无溶剂钆造影剂的制作方法
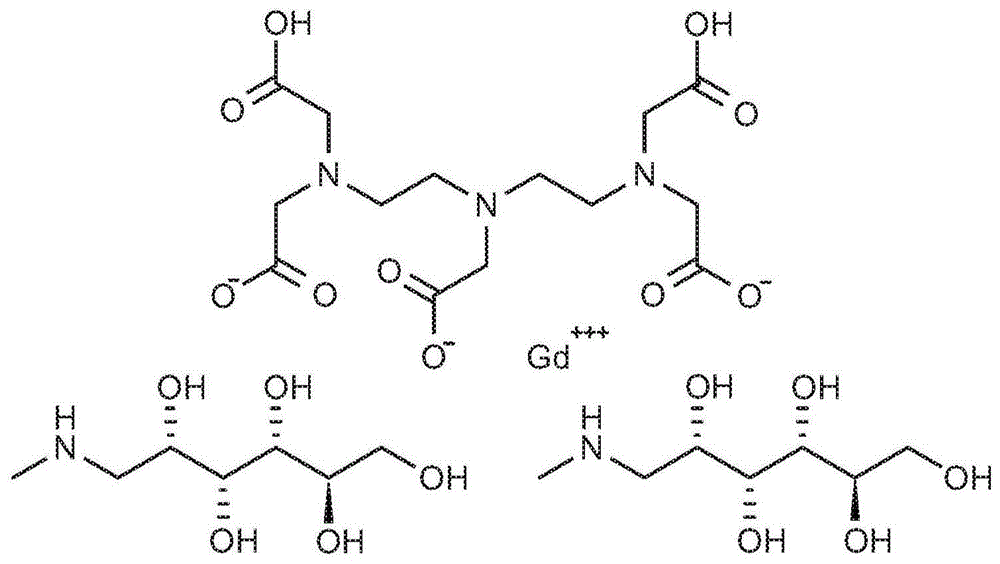
本申请要求美国临时申请第62/439,893号的权益,其在此通过引用整体并入本文。本公开总体上涉及金属螯合物,特别是镧系元素金属的那些金属螯合物,并且在一具体的实施方案中,涉及gd(iii)的那些金属螯合物,其可用作治疗和诊断应用以及临床和生物医学研究应用的磁共振成像中的造影剂。发明背景磁共振成像(mri)是产生体内身体组织的三维图像的一种强有力的诊断方法。得到的组织特征是这些组织中水的分布变化的结果。在成像前施用的mri造影剂改变了它们附近的质子弛豫时间,增强了图像的特定特征。mri造影剂提高了mri诊断的灵敏度和实用性。在临床环境中使用用于mri的造影剂已成为增加医学mri图像的分辨率和组织特异性的常规实践标准。顺磁性金属螯合物(诸如gd(iii)-二乙烯三胺五乙酸(gd(iii)-dtpa)(马根维显)、gd(iii)-n,n',n',n”,n”'-四羧甲基-1,4,7,10-四氮杂环十二烷(gd(iii)-dota),及它们的类似物)已被证明能够增加周围质子的弛豫速率,并已被广泛用作mri造影剂。钆络合物的热力学稳定性强烈依赖于ph,并且虽然体内ph不是高度可变的,但是目前的制造方法产生的钆络合物的组分的ph变化很大。降低的热力学稳定性可导致从配体中释放出有毒的gd(iii)离子,并且可能与肾源性系统性纤维化有关。虽然gd(iii)离子的形成由于产品ph的制造变化而发生,并且该产品ph在被注入时最终与体内ph平衡,但是与ph平衡相比,由于注射而发生的稀释足够快以分离gd(iii)离子和配体(例如,喷替酸),使得当达到有利的ph时,金属离子和配体被充分分离,以至它们不会重新组合成配体和钆的缀合物。因此,在产品说明书中具有大ph范围的医学造影剂存在关于产品稳定性和从络合物形成释放毒性的gd(iii)的可能性的安全问题。药物产品中大ph范围与药物纯化过程中溶剂的使用有关,所述溶剂的使用倾向于以不可预测的方式去除配体。过去尝试制备顺磁性金属螯合物的合成方法有一个或多个缺点,诸如使用大量过量的配体来减少游离的gd(iii)离子;或者由于原始试剂中的杂质而需要进行大量的溶剂纯化。具有讽刺意味的是,用于纯化药物产品的生物相容性溶剂可以与它们打算去除的杂质络合。如果最初使用纯的成分,则除去了对溶剂纯化的需要。然而,仍然需要溶剂,因为钆络合物必须在无水状态下沉淀,以便以治疗效力配制药物产品。在水中络合gd(iii)和配体、干燥,然后在水中重新配制的顺序是一个多步骤过程,其导致钆离子和配体之间微妙平衡的显著变化。最后,这个多步骤过程是引起溶剂络合的杂质、ph和钆-配体平衡的变化的原因。因此,在工业中允许大范围的ph和葡甲胺含量已经成为标准。特别地,在目前的mri造影剂马根维显中有意配制了显著过量的配体(例如喷替酸)。在马根维显中,在过量喷替酸存在下,gd(iii)离子的形成减少。gd(iii)离子的形成主要是在溶剂存在下喷替酸配体和钆的大分子缀合物的热力学稳定性变化的结果。通过钆缩短质子弛豫时间是由钆的未配对价电子和相邻水质子之间的偶极-偶极相互作用所介导的。钆磁偶极相互作用的大小作为它与这些质子的距离的函数(作为半径的六次方)非常快速地下降。因此,有效弛豫的唯一质子是那些能够进入钆金属的质子。质子可以进入钆金属和金属络合物的第一或第二配位层。在配位化学中,金属离子被描述为由两个同心配位层组成。第一配位层是指中心原子或离子(在这种情况下是钆)。第二配位层可以由离子(特别是在带电的络合物中)、分子(特别是那些与第一配位层中的配体氢键键合的分子)和配体骨架的部分组成。与第一配位层相比,第二配位层对金属络合物的反应性和化学性质的直接影响较小。尽管如此,第二配位层与理解金属络合物的反应有关,包括配体交换和催化的机理。在rf脉冲和信号检测之间的间隔期间,质子进入钆金属络合物的第一或第二配位层。该间隔持续范围为105至106个质子/秒(brown(1985)mag.res.imag.v3,p3)。钆在它的4f轨道中具有七个未配对的价电子,并因此具有最大的顺磁偶极性(7.9玻尔磁子),并且展现出任何元素的最大顺磁弛豫率(runge等人,(1983)am.j.radiolv141,p1209和weinman等人(1984)am.j.radiolv142,p619)。因此,钆具有用于增强磁共振成像的任何元素的最高潜力。为了利用钆的大的顺磁偶极性,必须认识到体内游离钆金属离子(gd(iii))的毒性。因此,体内使用钆金属(例如氯化钆或氧化钆)是不安全的,并且必须使用钆的水溶性螯合物。虽然基于水溶性螯合的钆的造影剂在患者中注射更安全,但是毒性问题并未完全被解决。潜在毒性部分是钆的沉淀的结果,其可以在身体ph下作为氢氧化钆而发生。然而,gd(iii)离子,即使它不形成水不溶性化合物,仍然可能是有毒的,因为gd(iii)的反应性与ca(ii)非常相似,并且ca(ii)在哺乳动物身体的化学通路中无处不在。为了增加溶解度并降低毒性,钆已经被小的有机分子化学螯合。迄今为止,从一般效用、活性和毒性的观点来看,最令人满意的螯合剂是二乙烯三胺五乙酸(dtpa)(runge等人,(1983)am.j.radiolv141,p1209和weinman等人(1984)am.j.radiolv142,p619)。根据gries、rosenberg和weinmann在西德递交的专利申请(de-os3129906a1(1981)),schering-berlexag开发了进行广泛临床试验的该螯合物的第一个制剂。螯合物由gd-dtpa组成,gd-dtpa是ph中性的并且被用有机碱n-甲基-d-葡糖胺(葡甲胺或甲基葡甲胺)稳定。x射线衰减子的浓度与其在对比度增强中的功效之间存在直接关系。对于mri造影剂,浓度和对比效应之间的关系不是线性的,其中需要顺磁性实体的阈值浓度来影响正在成像的生理区域中的质子弛豫速率。超过该阈值浓度,浓度的任何进一步增加均会导致很少的对比增强的提高。因此,mri造影剂被配制为尽可能接近阈值浓度,以帮助减少螯合作用无法减轻的毒性作用。然而,如果钆络合物不稳定,则必须对制剂进行限制,并使螯合物浓度大于阈值。三价镧系阳离子的离子半径范围从la(iii)的1.1a到lu(iii)的而恰好位于镧系元素系列中心的gd(iii)具有的离子半径,几乎等于二价ca(ii)的离子半径。gd(iii)可以在生物系统的化学通路中与ca(ii)竞争,并且这种取代潜力导致对生物体的钆毒性。事实上,钆的三价离子以比钙的二价离子高得多的亲和力结合。当与ca(ii)结合酶结合时,镧系离子取代通常改变由该酶催化的生物过程的动力学。钆的毒性强调了钆-配体(gdl)络合物的稳定性,因为络合物形式的毒性显著低于金属离子形式的毒性。络合物的热力学稳定性简单地描述了如下式所示的平衡时溶液中存在的所有种类的浓度:kst=[gdl][gd][l]其中gd为钆离子,l为配体,k为稳定性常数,gdl为钆-配体络合物,[l]为配体质子化常数,[gdl]为络合物的热力学稳定常数以及[gd]为gd(iii)离子形成常数。当将溶剂引入平衡方程式中时,络合物的热力学稳定常数减小,并且gd(iii)离子形成常数增加。游离钆金属离子具有8个水的内球体(inner-sphere)位点,并且络合物形式仅具有1个水的内球体。络合物与游离金属离子之间的平衡过程的吉布斯自由能将由于释放八个水的内球体中的七个而对络合物形式具有大的有利熵。该熵贡献被称为“螯合效应”。可以与溶剂而不是水形成结合球体的溶剂的存在会损害这种螯合效应。此外,钆离子-配体相互作用具有大的静电成分,其有助于产生有利的焓项。结果是总体自由能变化变得对络合物形式相当有利。由于这些原因,溶剂化的gd(iii)离子与具有更多碱性供体原子的配体形成非常稳定的络合物。稳定性因不存在溶剂而增强。对配体中最大碱性基团的需求导致普遍选择由胺类组成的配体。该考虑还解释了为什么具有含酰胺侧链的胺基团比具有乙酸侧链的胺基团(例如二乙烯三胺五乙酸(dtpa)或喷替酸)的碱性低得多。络合物的较高热力学稳定性由较大的热力学稳定常数kst来表示。应当理解,配体质子化常数中小的差异可能对产生的gdl络合物的热力学稳定性具有显著的影响。与配体的log[l]值的相对小的变化不同,络合物的logkst值可以变化超过10个数量级。稳定常数被广泛用于比较造影剂,因为它减少了与单个方便数字的比较。热力学稳定常数描述了其中配体完全去质子化条件下的平衡。在生理ph值下,配体将被部分质子化,因此人们可以争辩说,比较gdl稳定性的更好方法是使用表1中列出的所谓的条件稳定常数。表1:常见gd络合物的热力学和条件稳定常数(schmitt-willichh,brehmm,ewerscl,michlg,mueller-fahrnowa,petrovo等人,inorgchem1999;38:1134–44.)表1比较了在ph14下的钆和各种配体之间形成的络合物的稳定性常数(去质子化的和标准的“热力学稳定常数”),以及在ph7.4下的条件稳定常数。更强的酸性条件明显导致更低的络合物稳定性。除了质子之外还有其他离子竞争者能够影响络合物的稳定性。例如,像锌、铜和铁的离子与这些配体形成非常稳定的络合物,并且可以在恰当的活化能下促使钆退出较少毒性的络合物状态。同时,钆对一些污染物具有高亲和力,并且将会为了可能存在于溶液中的磷酸盐、柠檬酸盐和碳酸盐离子而离开络合物。这些污染物的影响程度通常由产品ph下的热力学稳定常数来确定。钆络合物的转金属化马根维显在其标签上列出的ph范围为6.5-8ph。仅基于存在的稀土金属,报道的在马根维显制备中使用的氧化钆杂质通常为99.9%的纯度。因此,没有评估可能是黄色来源的铁的存在。铁-dtpa络合物为黄色。zn(ii)与钆造影剂反应并取代钆的可能性已经受到了很多关注。此类将一种金属交换成另一种金属被称为转金属化。在化学化合物中常见的金属离子污染物中,na(i)、k(i)、mg(ii)和ca(ii)都与造影剂中使用的螯合剂形成非常弱的络合物,并且在热力学上不利于此类转金属化反应。造影剂螯合剂对其他内源性离子的亲和力顺序为fe(iii)>cu(ii)>zn(ii)。钆络合物的转金属化导致更不稳定的金属络合物的形成这一事实意味着合成方法可以影响转金属化的程度。特别地,如果在络合过程期间反应温度保持尽可能低,则该过程可以显著降低非钆金属络合物形成的发生率。因此,需要改善mri造影剂的安全性。本公开通过提供用于注射的钆络合物制剂解决了这种需要,其中不存在溶剂,并且消除了与配体的竞争性相互作用。发明概述总体上,本发明的一个目标是提供能够增强身体器官和组织的磁共振成像的药物产品和合成造影剂的方法,并且特别地,具有改善的安全性、减少的产品可变性和不存在溶剂污染物的配体-钆络合物。其他目标和特征将在下文中部分变得显而易见,并部分被指出。在本发明的几个目标中,可以注意到的是通过平衡离子(例如葡甲胺)平衡的配体和钆的有机无溶剂络合物的合成,仅具有钆一个中心金属离子以用于增强身体器官和组织的磁共振成像。本发明的另一个目标是提供在一步过程中形成配体和钆的络合物的方法,其以络合物形成开始并以药物产品制剂结束,且有利地消除溶剂的使用。本发明的另一个目标是提供具有小于当前可用的制剂的ph范围的钆造影剂(诸如钆喷酸二葡甲胺或钆特酸葡甲胺)。本发明的另一个目标是提供制备钆造影剂制剂的方法,其中非钆和配体络合物的形成被显著减少或消除。特别地,通过本发明的方法排除了溶剂-配体络合物的形成。本发明的另一目标是提供制备具有增强的权度的钆造影剂制剂的方法,使得制剂的颜色基本上可再现并且优选为无色的,而目前市售的钆喷酸二葡甲胺颜色范围从无色至黄色。本发明的另一个目标是组合上述的提供,使得结果为具有低可变的热力学稳定性和增强的稳定性的钆的配体络合物(例如钆喷酸二葡甲胺)。本发明的另一个目标是提供无溶剂配体-钆络合物,其具有降低的热力学稳定常数的可变性,并且具有降低的引起或有助于肾源性系统性纤维化的病因学的倾向性。因此,本公开提供了包含在适于哺乳动物注射的制剂中的gd(iii)离子、配体和葡甲胺的络合物的钆造影剂,其中所述络合物包含少于百万分之五十的非水性溶剂。在一些实施方案中,络合物少于百万分之一的非水性溶剂。在一些实施方案中,非水性溶剂选自丙酮、甲醇、乙醇、庚烷、己烷、乙腈、甲苯或它们的组合。在更具体的实施方案中,溶剂为甲醇、乙醇或它们的组合。在一些实施方案中,钆对比剂为钆喷酸二葡甲胺或钆特酸葡甲胺。在某些实施方案中,制剂包含少于0.025重量%的游离配体,并且更具体地,少于0.020%或0.010%。在其他实施方案中,制剂的ph范围为约7.2至约7.5。在其他实施方案中,络合物的热力学稳定常数范围为约18.1至约18.6。本公开的钆造影剂有利地具有减少的杂质。在一些实施方案中,造影剂包含少于百万分之一的游离gd(iii)离子。在一些实施方案中,造影剂包含少于百万分之十的非gd喷替酸络合物。本公开还提供了合成包含在适于哺乳动物注射的制剂中的gd(iii)离子、配体和葡甲胺的络合物的钆造影剂的方法,其中所述方法不使用非水性溶剂。本发明的方法有利地在所有过程步骤中维持水合状态,意味着不需要或不期望去除水。在一些实施方案中,在每一方法步骤期间,络合物处于至少1重量%的水的水合状态。在具体的实施方案中,所述方法包括以下步骤:i)制备dota的水性溶液,ii)在水中制备钆:dota络合物,iii)核查络合物中的游离钆的含量,iv)核查钆:dota络合物的形成,v)制备钆特酸葡甲胺溶液,和vi)过滤钆特酸葡甲胺溶液。在其他实施方案中,所述方法包括以下步骤:i)制备喷替酸的水性溶液,ii)在水中制备钆:喷替酸盐络合物,iii)核查络合物中的游离钆的含量,iv)核查钆:喷替酸盐络合物的形成,v)制备钆喷酸二葡甲胺溶液,和vi)过滤钆喷酸二葡甲胺溶液。本公开还提供了降低接受钆造影剂的患者中肾源性系统性纤维化的风险的方法,其包括向所述患者施用权利要求1所述的钆造影剂。附图的几个视图的简要说明图1示出了gd(iii)、dtpa和葡甲胺之间形成钆喷酸二葡甲胺的反应。图2示出了二乙烯三胺五乙酸的分子结构。图3示出了二乙烯三胺五乙酸二酐的分子结构。图4为比较可商购的钆喷酸二葡甲胺(gadopentatedimeglumine)制剂和根据本公开制备的钆喷酸二葡甲胺(gadopenteatedimeglumine)制剂中甲醇和乙醇的量的图。图5为比较可商购的钆喷酸二葡甲胺制剂与根据本公开制备的钆喷酸二葡甲胺制剂的杂质含量的图。发明详述本文中公开了合成无溶剂钆络合物的方法,其可以显著减少或消除可能与不良临床结果有关的次优产品特征的出现。这些次优产品特征是:1)溶剂杂质的存在,2)产品ph的变化,3)产品颜色的变化,4)产品热力学稳定常数的变化,5)游离gd(iii)离子的形成,以及6)与喷替酸形成非钆络合物。图1中提供了本发明的增强mri造影剂的实例的结构。图1显示了一个gd(iii)原子、两个dtpa分子(二乙烯三胺五乙酸)和两个葡甲胺(n-甲基葡糖胺)分子之间的化学反应。在本方法的步骤1-20中,形成了gd和dtpa的络合物。同时在络合物形成过程期间,将共配体(葡甲胺)与络合物缀合以获得更高的稳定性。结果是产生钆喷酸二葡甲胺(gd-dtpa-葡甲胺)。商业销售的钆喷酸二葡甲胺含有0.027-0.04%的非络合的(过量的)喷替酸污染物(sourcesofcontaminationinmedicinalproductsandmedicaldevices,p.157,denisebohrer)。没有理论要求在钆喷酸二葡甲胺的商业制剂中应该存在这种程度的过量喷替酸。过量的喷替酸的常见解释是它作为防止gd(iii)离子形成的安全特征提供。实际上,过量的存在部分是由于当干燥的钆络合物再水化时ph的变化。热力学稳定常数描述了一方面gd-络合物(gdl)的浓度与另一方面游离gd(iii)和游离配体(l)的解离浓度之间的平衡。由于游离配体比游离gd(iii)安全得多,增加游离配体的浓度可以抑制游离gd(iii)的形成。从这个意义上说,过量的游离配体可以被视为一种安全措施。实际上,发现在封闭环境中,在基于钆的造影剂的制剂中游离配体浓度的增加导致游离gd(iii)浓度的降低。这种保护效应在线性配体组中更为显著,其中钆喷酸二葡甲胺是一员,并且在循环配体中效果较差。通过使用过量配体将gdl络合物的浓度和各个络合物配偶体的浓度之间的平衡移至gdl络合物一侧,以维持通过热力学稳定常数描述的平衡。这种安全特征有点可疑,这在于一种剧毒成分被存在的毒性较低的成分取代,但是当gd(iii)和配体完全平衡且仅以gdl形式存在时,可以实现最高的安全程度。认为产品ph的变化取决于喷替酸与钆形成络合物的方式和程度是很自然的。然而,实际上,最终ph的变化主要起因于在溶剂沉淀步骤期间去除葡甲胺。其次,葡甲胺与螯合物的缔合是有意义的。并且这种缔合也可能受到存在的残留溶剂的影响。为了实现产品ph的低变化,控制其中形成钆-配体络合物的环境是重要的,并且特别地将钆络合物保持在水合状态(即在水性溶液中),可用于减少配体的酸酐状态的演变。喷替酸不是惰性化合物。对材料安全数据表(thematerialsafetydatasheet)的审查表明,喷替酸(二乙烯三胺五乙酸)具有多种潜在的健康影响:潜在的健康影响吸入,吸入有毒。引起呼吸道刺激。皮肤,如果通过皮肤吸收时可能有害。引起皮肤刺激。眼睛,引起眼睛刺激。吞食,摄入可能有害。本发明减少了预期的过量喷替酸的量,而不增加产品中gd(iii)离子的浓度。实现这一目标的一般方法是减少可以使平衡向gd(iii)转移的参数的可变性。向gd(iii)的转移可能起因于以下中的一种或多种:1)金属污染物导致转金属化,2)离子化污染物导致拉动配体远离gd(iii),以及3)热力学稳定常数本身的变化。通过在尽力阻止它们形成螯合物,可以减少或消除产品中的金属污染物。在这种情况下,可以利用含胺配体的优选性来在其它金属之前与钆形成络合物。因此,控制反应温度的精确性,并且特别是注意避免常见的转金属化反应的活化能特性,可以显著降低污染物诱导的转金属化的影响。可以通过引入能够与污染物结合并且比污染物部分更容易从反应中去除的清除剂种类(例如碳过滤),来减少配体中存在的倾向于通过排除gd(iii)而与配体络合的离子污染物。随着ph升高,热力学稳定常数增加(更高的稳定性)。对于热力学稳定常数的给定值,特定比例的gd(iii)和dtpa是最佳的。有意添加过量配体的实践产生于这样的需要,即补偿在通常的溶剂干燥过程期间由产品ph的制造可变性和配体损失引起的热力学稳定性的变化。当从结晶的药物产品中倒出过量的溶剂时,配体会损失。在基于钆的造影剂中使用过量配体的临床基本原理部分是基于接受造影剂的患者中nsf(肾源性系统性纤维化)的发生率增加。nsf是一种非常罕见的疾病,其迄今为止主要在具有严重肾功能损害的患者中观察到。nsf的病因尚不清楚,但被认为是多因素的。迄今尚未阐明引发nsf发展所必需的辅因子的特定组合和严重性。暴露于基于gd的造影剂(gbca)已被认定为获得这种严重和致残性疾病的潜在风险因素。该理论最初于2006年提出。先前还提出了许多其他的机制和潜在的风险因素,包括手术和/或血栓形成或其他血管损伤的发生、促炎状态以及高剂量促红细胞生成素的施用。医学文献中发表的研究表明,马根维显施用后nsf的发生率低于非离子线性gbca(欧乃影(omniscan))。由于离子线性gbca比非离子更稳定,这表明nsf可能与gbca中包含的金属-配体络合物的热力学稳定性有关。通过考虑二乙烯三胺五乙酸的酸酐状态,可以理解水在药物制备过程的所有步骤中的重要性。图2示出了二乙烯三胺五乙酸分子。在无水条件下位点a可以与位点b结合以形成两个六环结构并释放两个水分子。所得分子是二乙烯三胺五乙酸酐,或饱和的二乙烯三胺五乙酸二酐(如图3中所示)。二乙烯三胺五乙酸的酸酐的酸性低于二乙烯三胺五乙酸。这可以是观察到的当前产物ph的ph变化的来源,并且最终是热力学稳定常数变化的原因。例如,看二乙烯三胺五乙酸二酐(sigma-aldrich,st.louis,mo)的商业规格,可以看出,根据脱水程度,二乙烯三胺五乙酸二酐的颜色从无色变到很深的黄色。该观察结果与马根维显的标记一致,并且表明在马根维显中存在一些二乙烯三胺五乙酸二酐。此外,该污染物的来源可能与溶剂中钆络合物的无水沉淀有关。考虑到它的酸值:用naoh滴定97.5-102.5%,可以看出存在的二乙烯三胺五乙酸二酐的影响。在产生含有二乙烯三胺五乙酸二酐的产物的制造过程中,可以通过在缀合步骤之后过量加入二乙烯三胺五乙酸来滴定降至ph规格。当配制商业产品时,其以含水状态配制。因此,相对碱性的酐转变为更酸的二乙烯三胺五乙酸,这降低了产品的ph,降低了热力学稳定常数,并导致gd(iii)离子的形成。如果在所得络合物盐中仍存在酸性基团,则通常有利的是通过与形成生理上生物相容的阳离子的无机和/或有机碱或氨基酸反应来将酸性络合物盐转化为中性络合物盐,并分离它们。在许多情况下,该程序是不可避免的,因为在制备期间络合物盐的解离通过ph值的变化向中性移动到此类程度使得这是分离同质产品或至少可能进行其纯化的唯一方式。用有机碱或碱性氨基酸有利地进行生产。然而,通过钠、钾或锂的无机碱(氢氧化物、碳酸盐或碳酸氢盐)的方式进行中和也可能是有利的。本发明的新的合成钆喷酸二葡甲胺的方法涵盖所有上述考虑以提供产品,其:1)不包含溶剂残留物,2)具有7.2至7.5的产品ph变化范围,3)无色,4)具有18.2-18.6的产品热力学稳定常数变化范围,5)少于1ppm的游离gd(iii)离子,以及6)少于10ppm的非钆络合物与喷替酸。本发明的方法有利地将水性溶液用于药物产品浓度和纯度,而不需要获得钆络合物的干粉,这是有利的。在药物产品中不存在有机溶剂是本发明的新药化合物的明显特征。实际上,当在合成过程期间使用除水以外的溶剂时,不可能从钆造影剂中去除所有溶剂。所有目前的钆造影剂都具有可测量的溶剂污染物;并且在大多数商业产品中,一种或多种溶剂代表最多的杂质。通常,本发明的造影剂可以预先形成,或者可以可替代地通过在水性溶液中将螯合剂与含有顺磁性金属的可溶性化合物与生理上可接受的平衡离子混合来在施用前直接制备。通常,螯合实体本身是盐的形式。平衡离子也应该是生理上可接受的,并且可以例如是葡甲胺。此外,诸如丙酮、各种醇、庚烷等的溶剂与配体中存在的天然存在的杂质形成络合物。旨在产生结晶纯药物化合物的溶剂具有讽刺性地与药物产品中的杂质结合。最终,溶剂主要用作去除水的共沸剂而不是纯化剂。在本发明中未使用溶剂。产品钆喷酸二葡甲胺是吸湿的,并且难以与合成中使用的水分离。最终产品形式被配制成水性溶液。因此,重要的是确定钆喷酸二葡甲胺在批量产出中的最终质量或质量比例。这可以使用本文中公开的hplc分析技术来完成。确定将多少额外的水加入特定的水合批量产品中以获得所期望的产品制剂规格是一件简单的事情。实施例在下文中,成分的绝对重量和所用设备的体积的仅是说明性的,并且应该理解,成分之间的质量比是本发明的重要方面。在下表中给出了那些成分。成分摩尔质量量当量实施例中的量dtpa393.351摩尔393.35g/mol393.35kggd(iii)氧化物362.501/2摩尔181.25g/molgd181.25kgn-甲基葡糖胺195.212摩尔390.42g/mmol390.42kg以下程序是复杂平衡计算的结果,涉及质子化常数、热力学稳定常数和平衡形态图的计算。配体的纯化作为一个实施例,提供了用于纯化dtpa的程序。应该理解,该程序适用于形成用作医学成像中的造影剂的钆的络合物中使用的所有配体。在25-30℃下,在配备有机械搅拌器、冷凝器和加热套的5.0升四颈圆底烧瓶中,填充3000ml的蒸馏水(ph7)和500g的dtpa。搅拌10分钟后,将反应混合物的温度升至95-100℃,搅拌直至混合物形成澄清溶液,并在形成澄清溶液后在该温度下保持0.5hrs。将反应混合物冷却至35-40℃并搅拌1h。dtpa应作为微晶从溶液中沉淀出来。通过布氏漏斗过滤浆液并用蒸馏水洗涤。进行hplc并定量杂质,总杂质应少于0.5%n/n。若非如此,返回至步骤1。将纯化的dtpa在55-60℃减压下干燥1h。确定游离配体%重要的是确定钆和配体之间络合合成中的游离配体的量。在该实施例中,提供hplc程序以用于确定钆-dtpa络合物溶液中游离dtpa相比络合物重量的%w/w。色谱系统:仪器:agilent1200系列(或)等同物柱子:hypersilmos-1,150x4.6mm,5μ波长:195nm流动相:泵‘a’:10mm磷酸二氢钾水溶液,ph3.0,用正磷酸(0.1m)泵’b’含1.5mm四正丁基高氯酸铵的乙腈:水(20:80)等度a:b(30:70)流速1.5ml/min柱温箱:25℃注射体积:20μl运行时间:20分钟稀释剂:水定义:纯度:钆喷酸二葡甲胺相对于其他hplc峰的质量数量。纯度不是稀释特异性的,在取样的api中水的量不会改变纯度。湿api样品效力:钆喷酸二葡甲胺api的质量数量为在稀释剂(通常为水)中的质量数量。适用于进程内/湿api。一般注意事项:只要保持每种溶液的最终浓度,可以调整以下烧瓶尺寸和标准重量。可以使用超声处理或其他适当的方法来帮助溶解。混合每种溶液直至所有固体溶解。dtpa标准物制备:在稀释剂中制备含有0.0033mg/ml纯化的dtpa的溶液。在100ml容量瓶中称取约0.33mgdtpa,加入10ml稀释剂,在水浴中温和升温至60度并冷却。旋转溶解。用水来达到体积。api样品制备(湿的或干的):制备溶液,其产生在水中的产品值=(api浓度(mg/ml)x湿api效力)为5-10mg/ml的范围。换言之,当制得溶液时,其应满足:5<(api浓度(mg/ml)x湿api效力)<10mg/ml的api,在水中在100ml容量瓶中称取约500.0x(1/效力)mg的api,加入10ml稀释剂,充分混合。用稀释剂达到体积。通过hplc评估钆喷酸二葡甲胺的浓度满足目标规格。系统适用性的评估:dtpa保留时间应为约4.5分钟。色谱程序如下表中所示:描述#注射空白3dtpa标准物5api溶液2在api样品中,钆喷酸二葡甲胺峰将相当大,应调整hplc的mv比例,以使dtpa峰可辨别。应该视觉上验证api样品中的dtpa峰和钆喷酸二葡甲胺峰不重叠。重叠将会产生错误的积分面积。报告标准:仅对dtpa标准物和api中的dtpa峰积分。使用以下方程式中的一个计算相对于api的游离dtpa的%(w/w):(1)游离dtpa/湿api样品%(w/w)=apidtpa峰的面积×标准dtpa浓度(mg/ml)xdtpa的纯度x100%/dtpa标准物(n=5)的平均峰面积×api浓度(mg/ml)(2)游离dtpa/api%(w/w)=(1)x(1/湿api效力)从apihplc效力程序获得效力。确定钆-配体络合物的效力。在本实施例中,提供hplc程序用于测定钆-配体络合物相比钆-dtpa络合物的溶液的重量的%w/w。该方法通过具有uv探测器的hplc利用测定钆喷酸二葡甲胺药物的色谱效力:色谱系统:仪器:agilent1200系列(或)等同物柱子:hypersilmos-1,150x4.6mm,5μ波长:195nm流动相:泵‘a’:于水中的10mm磷酸二氢钾水溶液,ph3.0,用稀释的正磷酸(0.1m)泵’b’等度:a:b(30:70)流速:1.5ml/min柱温箱:25℃注射体积:20μl运行时间:20分钟含1.5mm四正丁基高氯酸铵的乙腈:水(20:80)只要保持每种溶液的最终浓度,可以调整以下烧瓶尺寸和标准重量。可以使用超声处理或其他适当的方法来帮助溶解。混合每种溶液直至没有固体残留。储备标准物制备:在稀释剂中制备含有0.5mg/ml钆喷酸二胺标准物的溶液。例如,在100ml容量瓶中称取约50.0mg的钆喷酸二葡甲胺标准物,加入30ml稀释剂,并轻轻旋转溶解,并用稀释剂稀释至体积。工作标准物制备:在稀释剂中制备含有0.005mg/ml的钆喷酸二葡甲胺标准物的溶液。移取2.0ml至200ml容量瓶中,加入稀释剂至标记并混匀。样品制备(api):在稀释剂中制备含有5.0mg/ml样品的溶液。在20ml容量瓶中称取约100.0mg的样品,加入10ml的稀释剂并轻轻旋转以溶解。用稀释剂达到体积。系统适用性的评估:标准溶液的前5次注射的保留时间的rsd%不大于2.0%。前5次注射和整个标准溶液运行期间的峰面积的rsd%不大于15%。在钆喷酸峰的保留时间处,稀释峰不应该显示大于来自工作标准物的分析的钆喷酸峰的峰面积响应的5%的任何干扰峰。工作标准溶液的拖尾因子不应大于2,基于工作标准溶液的第一次注射评估。仅对样品和工作标准物中的钆喷酸二葡甲胺峰积分。使用以下方程式计算湿api样品中的钆喷酸二葡甲胺api的效力(%w/w):api的效力/湿api样品(%w/w)=样品中钆喷酸二葡甲胺的峰面积×标准物浓度(mg/ml)×100%/工作标准物(n=5)的平均峰面积×样品浓度(mg/ml)确定钆-配体络合物的纯度在本实施例中,提供hplc程序用于确定非络合物部分(杂质)相比钆-dtpa络合物溶液中的络合物的重量的%w/w。该方法通过具有uv探测器的hplc测定钆喷酸二葡甲胺药物的色谱纯度:柱子:hypersilmos-1,150x4.6mm,5μm,部件#30205-154630纯水或hplc级,fisherscientificcat#w5-4或等同物乙腈,hplc级,fisherscientificcat#a996-4或等同物四正丁基高氯酸铵,alfa-aesarcat#30801或等同物磷酸二氢钾,acs级,bdhcat#bdh9268或等同物磷酸,hplc级,emdcat#px0996-6或等同物葡甲胺(n-甲基葡糖胺),acrosorganicscat#126841000或等同物钆喷酸二葡甲胺参考标准能够阅读0.01mg的分析天平标刻度的ph计“a”类玻璃量具溶液制备稀释的磷酸:移取5.0ml的磷酸至50-ml容量瓶中,并用纯水稀释至体积,混匀。流动相a:(10mm磷酸二氢钾水溶液)准确称取1.36g的磷酸二氢钾(kh2p04)并转移至已含有1000ml水的1lhplc瓶中,并混匀。根据需要按比例放大体积。使用稀释的磷酸调整至ph3.0。流动相b:(含1.5mm四正丁基高氯酸铵的乙腈:水/20:80)准确称取并转移0.51g的四正丁基高氯酸铵至1lhplc瓶中。加入200ml乙腈并溶解固体。加入800ml的纯水并将其混匀。根据需要按比例放大体积。稀释剂:纯的或hplc级水。葡甲胺41.5%的溶液:称取约41.5mg的葡甲胺至20ml容量瓶中。用稀释剂溶解并稀释至标记,并混匀。注意:在称量钆喷酸二葡甲胺标准物和样品时,使用防静电枪从小铲、手套、样品标准物瓶和称量盘或容量瓶去除电荷。储备标准物制备(0.5mg/ml)准确称取50mg±1mg的钆喷酸二葡甲胺参考标准物至100ml容量瓶中。向烧瓶中加入大约30ml稀释剂,并轻轻旋转或涡旋以溶解。用稀释剂达到体积。如有必要,超声处理约5分钟,以确保物质完全溶解。允许冷却至室温并充分混合溶液。储备标准物浓度(mg/ml)=标准物重量(mg)x十进制纯度/100ml。工作标准物制备(0.005mg/ml)准确移取2.0ml的钆喷酸二葡甲胺储备标准物至200ml容量瓶中。加入稀释剂至标记并混匀。转移标准溶液至hplc小瓶中,并密封用于分析。工作标准物浓度(mg/ml)=储备标准物浓度(mg/ml)x2.0ml/200ml。样品制备称取100mg±1mg的钆喷酸二葡甲胺样品至20rnl容量瓶中。向烧瓶中加入大约10ml的稀释剂至,并轻轻旋转或涡旋以溶解。用稀释剂达到体积。如有必要,超声处理约5分钟,以确保物质完全溶解。允许冷却至室温并充分混合溶液。将样品溶液转移至hplc小瓶中,并密封用于分析。样品浓度(mg/ml)=样品重量(mg)/20ml。仪器操作条件典型的起始柱压力为大约96巴(bar)。操作程序注射顺序:(在每5次样品制剂注射后注射标准溶液。)1.空白(3x,至少以确保干净的基线)2.工作标准溶液(5x)3.葡甲胺41.5%溶液(1x)4.空白(1x)5.样品溶液(2x,对于每个样品)6.空白(1x)7.标准溶液(1x)系统适用性1.在钆喷酸峰的保留时间处,稀释剂空白不显示大于来自工作标准溶液的分析的钆喷酸峰的峰面积响应的5%的任何干扰峰。2.工作标准溶液的拖尾因子为nmt2;基于工作标准溶液的第一次注射评估。3.标准溶液的前5次注射的保留时间的rsd%为nmt15%。4.第一次注射和整个标准溶液运行期间的峰面积的rsd%为nmt15。计算1.积分所有峰,其不包括空白和葡甲胺41.5%溶液注射中存在的峰(葡甲胺和相关的峰)。2.使用以下方程式计算钆喷酸二葡甲胺相关物质的%重量:%wt/wt=相关物质的峰面积x标准物浓度(mg/ml)x100%/工作标准物(n=6)的平均峰面积x样品浓度(mg/ml)实施例1:无溶剂钆喷酸二葡甲胺原材料原材料摩尔比1dtpa12gd2o30.4963水2.2v1(dtpa)4葡甲胺0.992制备dtpa溶液1.加热反应器至25-30℃2.加水3.开始搅拌(除非另有说明,搅拌,标称速率300rpm)4.加10%的dtpa5.搅拌直至在水中均匀分布6.如果加所有dtpa,则转到步骤87.转到步骤48.搅拌10min制备钆:dtpa络合物9.加25重量%的氧化钆10.搅拌直至均匀分布11.如果加所有氧化钆,则转到步骤1312.转到步骤913.搅拌10min14.升温至95+/-2℃15.搅拌3hrs。16.检查澄清度17.如果不澄清,则继续进行1hr,转到步骤1618.如果澄清,则继续1h,然后冷却至40-45℃19.如果形成沉淀,则加热至95+/-2℃并搅拌1hr,转到步骤16验证络合物制剂20.使用二甲酚橙验证不存在游离钆21.如果检测到游离钆,则加入0.05%额外的dtpa,升温至95+/-2℃,搅拌1hr并继续进行步骤1622.否则,继续进行步骤23制备钆喷酸二葡甲胺溶液23.在40-45℃下加入90%的葡甲胺24.搅拌直至溶液~1hr25.测量ph–内联(inline)探针校准至25℃(usp)26.如果ph为>7.5,则丢弃27.如果ph为7.0至7.5,则转到步骤2928.如果ph<7.0,则加入1%的葡甲胺,搅拌10min,转到步骤2529.40-45℃下搅拌1hr30.检查溶液澄清,若是则继续进行31,若否则重复29钆喷酸二葡甲胺溶液过滤31.测量ph–内联探针校准至25℃(usp)32.如果ph为7.0至7.5,则转到步骤3433.如果ph<7.0,则加入葡甲胺,搅拌10min,转到步骤3134.使用碳过滤器过滤溶液35.使用1/4v的20-25℃的水冲洗反应器36.使冲洗物通过过滤器37.重复冲洗步骤35&36,进行总共2次冲洗38.将滤液和冲洗物放回反应器中39.在40-45℃下搅拌10min40.使用二甲酚橙验证不存在游离钆41.如果检测到游离钆,加入0.05%额外的dtpa,搅拌1hr,并转到步骤4042.若是,则继续进行步骤4343.测量ph–内联探针校准至25℃(usp)44.如果ph为6.0至6.6,则转到步骤4645.如果ph<6.0,则加入葡甲胺。搅拌10min。转到步骤43。46.搅拌1/2hr。47.检查溶液澄清,若是则继续进行48,若否则转到步骤46验证纯度48.通过hplc测量纯度49.如果个别杂质>0.05%,则转到步骤31最终api调整50.通过hplc测量游离dtpa51.如果游离dtpa>0.06%ww,则转到步骤3152.如果游离dtpa为0.01-0.06%ww,则继续进行5653.如果游离dtpa<0.01%ww,则加入0.05%的dtpa54.搅拌1hr55.转到步骤5056.测量ph57.如果ph为6.0-6.6,则转到步骤5958.如果ph<6.0,则加入葡甲胺。搅拌10min。转到步骤56最终的api测试59.进行完全的api测试:钆含量;葡甲胺含量;测定;水含量;重金属比较:本发明和马根维显钆喷酸二葡甲胺的商标名称是马根维显。使用hplc,可以直接进行马根维显的溶剂含量与本发明的比较。图4描绘了可商购的马根维显中代表性样品的溶剂的百万分率。通过在制造过程中不使用溶剂,改善了所有杂质(包括非溶剂杂质)的量。在图5中,将马根维显(gd参考标准物)的杂质含量与用溶剂(gd溶剂样品)和使用无溶剂程序(gd无溶剂样品)“纯化”的本发明的络合物进行比较。这些数据说明了当从药物产品过程中去除溶剂时,杂质水平的总体改善。实施例2:无溶剂钆特酸葡甲胺制备dota溶液1.加热反应器至25-30℃2.加水3.开始搅拌(除非另有说明,搅拌,标称速率300rpm)4.加10%的dota(1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸)5.搅拌直至在水中均匀分布6.如果加所有dota,则转到步骤87.转到步骤48.搅拌10min制备钆:dota络合物9.加25重量%的氧化钆10.搅拌直至均匀分布11.如果加所有氧化钆,则转到步骤1312.转到步骤913.搅拌10min14.升温至95+/-2℃15.搅拌3hrs。16.检查澄清度17.如果不澄清,则继续1hr,转到步骤16(该步骤耗时约12小时,比马根维显合成慢)18.如果澄清,则继续1hr,然后冷却至40-45℃19.如果形成沉淀,则加热至95+/-2℃并搅拌1hr,转到步骤16验证络合物制剂20.使用二甲酚橙验证不存在游离钆21.如果检测到游离钆,则加入xdota,升温至95+/-2℃,搅拌1hr并继续进行步骤1622.若否,则继续进行步骤23制备钆:dota络合物23.在40-45℃下加入90%的葡甲胺24.搅拌10分钟25.测量ph–内联探针校准至25℃(usp)26.如果ph为>7.5,则丢弃27.如果ph为7.0至7.5,则转到步骤2928.如果ph<7.0,则加入2%的葡甲胺,转到步骤2429.在40-45℃下搅拌1hr30.检查溶液澄清,若是则继续进行31,若否则重复29钆特酸葡甲胺溶液过滤31.冷却溶液至20-25℃32.使用碳过滤器过滤溶液33.使用1/4v的20-25℃的水冲洗反应器34.将冲洗物通过过滤器35.重复冲洗步骤33&34,进行总共2次冲洗36.将滤液和冲洗物放回反应器中37.在25-30℃下搅拌10min38.通过hplc测量游离dota39.如果游离dota为0.01-0.06%ww,则继续进行4240.如果游离dota<0.01%ww,则加入0.03%ww当量的dota41.搅拌1/2hr并转到步骤3842.测量ph–内联探针校准至25℃(usp)43.如果ph为7.0至7.5,则转到步骤4544.如果ph<7.0,则加入葡甲胺。搅拌10min。转到步骤42。45.搅拌1/2hr。46.检查溶液澄清,若是则继续进行47,若否则重复45验证纯度47.通过hplc测量纯度48.如果个别杂质>0.05%,则转到步骤32最终api调整49.通过hplc测量游离dota50.如果游离dota>0.06%ww,则重复步骤32-4251.如果游离dota为0.01-0.06%ww,则继续进行5552.如果游离dota<0.01%ww,则加入0.03%ww当量的dota53.搅拌1/2hr54.转到步骤4955.测量ph56.如果ph为7.0-7.5,则转到步骤5357.如果ph<7.0,则加入葡甲胺。搅拌10min。转到步骤55最终api调整58.进行完整的api测试:钆含量;葡甲胺含量;测定;水含量;重金属可以以本领域已知的方式临床配制根据本发明的医学造影剂产品。例如,将钆喷酸二葡甲胺溶液在水性介质中稀释,然后将溶液或悬浮液灭菌。合适的添加剂包括,例如生理上生物相容的缓冲剂(如例如盐酸氨丁三醇),少量添加的络合剂(如例如dtpa),或如果必要,电解质类(例如氯化钠)。在本文中描述了本发明的优选实施方案。一经阅读前面的描述,对于本领域普通技术人员而言,那些优选实施方案的变化可以变得显而易见。发明人期望技术人员适当地利用此类变化,并且发明人希望本发明以不同于本文中具体描述的方式实施。因此,本发明包括适用法律所允许的在此所附权利要求中叙述的主题的所有修改和等同物。此外,除非本文中另有说明或上下文明显矛盾,否则本发明涵盖上述元素的所有可能变化的任何组合。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1