用serm、雌激素、双磷酸盐类或降钙素无法实现的效果)和进一步减少骨再吸收,超过单独用em-652实现的效果。重要地,在切除卵巢的大鼠中用em-800和dhea联用治疗12个月对骨形态计量学具有有益的效果。小梁骨的体积对于骨强度和预防骨折是特别重要的。因此,在上面提及的研究中,单独用dhea时在切除卵巢的大鼠中胫骨的小梁骨的体积从4.1±0.7%增加至11.9±0.6%(p<0.01),而向dhea中加入em-800进一步使小梁骨体积增加至14.7±1.4%,该值类似于在完好的对照组中发现的值(图6)。在切除卵巢的大鼠中,从0.57±0.08/(每)mm的值,用dhea治疗导致与切除卵巢的对照组相比小梁骨数目增加137%。由此,dhea的刺激效应达到1.27±0.1/(每)mm,而同时用em-800和dhea治疗导致小梁骨数目与单独用dhea相比增加另外的28%(p<0.01)(图7)。类似地,向dhea的治疗中加入em-800导致小梁骨分离与单独用dhea治疗相比,另外减少15%(p<0.05),由此导致该值与完好对照组没有区别。作为对图6和图7中给出的数据的补充,图8举例说明了在切除卵巢的治疗动物(c)中由dhea诱导的在近端胫骨干骺端小梁骨体积与切除卵巢的对照组(b)相比时的增加,以及在向dhea治疗(d)中加入氟他米特(flu)之后dhea的刺激效应的部分抑制。另一方面,组合给予dhea和em-800导致对卵巢切除术-诱导的骨质减少的彻底预防(e),小梁骨体积可与完好的对照组(a)的相比较。在提及的研究(图5-8)中,在几乎所有的所研究的骨组织形态测定术参数上都观察到dhea的雄激素性刺激效应。由此dhea导致小梁骨体积的显著增加,以及小梁数目的增加,然而它降低了小梁间隙面积。为了实现更彻底的雌激素丧失,为了中和在用lhrh激动剂治疗的头2周期间卵巢雌激素分泌的“潮红”,以及中和来源于卵巢雄烯二酮以及肾上腺和卵巢dhea的雌激素的作用,在目前研究向lhrh激动剂或拮抗剂加入了单纯的抗雌激素药(labrie,martel等人,2011)。事实上,肾上腺和卵巢dhea转变为子宫内膜异位,而不是正常子宫内膜组织,中的雌激素。相应地,由肾上腺和dhea产生的卵巢起源的雌激素可在由lhrh激动剂或拮抗剂引起的卵巢雌激素分泌停止之后继续刺激子宫内膜异位细胞。重要的是提到出虽然正常子宫内膜由于没有芳香酶不能由dhea合成雌激素,而子宫内膜异位的组织具有能够将dhea转变为雌激素的酶。(bulun,lin等人,2005)。这种数据表明,由lhrh激动剂或拮抗剂阻断卵巢雌激素分泌仅仅是对子宫内膜异位症的部分治疗,因为雌激素是由dhea在子宫内膜异位组织中局部产生的,并由此刺激子宫内膜异位的细胞的增殖,而正常子宫内膜由于没有雌激素的局部形成而不被dhea刺激。具有单纯的和有效的雌激素拮抗活性的serm在子宫内膜组织中特别高的效能,将会阻断任何由dhea在子宫内膜异位的组织产生的任何雌激素活性。这种彻底的雌激素阻断将导致更彻底和更快速的细胞程序死亡,并由此降低停止治疗后子宫内膜异位症复发的发病率。另一方面,尽管em-652·hcl减少骨损失和dhea刺激骨形成,由此更有效地保护骨功能(bonefunction),但加入dhea将预防热潮红,这是单独用lhrh激动剂治疗的主要限制。在图9,10和11中举例说明了推荐的组合对hrt中反向加入治疗的预期效果的比较。为了有利于本发明的联合治疗方面,对于本文所讨论的任何适应症,本发明提供了试剂盒,其包括分离的或在一个容器中的一或多种serm和性甾体前体物,和在另一个容器包括卵巢激素分泌的抑制剂。该试剂盒可以包括用于口服,例如片剂,胶囊剂,糖浆剂等等,和用于透皮给予,例如,软膏剂,洗剂,凝胶剂,乳膏剂,持续释放贴片等等,用于阴道内给予,例如栓剂,乳膏剂,软膏剂,片剂,凝胶剂等等,和用于皮下注射和肌内注射的合适的材料。可以阴道内给予阿考比芬和dhea。申请人相信在给予或不给予性甾体前体物的情况下给予serm和卵巢激素分泌抑制剂,可用于治疗和/或预防子宫内膜异位症及其它雌激素-相关疾病的进展。本发明的选择性雌激素受体调节剂具备具有下列特征的分子式:a)由1至2个介于其间的碳原子间隔的两个芳香环,两者芳香环是未取代的,或被羟基或可在体内转变为羟基的基团、或卤素或c1-c6烷基或c1-c6烷基磺酰基取代的;和b)一个具有芳香环和叔胺、羧酸或醇官能团的侧链,或其盐。本发明的选择性雌激素受体调节剂的优选的侧链选自:。本发明的优选的选择性雌激素受体调节剂选自:苯并噻吩衍生物,三苯乙烯衍生物,吲哚衍生物,苯并吡喃衍生物,色满衍生物,萘衍生物,二氢萘衍生物,四氢化萘衍生物,苯并硫吡喃衍生物,二氢苯并噻喃(thiochroman)衍生物,喹啉衍生物,二氢喹啉衍生物,和四氢喹啉衍生物。本发明优选的选择性雌激素受体调节剂具备选自下列的化学式之一:其中r1和r2独立地选自:氢,羟基,可在体内转变为羟基的部分,卤素,c1-c6烷基和c1-c6烷基磺酰基;其中r3和r4独立地选自:c1-c4烷基,或与它们结合的氮原子组合形成的部分,选自吡咯烷基,2,2-二甲基吡咯烷基,2-甲基吡咯烷基,哌啶基,六亚甲基亚氨基和吗啉代基;其中a选自:-co-,-choh-,-o-,和-ch2-;其中b选自亚苯基,吡啶亚基,和-环c4h2n2-;其中d是-och2ch2n(r3)r4,-och2ch2oh,-och2ch2och2ch2oh或-ch=ch-cooh(其中r3和r4独立地选自:c1-c4烷基,或与它们结合的氮原子组合形成的部分,选自吡咯烷基,2,2-二甲基吡咯烷基,2-甲基吡咯烷基,哌啶子基,六亚甲基亚氨基和吗啉代基);其中e和k独立地是氢,羟基,可体内转变为羟基的部分或卤素;其中j是氢或卤素;其中m是氢或c1-c6烷基;其中d选自:-och2ch2n(r7)r8,-ch=ch-con(r7)r8,-cc-(ch2)n-n(r7)r8(r7和r8二者之一独立地选自:c1-c4烷基,或与它们结合的氮原子组合形成的部分,选自吡咯烷基,2,2-二甲基吡咯烷基,2-甲基吡咯烷基,哌啶子基,六亚甲基亚氨基和吗啉代基);其中x选自氢和c1-c6烷基;其中r1,r2,r3,r4,r5,和r6独立地选自氢,羟基,c1-c6烷基,卤素,和可在体内转变为羟基的部分;其中r1和r2独立地选自:氢,羟基,卤素,c1-c6烷基和c1-c6烷基磺酰基,和可在体内转变为羟基的部分;其中r5和r6独立地是氢或c1-c6烷基;其中d是-och2ch2n(r3)r4(其中r3和r4独立地选自:c1-c4烷基,或与它们结合的氮原子组合形成的部分,选自吡咯烷基,2,2-二甲基吡咯烷基,2-甲基吡咯烷基,哌啶子基,六亚甲基亚氨基和吗啉代基);其中x选自:-o-,-ch2-,-s-,-ch=,-n=,和-nr7-(r7是氢或c1-c6烷基);其中y选自:-o-和-ch2-或直接键;其中r1和r2独立地是:氢,羟基,卤素,c1-c6烷基,和可在体内转变为羟基的部分;其中z选自:-o-,-ch2-,-s-,和-nr7-(r7是氢或c1-c6烷基);其中r100是将l与b-环通过4-10个介于其间的原子隔离的二价部分;其中l是选自下列的二价或三价极性部分:-so-,-con<,-n<,和-son<;其中g1选自氢,c1至c5烃,与g2组合的二价部分,和l是5-至7-元杂环,和卤代或前述的不饱和衍生物;其中g2或者不存在,或者选自氢,c1至c5烃,与g1组合的二价部分,和l是5-至7-元杂环,和卤代或前述的不饱和衍生物;其中g3选自氢,甲基,乙基和三氟甲基;或其可药用盐;其中d是-och2ch2n(r3)r4(其中r3和r4独立地选自:c1-c4烷基,或与它们结合的氮原子组合形成的部分,选自吡咯烷基,2,2-二甲基吡咯烷基,2-甲基吡咯烷基,哌啶子基,六亚甲基亚氨基和吗啉代基);其中r1和r2独立地选自:氢,羟基,卤素,c1-c6烷基,和可在体内转变为羟基的部分;其中g3选自氢,甲基,乙基和三氟甲基;或其可药用盐;其中苯并吡喃衍生物是光学活性的,由于其立体异构体的大部分在碳2上具有s绝对构型,而基本上没有(2r)-对映体;其中r1和r2独立地选自:羟基,卤素,c1-c6烷基,和在体内可转变为羟基的部分;其中r3是选自下列的种类:饱和、不饱和或取代的吡咯烷基,饱和、不饱和或取代的哌啶子基,饱和、不饱和或取代的哌啶基,饱和、不饱和或取代的吗啉代基,含有氮的环状部分,含有氮的多环部分,和nrarb(ra和rb独立地氢,直链或支链c1-c6烷基,直链或支链c2-c6烯基,和直链或支链c2-c6炔基);其中g3选自甲基和三氟甲基;其中任选的酸的盐选自下列:乙酸,己二酸,苯磺酸,苯甲酸,樟脑磺酸,枸橼酸,富马酸,氢碘酸,氢溴酸,盐酸,双氢氯噻嗪酸(hydrochlorothiazideacid),羟基-萘酸,乳酸,马来酸,甲磺酸,甲基硫酸,1,5-萘二磺酸,硝酸,棕榈酸,特戊酸,磷酸,丙酸,琥珀酸,硫酸,酒石酸,对苯二甲酸,对甲苯磺酸,和戊酸。本发明的一个优选的serm是em-800,其在pct/ca96/00097(wo96/26201)中报道。em-800的分子结构是:本发明的另一个优选的serm是em-652·hcl,其在us专利6,710,059b1中报道:em-652·hcl(也称为em-1538或阿考比芬)是有效的抗雌激素药em-652的盐酸盐。与em-800相比,em-652·hcl是简单的和容易合成的盐。它还便于分离,纯化,结晶,并显示了良好的固态稳定性。给予em-800或者em-652·hcl,导致具有相同的体内活性d相同的活性化合物。因为两种前体物导致活性化合物em-652的类似的血液水平。另一个优选的serm是巴多昔芬(tse-424;way-tse424;way140424;1-[[4-[2-(六氢-1h-氮杂-1-基)乙氧基]苯基]甲基]-2-(4-羟基苯基)-3-甲基-1h-吲哚-5-醇,乙酸酯),其由wyethayers(usa)研制,公开在jp10036347中(americanhomeproducts公司),且在美国批准用于预防经绝期后骨质疏松,且非甾体雌激素衍生物描述在wo97/32837中。其它本发明优选的serm包括它莫西芬((z)-2-[4-(1,2-二苯基-1-丁烯基)苯氧基]-n,n-二甲基乙胺)(得自于zeneca,英国),托瑞米芬((z)-2-(4-(4-氯-1,2-二苯基-1-丁烯基)苯氧基)-n,n-二甲基乙胺),得自于orion,芬兰,商标为fareston或schering-plough),屈洛昔芬((e)-3-(1-[4-[2-(二甲基氨基)乙氧基]苯基]-2-苯基-1-丁烯基]苯酚)和,得自于elilillyandco.公司,美国:雷诺昔酚[(2-(4-羟基苯基)-6-羟基苯并[b]噻吩-3-基][4-[2-(1-哌啶基)乙氧基]苯基]-甲酮盐酸盐),ly335124,ly326315,ly335563(去甲阿佐昔芬)(6-羟基-3-[4-[2-(1-哌啶基)乙氧基]苯氧基]-2-(4-羟基苯基)苯并[b]噻吩盐酸盐)和阿佐昔芬(ly353381,6-羟基-3-[4-[2-(1-哌啶基)乙氧基]苯氧基]-2-(4-甲氧基苯基)苯并[b]噻吩盐酸盐)。其它优选的serm是拉索昔芬(cp-336,156)(顺式-1r-[4’-吡咯烷基乙氧基苯基]-2s-苯基-6-羟基-1,2,3,4-四氢化萘d-(-)-酒石酸盐)(辉瑞公司,美国,描述在us5,889,042中),艾多昔芬((e)-1-[2-[4-[1-(4-碘代苯基)-2-苯基-1-丁烯基]苯氧基]乙基]吡咯烷)(smithklinebeecham,usa),左美洛昔芬(3,4-反式-2,2-二甲基-3-苯基-4-[4-(2-(2-(吡咯烷-1-基)乙氧基)苯基]-7-甲氧基色满)(novonordisk,a/s,丹麦),其公开在shalmi等人,wo97/25034,wo97/25035,wo97/25037,wo97/25038;和korsgaard等人,wo97/25036中),gw5638(描述在willson等人,1997中)和吲哚衍生物(描述在miller等人,ep0802183a1中)。还包括iproxifen(tat59;(e)-4-[1-[4-[2-(二甲基氨基)乙氧基]苯基]-2-[4-(1-甲基乙基)苯基]-1-丁烯基]苯酚的磷酸二氢盐)得自于taiho(日本),奥培米芬(fc1271;((z)-2-[4-(4-氯-1,2-二苯基-1-丁烯基)苯氧基]乙醇),得自于orion-farmospharmaceutica,芬兰,serm3471,hmr3339和hmr3656,得自于sanofi-aventis(法国),哌喷昔芬(era-923),由wyeth-ayers研制,非甾体雌激素衍生物,描述在wo97/32837中,非培米芬,由quatrx(美国)研制,和cc8490,由celgene,美国研制。可以使用任何为效果所需要的、如厂家所推荐的serm。合适的剂量是本领域已知的。可以使用按照本发明的可商业购买的任何其它的非甾体抗雌激素药。可以使用具有类似于serm活性的任何化合物(例如:雷诺昔酚)。根据本发明给予的serms,对于平均体重的人,当口服给予时,优选是以每天0.01至10mg/kg体重的剂量范围(优选0.05至1.0mg/kg)给予的,每天60mg,特别是每天20mg,优选以一或两个同样的分开剂量给予,或当胃肠外给予(即肌内,皮下或经皮或阴道内给予)时,对于平均体重的人,以每天0.003至3.0mg/kg体重的剂量范围(优选0.015至0.3mg/kg体重),每天20mg,特别是每天10mg,优选以两个同样的分开剂量给予。优选serms是与如下所述的可药用稀释剂或载体一起给予。就本文推荐的所有剂量而论,护理临床医师将监测个体患者的响应和相应地调节剂量。优选的性甾体前体物是脱氢表雄甾酮(dhea)(得自于proquina,墨西哥),其前体药物(得自于steraloids,wilton,newhampshire,美国),5-雄烯-3β,17β-二醇和其前体药物雄甾-5-烯-3β,17β-二醇3-乙酸酯和雄甾-5-烯-3β,17β-二醇二半琥珀酸酯(得自于steraloids,wilton,newhampshire美国)。雄甾-5-烯-3β,17β-二醇3-乙酸酯雄甾-5-烯-3β,17β-二醇二半琥珀酸酯本发明的活性组分(不论是否是serm或前体物或卵巢激素分泌的抑制剂)可以以各种方式配制和给药。根据本发明给予的性甾体前体物优选以下面的剂量范围给药:(1)当阴道内给药时,每天0.5至100mg,(优选每天3至50mg);(2)当皮肤上给药时,剂量范围在每天15至200mg之间(优选每天30mg至100mg);(3)当口服给药时,剂量范围在每天10至200mg之间(优选每天25mg至100mg),例如每天75mg;或(4)当肠胃外给药时(即肌内或皮下给药),剂量范围在每天1.0至25mg之间(优选每天3.25至20mg)。用于透皮或透粘膜的活性组分优选以相对于药物组合物总重量0.5%至20%重量,更优选在0.1至10%之间存在。或者,可以把活性组分放入具有本领域已知结构的透皮贴片中,例如在e.p.专利no.0279982中列出的结构。当配制成软膏剂,洗剂,凝胶或乳膏剂等等时,可以将活性化合物与合适的载体混合,该载体与人皮肤或粘膜相容,其可提高化合物透皮渗透到皮肤或粘膜。合适的载体是本领域已知的,包括但不局限于klucelhf和glaxal碱。某些是可商业购买的,例如,glaxal碱得自于glaxal加拿大有限公司。其它合适的赋形剂可能在以下文献中发现:koller和buri,s.t.p.pharma3(2),115-124,1987。优选的载体是可在环境温度、在所使用的活性组分的浓度下可使活性组分溶解在其中的载体。载体应具有足够的粘度,以使抑制剂保持在组合物所应用的皮肤或粘膜的限定区域,在一段时间内不移动或蒸发,以使前体物足以基本上渗透通过皮肤或粘膜的限定区域并进入到血液中,其在此处将会导致合乎需要的临床效果。载体典型地是若干组分例如可药用溶剂和增稠剂的混合物。有机和无机溶剂的混合物可有助于亲水性和亲脂性的溶解度,例如水和醇例如乙醇的混合物。在另一个方面,本发明提供了药物组合物,其包括性甾体前体物,选自:脱氢表雄甾酮,脱氢表雄甾酮-硫酸酯,雄甾-5-烯-3β,17β-二醇,和4-雄甾烯-3,17-二酮;和进一步包括可药用赋形剂,稀释剂或载体,选自:饱和脂肪酸c12-c18的甘油三酯,具有不同部分的相应的偏甘油酯(硬脂,witepsol),黄油,油酸、棕榈酸和硬脂酸的混合甘油三酸酯(可可脂),部分氢化的棉子油(cotomar),氢化脂肪醇和酯(dehydag碱i,碱ii或碱iii,还可以含有c12-c16饱和脂肪酸的甘油酯),得自于棕榈、棕仁和椰油的甘油三酯,具有自乳化的单硬脂酸甘油酯和聚氧乙烯硬脂酸酯(fattibase),hexaride碱95,较高的融化馏分的椰油和棕榈坚果油(hydrokote),重排氢化植物油(s-70-xx95和s-070-xxa),来源于天然植物油的单-、二-、三甘油酯的低共熔混合物(suppocire),tegester甘油三酯,吐温61,来源于椰子油(wecobee)、可可脂、半合成甘油酯的甘油三酯(japocire,ovucire),饱和脂肪酸三-、二-、和一甘油酯的混合物(massaestarinum)和上述的组合(见allen等人,2008)。可将dhea及其它前体物溶解在其中的包括液体的任何赋形剂都被本发明所涵盖。优选将性甾体前体物配制成含有2.0至10%辛酸-癸酸甘油三酯(neobeem-5);10至20%的己二醇;2.0至10%的二乙二醇一甲醚(transutol);2.0至10%的环甲硅油(dowcorning345);1.0至2%的苯甲醇和1.0至5.0%的羟丙基纤维素(klucelhf)的醇凝胶。载体还可以包括通常在软膏剂和洗剂中使用的,和在美容和医学领域众所周知的各种添加剂。可以给出的是例如,香料,抗氧化剂,香料,凝胶剂,增稠剂例如羧甲纤维素,表面活化剂,稳定剂,软化剂,着色剂及其它类似的试剂。当在治疗全身性疾病使用时,为了避免活性组分的过量的局部浓度和可能由性甾体前体物的雄激素性代谢物引起的皮肤和皮脂腺的过度兴奋,其在皮肤上的应用部位应有所改变。在口服药物组合物中,dhea或其它前体物优选是以相对于组合物总重量浓度在5和98%重量之间、更优选在50和98%之间,特别是在80和98%之间的浓度存在。单一的前体物例如dhea可以是唯一活性组分,或者可以使用多个前体物和/或它们的类似物(例如,dhea、dhea-s,5-二醇的组合,或体内可转变为dhea、dhea-s或5-二醇的两种或多种化合物的组合,或dhea或5-二醇和其可体内转变为dhea或5-二醇的一或多种类似物的组合,等等。dhea的血液水平是充足剂量的最终标准,其考虑了在吸收和代谢机制中的个体差异。优选,特别是在治疗开始时,护理临床医师将会监测个体患者的总响应和dhea血清水平(与上述讨论的优选血清浓度相比),和监测患者对治疗的总响应,以在给定患者的代谢或对治疗的反应是非典型性的情况下根据需要调节剂量。按照本发明的治疗适合于不限定的继续治疗。人们预期dhea和/或5-二醇治疗将会简单地将dhea水平保持在与绝经之前女性自然出现的dhea水平类似的范围之内(血清浓度在每升4和10微克之间)。还可以通过口服途径给予serm化合物或性甾体前体物,且可以用常规的药物赋形剂,例如喷雾干燥的乳糖,微晶纤维素,和硬脂酸镁配制成用于口服的片剂或胶囊剂。可以将活性物质(serm化合物或性甾体前体物)通过与固体,粉末状的载体物质,例如枸橼酸钠,碳酸钙或磷酸氢钙,和粘合剂例如聚乙烯吡咯烷酮,明胶或纤维素衍生物混合加工成片剂或糖锭的芯,还可能加入润滑剂例如硬脂酸镁,月桂基硫酸钠,“carbowax”或聚乙二醇。当然,在口服给药形式的情况下可以加入改善味道的物质。作为进一步的形式,可以使用栓塞胶囊剂(plugcapsules),例如硬明胶,以及封闭的软明胶胶囊包括软化剂或增塑剂,例如甘油。填料胶囊剂含有优选以颗粒形式的活性物质,例如以与填料,例如乳糖,蔗糖,甘露糖醇,淀粉,例如马铃薯淀粉或支链淀粉(amylopectin),纤维素衍生物或高分散的硅酸的混合物形式。在软明胶胶囊中,优选将活性物质溶解或悬浮在合适液体中,例如植物油或液体聚乙二醇中。应该将洗剂,软膏剂,凝胶或乳膏剂彻底地擦拭到皮肤中,以便没有容易看出的过量,且不应该洗涤该区域的皮肤,直到出现绝大部分透皮渗透为止,优选至少4小时和更优选至少6小时。可按照已知的技术用透皮贴片来递送前体物或lhrh激动剂或拮抗剂。典型地施用很长的时间,例如1至4天,但典型地使活性组分与较小的表面积接触,以允许缓慢和恒定的活性组分递送。已经研制和使用的许多透皮给药系统,都适合于递送本发明的活性组分(serm,性甾体前体物,和lhrh激动剂或拮抗剂)。释放速率典型地通过基体扩散控制,或通过使活性组分通过控制膜来控制释放速率。在大鼠中透皮装置的机械性方面是众所周知的,且在例如下列美国专利中解释说明:us5,162,037,5,154,922,5,135,480,4,666,441,4,624,665,3,742,951,3,797,444,4,568,343,5,064,654,5,071,644,5,071,657,本文以引证的方式包括其公开内容。另外的背景由欧洲专利0279982和英国专利申请2185187提供。装置可以是本领域已知的任何常规类型,包括粘附剂基质和储罐型透皮输送装置。装置可以包括含有药物的基质,其包括吸附了活性组分和/或载体的纤维。在储罐型装置中,储罐可以定义为载体和活性组分不能透过的聚合物膜。在透皮装置中,装置本身可保持活性组分与希望定位的皮肤表面接触。在这种装置中,与乳膏剂或凝胶相比更少关注用于活性组分的载体的粘度。用于透皮装置的溶剂系统可以包括,例如,油酸,直链醇乳酸酯和二丙二醇,或其它本领域已知的溶剂体系。可以将活性组分溶解或悬浮在载体中。为了与皮肤粘附,可以将透皮贴片安装在外科胶带上,其在中部打个洞。粘附剂优选在使用之前用保护它的释放衬料覆盖。适合于释放的典型材料包括聚乙烯和聚乙烯-涂覆的纸,和优选用硅氧烷涂覆,便于除去。为了应用该装置,将释放衬料简单地剥离开,并将粘附剂附着在患者皮肤上。在美国专利us5,135,480中,以引证的方式包括其公开内容,bannon等人描述了具有无粘着性装置的替代装置,以保证装置贴到皮肤。本发明的经皮或透粘膜递送系统还可以作为新的和改善的递送系统使用,以预防和/或治疗子宫内膜异位或其它疾病,这些疾病对用雄激素和/或雌激素治疗有利地做出响应。lhrh激动剂或拮抗剂是胃肠外给予的,即通过注射液肌内注射、皮下或静脉内给予,或通过滴鼻剂或栓剂输注。还可以将lhrh激动剂或拮抗剂微囊密封或贴附到生物相容的可生物降解的聚合物中,例如聚(d,1-交酯(lactide)-共聚-乙交酯),和通过称为皮下或肌内储存(depot)技术进行皮下或肌内注射,以提供lhrh激动剂或拮抗剂在30天或更久期间内的持续缓慢释放。lhrh激动剂或拮抗剂的最优选的给药途径是皮下或肌内储存(depot)注射。lhrh激动剂或拮抗剂可以以每天大约10至1500μg给予,和对于lhrh激动剂大约250(优选每天以50μg至500μg)给予,和对于lhrh拮抗剂优选以每天大约100至2000μg给予。lhrh激动剂或拮抗剂可以在头30天以500μg的日剂量皮下给予,此后以250μg的日剂量皮下给予,不考虑患者的体重。当给予lhrh激动剂或拮抗剂时,每30天周期使用一次,优选每30天周期的剂量为750至15,000μg。对于较长限期的控释制剂可使用类似的每日递送剂量。优选的lhrh激动剂是醋酸亮丙瑞林(leuprolideacetate),其得自于abbottlaboratoriesltd.,商标为“lupron”;得自于bayerag,商标为“viadur”;得自于sanofi-aventis,商标为“eligard”;得自于takedauk,商标为“prostapsr”和“prostap3”;戈舍瑞林乙酸酯,其得自于astrazeneca,商标为“zoladex”和“zoladexla”;那法瑞林,其得自于searle(现在是辉瑞的部分),商标为“synarel”;布舍瑞林乙酸酯,其得自于sanofi-aventis,,商标为“suprefact”或“suprefactdepot”,和得自于cinnagen,商标为“cinnafact”;组氨瑞林乙酸酯,其得自于endopharmaceuticals,商标为“vantas”和“supprelinla”;曲普瑞林乙酸酯或双羟萘酸酯,其得自于ipsen,商标为“decapeptyl”;得自于ferringpharmaceuticals,商标为“diphereline”和“gonapeptyl”;和得自于watson,商标为“trelstar”。可以使用任何lhrh激动剂或拮抗剂。lhrh激动剂或拮抗剂的典型的药物组合物包括lhrh激动剂或拮抗剂或其可药用酸式盐,苯甲醇,磷酸盐缓冲液(ph值6.0-6.5)和无菌水。用于肌内或皮下储存(depot)注射液的lhrh激动剂或拮抗剂可以通过相分离方法微囊密封在生物相容的、可生物降解的聚合物中,例如,聚(d,l-交酯-共聚-乙交酯),或加工形成颗粒(pellet)。然后可以将微球体悬浮在载体中,以提供注射制剂或储存(depot)制剂以颗粒(pellet)形式注射。参见欧洲专利申请epano.58,481,1982年8月25日公开,用于皮下注射液或植入法的固体组合物,或用于肌内或皮下注射的液体制剂,含有生物相容的、可生物降解的聚合物例如交酯-乙交酯共聚物和lhrh激动剂,例如d-ser-t-buo6,azgly10-lhrh。这些制剂允许肽的控制释放。术语“lhrh激动剂”是指天然促黄体生成激素释放激素(lhrh)的合成类似物,例如下列结构的十肽:l-焦谷氨酰基-l-组氨酰-l-色氨酰-l-丝氨酰-l-酪氨酰-甘氨酰-l-亮氨酰-l-精氨酰l-脯氨酰甘氨酰-nh2。合适的lhrh激动剂包括由下式代表的九肽和十肽:l-焦谷氨酰基-l-组氨酰-l-色氨酰-l-丝氨酰-l-酪氨酰-x-y-精氨酰-l-脯氨酰-z,其中x是d-色氨酰,d-亮氨酰,d-丙氨酰,亚氨基苄基-d-组氨酰,3-(2-萘基)-d-丙氨酰,o-叔丁基-d-丝氨酰,d-酪氨酰,d-赖氨酰,d-苯丙氨酰基,1-苄基-d-组氨酰或n-甲基-d-丙氨酰,和y是l-亮氨酰,d-亮氨酰,nα-甲基d-亮氨酰,nα-甲基-l-亮氨酰或d-丙氨酰,和其中z是甘氨酰-nhr1,(氮杂)甘氨酰-nhr1或nhr1,其中r1是h,低级烷基或低级卤代烷基。低级烷基包括具有1至6个碳原子的直链-或支链烷基,例如,甲基,乙基,丙基,戊基或己基,异丁基,新戊基等等。低级卤代烷基包括具有一个卤素取代基的1至6个碳原子的直链-和支链烷基,例如,-cf3,-ch2cf3,-cf2ch3。卤素是指f,cl,br,i,优选cl。在优选的九肽中,y是l-亮氨酰,x是光学活性的d-形式的色氨酸,丝氨酸(t-buo),亮氨酸,组氨酸(亚氨基苄基),和丙氨酸。优选的十肽包括[d-trp6]-lhrh,其中x=d-trp,y=l-亮氨酰,z=甘氨酰-nh2,[d-phe6]lhrh,其中x=d-苯丙氨酰基,y=l-亮氨酰和z-甘氨酰-nh2),或[d-nal(2)6]lh-rh,其是[(3-(2-萘基)-d-ala6]lhrh,其中x=3-(2-萘基)-d-丙氨酰,y=l-亮氨酰和z=甘氨酰-nh2)。在本发明范围内的其它有用的lhrh激动剂是天然lh-rh的α-氮杂类似物,特别是,[d-phe6,azgly10]-lhrh,[d-tyr(me)6,azgly10]-lhrh,和[d-ser-(t-buo)6,azgly10]-lhrh,公开在(dutta,furr等人,1978)和u.s.pat.no.4,100,274中,以及公开在u.s.pat.nos.4,024,248和4,118,483中的那些。优选的lhrh拮抗剂是得自于specialityeuropeanpharma以商标“plenaxis”可得到的阿倍瑞克,ardana研制的替维瑞克(teverelix);得自于merckserono,商标为“cetrotide”的西曲瑞克乙酸酯;得自于organoninternational,商标为“antagon”的加尼瑞克乙酸酯;得自于serono,商标为“antide”的伊妥瑞克(iturelix),merrionpharmaceuticals研制的acyline;得自于ferringpharmaceuticals,商标为“firmagon”的地盖瑞利(degarelix),和oakwoodlaboratories研制的ornirelix。其它lhrh拮抗剂是azalineb(salkinstitute),ozarelix(spectrumpharmaceuticals),lxt-101(药物化学系,北京药理和毒理学研究所),elagolix(neurocrinebiosciences),和tak-013和tak-385(takeda)。典型的合适的lhrh拮抗剂包括[n-ac-d-p-cl-phe1,3,d-phe3,d-arg6,d-ala10]lhrh,公开在(erchegyi,coy等人,1981)[n-ac-d-p-cl-phe1,2,d-trp3,d-arg6,d-ala10]lhrh,公开在(coy,horvath等人,1982);[n-ac-d-(3-(2-萘基)-ala)1,d-p-cl-phe2,d-trp3,d-harg(et2)6,d-ala10]-lhrh和[n-ac-pro1,d-p-cl-phe2,(d-(3-(2-萘基(ala3,6]-lhrh,公开在(nestor,ho等人,1984);用作lhrh拮抗剂的lhrh的九-和十肽类似物,公开在uspatno4,481,190中,高束缚的环状拮抗剂类似物,环[δ3pro1,d-p-cl-phe2,d-trp3,5,n-me-leu7,β-ala10]lhrh,公开在(rivier,rivier等人,1984),和[n-ac-d-(3-(2-萘基)-ala)1,d-p-f-phe2,d-trp3,d-arg6]-lhrh,公开在(corbin,bex等人,1984)。其它的lhrh激动剂和拮抗剂类似物公开在lhrh和其类似物(b.h.vickery等人,editorsatpage3-10(j.j.nestor),11-22(j.river等人,)和23-33(j.j.nestor等人,)和gynecologicalendocrinology13(suppl.1)1999:见gnrhantagonist(t-98475),p.8,abst.#015。其它的lhrh激动剂是得自于peptech,商标为“ovuplant”的地洛瑞林乙酸酯。效果实施例a-原料和方法a.1-动物和治疗在治疗开始时使用重量大约235-250g的十至十二周雌性spraguedawley大鼠(crl:cd(sd)br)。一百二十个大鼠随机地分成5组,每组具有15个无损伤的动物如下:1)对照组;2)lhrh-a(0.002mg/动物);3)lhrh-a+em-652·hcl(2.5mg/kg);4)lhrh-a+dhea(100mg/kg);5)lhrh-a+em-652·hcl+dhea。通过口服填喂法每日给予一次em-652·hcl((s)-(+)-7-羟基-3-(4’-羟基苯基)-4-甲基-2-(4’’-(2’’’-哌啶子基乙氧基)苯基)-2h-1苯并吡喃盐酸盐)在0.4%甲基纤维素中的悬浮液(0.5ml/大鼠),给予3个月,在同样的周期内将dhea以50%乙醇-50%丙二醇溶液(0.5ml/大鼠)每日一次局部地应用于背部皮肤,同时每日一次皮下注射在磷酸盐缓冲剂中的lhrh-a(0.5ml/大鼠)。治疗大约3个月之后,使用boehringermannheimdiagnostichitachi911分析仪收集过夜禁食动物的颈静脉血样,用于测量全部血清胆固醇水平。骨矿物密度测量法治疗12周后,将异氟烷麻醉的单独的大鼠,使用双能x射线吸光测定法(dexa;qdr4500a,hologic,waltham,ma)和区域的高分辨率扫描(regionalhighresolutionscan)软件扫描它们的全身骨骼以及它们的右股骨。测定全身骨骼、腰脊椎和股骨的骨矿物含量(bmc),骨矿物密度(bmd)。同时测定体躯成分。骨碱性磷酸酶使用boehringermannheimdiagnostichitachi911分析仪(boehringermannheimdiagnosticlaboratorysystems)测定血清碱性磷酸酶的总活度。然后,将血清样品(0.1ml)与0.1ml的小麦胚芽植物血凝素溶液(6mg/ml,在水中)混合,在室温下培养30min,和在10000g下离心3min,沉淀出骨alp。使用boehringermannheimdiagnostichitachi911分析仪测定在获得的上清液中的alp活性,骨alp活性计算如下:骨alp=全部alp-(2x上清液的alp)。统计分析数据用平均值±sem来表示。按照duncan-kramer邓肯多重范围检验测定统计显著性。试剂盒实施例以下列出了举例来说和非限制性的若干试剂盒,使用优选的活性serm阿考比芬(em-652·hcl,em-1538),优选活性的性甾体前体物脱氢表雄甾酮(dhea,prasterone)和优选lhrh-激动剂醋酸亮丙瑞林(leuprolideacetate)(luprondepot)。可以使用其它本发明的化合物或其组合代替(或补充于)阿考比芬,脱氢表雄甾酮,和醋酸亮丙瑞林(leuprolideacetate)。可以使用lhrh-拮抗剂代替lhrh-激动剂。活性组分的浓度可以在较宽范围内变化,如本文所讨论。可以包括的其它组分的数量和类型是本领域众所周知的。实施例a以在相同的制剂(胶囊剂)中的方式口服一起给予serm和性甾体前体物,而胃肠外给予lhrh激动剂。用于口服的serm和性甾体前体物组合物(胶囊剂)组分重量%(以全部组合物的重量计)阿考比芬5.0dhea10.0含水乳糖70.0淀粉4.8微晶纤维素9.8硬脂酸镁:0.4+用于肌内储存(depot)注射的lhrh激动剂组分重量%(以全部组合物的重量计)醋酸亮丙瑞林(leuprolideacetate)(luprondepot®-3个月)0.7聚乳酸6.1d-甘露糖醇5.8羧甲基纤维素钠0.5聚山梨醇酯800.1冰醋酸(usp)以控制ph值注射用水(usp)86.8实施例b以在相同的制剂(片剂)中的方式口服一起给予serm和性甾体前体物,而胃肠外给予lhrh激动剂。用于口服的serm和性甾体前体物组合物(片剂)组分重量%(以全部组合物的重量计)阿考比芬5.0dhea15.0明胶5.0乳糖:58.5淀粉16.5+用于肌内储存(depot)注射的lhrh激动剂组分重量%(以全部组合物的重量计)醋酸亮丙瑞林(leuprolideacetate)(luprondepot®-3个月)0.7聚乳酸6.1d-甘露糖醇5.8羧甲基纤维素钠0.5聚山梨醇酯800.1冰醋酸(usp)以控制ph值注射用水(usp)86.8实施例c以在相同的制剂(乳膏剂)中的方式经皮一起给予serm和性甾体前体物,而胃肠外给予lhrh激动剂。用于经皮给予的serm和性甾体前体物组合物(乳膏剂)组分重量%(以全部组合物的重量计)dhea1.0阿考比芬0.2乳化蜡,nf18.0轻质矿物油,nf12.0苯甲醇1.0乙醇95%usp33.8纯化水,usp34.0+用于肌内储存(depot)注射的lhrh激动剂组分重量%(以全部组合物的重量计)醋酸亮丙瑞林(leuprolideacetate)(luprondepot®-3个月)0.7聚乳酸6.1d-甘露糖醇5.8羧甲基纤维素钠0.5聚山梨醇酯800.1冰醋酸(usp)以控制ph值注射用水(usp)86.8实施例d以在相同的制剂(栓剂或小卵)中的方式阴道内一起给予serm和性甾体前体物,而胃肠外给予lhrh激动剂。用于阴道内给予的serm和性甾体前体物组合物(栓剂或小卵)组分重量%(以全部组合物的重量计)dhea0.25至2.0阿考比芬0.25至3.0witepsolh-15基料95.0至99.5+用于肌内储存(depot)注射的lhrh激动剂组分重量%(以全部组合物的重量计)醋酸亮丙瑞林(leuprolideacetate)(luprondepot®-3个月)0.7聚乳酸6.1d-甘露糖醇5.8羧甲基纤维素钠0.5聚山梨醇酯800.1冰醋酸(usp)以控制ph值注射用水(usp)86.8实施例e以在相同的制剂(胶囊剂)中的方式口服一起给予serm和性甾体前体物。用于口服的serm组合物(胶囊剂)组分重量%(以全部组合物的重量计)阿考比芬5.0含水乳糖80.0淀粉4.8微晶纤维素9.8硬脂酸镁0.4+用于口服的dhea组合物(明胶胶囊剂)组分重量%(以全部组合物的重量计)dhea25.0含水乳糖27.2甘醇酸(glycolate)淀粉钠20.0微晶纤维素,胶体二氧化硅,无水二氧化硅胶体和无水轻硅酸27.2胶体二氧化硅0.1硬脂酸镁0.5+用于肌内储存(depot)注射的lhrh激动剂组分重量%(以全部组合物的重量计)醋酸亮丙瑞林(leuprolideacetate)(luprondepot®-3个月)0.7聚乳酸6.1d-甘露糖醇5.8羧甲基纤维素钠0.5聚山梨醇酯800.1冰醋酸(usp)以控制ph值注射用水(usp)86.8可以用其它serms代替上面制剂中的阿考比芬,以及可以用其它性甾体前体物代替dhea,和以及可以用其它lhrh激动剂或拮抗剂代替醋酸亮丙瑞林(leuprolideacetate)。可以包括一种以上的serm或一种以上的性甾体前体物或一种以上的lhrh激动剂,在这样的情况下,联用的重量百分数优选是在上面实施例中给出的单一性甾体前体物或单一serm或单一lhrh激动剂的重量百分数。实施例f口服给予serm,阴道内给予性甾体前体物,而胃肠外给予lhrh激动剂。用于口服的serm组合物(胶囊剂)组分重量%(以全部组合物的重量计)阿考比芬5.0含水乳糖80.0淀粉4.8微晶纤维素9.8硬脂酸镁0.4+用于阴道内给予的性甾体前体物组合物(栓剂或小卵)组分重量%(以全部组合物的重量计)dhea0.25至2.0witepsolh-15基料98.0至99.75使用witepsolh-15基料(paddocklaboratories,minneapolis,usa)制备dhea栓剂。可以使用任何其它亲脂性的基料例如硬脂(hardfat),fattibase,wecobee,可可脂,可可豆油或witepsol基料的其它组合。+用于肌内储存(depot)注射的lhrh激动剂组分重量%(以全部组合物的重量计)醋酸亮丙瑞林(leuprolideacetate)(luprondepot®-3个月)0.7聚乳酸6.1d-甘露糖醇5.8羧甲基纤维素钠0.5聚山梨醇酯800.1冰醋酸(usp)以控制ph值注射用水(usp)86.8实施例g阴道内给予serm和性甾体前体物,而胃肠外给予lhrh激动剂。用于阴道内给予的性甾体前体物组合物(栓剂或小卵)组分重量%(以全部组合物的重量计)dhea0.25至2.0witepsolh-15基料98.0至99.75+用于阴道内给予的serm组合物(栓剂或小卵)组分重量%(以全部组合物的重量计)阿考比芬0.3至3.0硬脂(hardfat)97.0至99.7使用硬脂(hardfat,witepsol)制备阿考比芬栓剂。可以使用任何其它基料例如fattibase,wecobee,可可脂,可可豆油或硬脂(hardfat)的其它组合。+用于肌内储存(depot)注射的lhrh激动剂组分重量%(以全部组合物的重量计)醋酸亮丙瑞林(leuprolideacetate)(luprondepot®-3个月)0.7聚乳酸6.1d-甘露糖醇5.8羧甲基纤维素钠0.5聚山梨醇酯800.1冰醋酸(usp)以控制ph值注射用水(usp)86.8实施例h口服给予serm,经皮给予性甾体前体物,而胃肠外给予lhrh激动剂。用于口服的serm组合物(胶囊剂)组分重量%(以全部组合物的重量计)阿考比芬5.0含水乳糖80.0淀粉4.8微晶纤维素9.8硬脂酸镁0.4+用于经皮给予的性甾体前体物组合物(凝胶)组分重量%(以全部组合物的重量计)dhea2.0辛酸-癸酸甘油三酯(neobeem-5)5.0己二醇15.0transcutol(二乙二醇一甲醚)5.0苯甲醇2.0环甲硅油(dowcorning345)5.0乙醇(绝对)64.0羟丙基纤维素(1500cps)(klucel)2.0或用于经皮给予的性甾体前体物组合物(乳膏剂)组分重量%(以全部组合物的重量计)制剂em-760-48-1.0%环甲硅油5.0%轻质矿物油3.0%2-乙基己基硬脂酸酯10.0%cutinae241.0%dc乳化剂103.0%bht0.09%丙二醇46.01%乙醇9510.0%dhea1.0%纯水(eaupurifiée)15.0%mgso40.65%乙醇955.25%总计100.0%+用于肌内储存(depot)注射的lhrh激动剂组分重量%(以全部组合物的重量计)醋酸亮丙瑞林(leuprolideacetate)(luprondepot®-3个月)0.7聚乳酸6.1d-甘露糖醇5.8羧甲基纤维素钠0.5聚山梨醇酯800.1冰醋酸(usp)以控制ph值注射用水(usp)86.8实施例i口服给予serm(胶囊剂),而胃肠外给予lhrh激动剂。用于口服的serm组合物(胶囊剂)组分重量%(以全部组合物的重量计)阿考比芬5.0含水乳糖80.0淀粉4.8微晶纤维素9.8硬脂酸镁0.4+用于肌内储存(depot)注射的lhrh激动剂组分重量%(以全部组合物的重量计)醋酸亮丙瑞林(leuprolideacetate)(luprondepot®-3个月)0.7聚乳酸6.1d-甘露糖醇5.8羧甲基纤维素钠0.5聚山梨醇酯800.1冰醋酸(usp)以控制ph值注射用水(usp)86.8实施例j口服给予serm(片剂),而胃肠外给予lhrh激动剂。用于口服的serm组合物(片剂)组分重量%(以全部组合物的重量计)阿考比芬5.0明胶5.0乳糖73.5淀粉16.5+用于肌内储存(depot)注射的lhrh激动剂组分重量%(以全部组合物的重量计)醋酸亮丙瑞林(leuprolideacetate)(luprondepot®-3个月)0.7聚乳酸6.1d-甘露糖醇5.8羧甲基纤维素钠0.5聚山梨醇酯800.1冰醋酸(usp)以控制ph值注射用水(usp)86.8可以用其它serm(托瑞米芬,奥培米芬,雷诺昔酚,阿佐昔芬,拉索昔芬,醋酸巴多昔芬(tse-424),era-923,gw5638)代替上面制剂中的阿考比芬,以及可以用其它性甾体前体物例如硫酸脱氢表雄甾酮(dhea-s),4-雄甾烯-3,17-二酮和雄甾-5-烯-3β,17β-二醇(5-二醇)代替dhea。可以用其它商用的lhrh激动剂代替醋酸亮丙瑞林(leuprolideacetate)。可以包括一种以上的serm或一种以上的性甾体前体物或一种以上的lhrh激动剂或拮抗剂,在这样的情况下,联用的重量百分数优选是在上面实施例中给出的单一性甾体前体物或单一serm或单一lhrh激动剂的重量百分数。本发明已经以优选实施方案和实施例形式描述,但不局限于此。本领域技术人员将容易地认识到本发明的更广泛的实用性和更宽的范围,其仅仅由本专利的权利要求限定。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1