一种基于复杂网络的药物疗效筛选方法与流程
本发明属于具体涉及一种基于复杂网络的药物疗效筛选方法。
背景技术:
目前,对复杂疾病的药物研发,面临药效不足、耐药严重、药物不良反应评估不全面及个性化用药带来的挑战。由此,药物研发由单靶标模式向基于复杂生命系统的多靶标模式转变。即从复杂系统的整体网络水平,来全面理解疾病的发生发展机制,进而评估药物的整体效应,因而急需发展新的网络研究方法和工具。
传统的“一药一靶”药物研发策略已经被证明存在一定的缺陷,比如容易产生耐药性和毒副作用等。大多数疾病的出现不能用单基因缺陷来解释,而是涉及不同基因组协调功能的分解。另外,有些疾病还尚未开发出有效的治疗药物,而且有些药物并不能完全治疗该疾病。疾病的治疗要求药物可以作用于多个靶点和多条信号通路,并从多个靶点和通路上阻止疾病相关信号的发生、传导和作用。因此,要取得成功,药物开发必须将其重点从携带疾病相关突变的个体基因转向基于网络的疾病机制视角。将药物的靶标基因与未知疗效疾病的致病基因建立联系,以寻找最有效治疗该疾病的几种药物。这种方法对现有但未知如何治疗的疾病中具有非常重要的现实意义,并且可以使我们进一步地理解复杂网络在药物疗效筛选上的作用。
现有的药物疗效筛选方法,节点间的相似性就只是用节点与节点之间的距离间隔数来表示,从而得出疾病与药物的关联程度。这种方法准确率不够高,结果信息不够全面并且不具有针对性。同时收集到的药物与疾病的信息不够丰富,没有有效地利用机器学习中的节点间的相似度去判断药物的疗效。
药物开发成本的增加以及新药批准数量的显着下降,提出了对目标识别和功效预测的创新方法的需求。因此,利用我们对基于网络的疾病起源的日益了解,引入药物-疾病接近度量,量化药物目标和疾病之间的相互作用。将基因网络与机器学习相结合,使用蛋白质-蛋白质相互作用,药物-疾病关联和药物-靶标关联数据,得到药物靶标与疾病蛋白质的相似度。从而量化药物的治疗效果,并提供无监督的方法来揭示现有药物的新用途。
技术实现要素:
本发明目的就是针对现有技术的不足,提供一种基于复杂网络的药物疗效筛选方法。
本发明的目的是解决:本发明将收集到的近36万对基因关系数据,采用node2vec机器学习方法进行网络结构特征提取,进而准确高效地计算出节点间的相似度。利用得出的相似度,再计算出所有药物治疗每一种疾病的可能性。这种方法在技术上实现了所有药物对某种疾病的一个疗效预测,解决了现有方法的数据不够充分,结果准确率低,计算效率低下等问题。
本发明解决其技术问题所采用的技术方案包括如下步骤:
步骤1、数据采集与数据处理;
步骤2、药物疗效计算;
步骤3、基于网络的药物与疾病之间的接近度计算;
步骤4、结果展示与评测;
步骤1所述的数据采集与数据处理,具体实现如下:
1-1.从omim基因库和gwas基因库中采集疾病—基因关联数据;
1-2.对疾病—基因关联数据作预处理:对于输入的疾病a,在基因库中查询该疾病a的所有致病基因;
1-3.每一种药物都有许多的靶标基因组成;在所有的药物与靶标基因的关系数据中,将药物与靶标基因进行分类和统计,统计出每一种药物对应的所有靶标基因;首先将不同的药物放入数组drug[]中,然后将同一药物对应的多个靶标基因以元素的形式存为一个数组,即代表同一种药物的所有靶标基因。然后再连续地放在数组target[]中,即代表所有药物的靶标基因;
步骤2所述的药物疗效计算,具体实现如下:
2-1.基于网络的基因与基因之间的相似性计算
使用node2vec方法将基因网络中的基因节点向量化,引入了“randomwalks”的邻居节点选择方式;通过调用gensim库中的word2vec进行模型训练,具体步骤如下:
2-1-1.采用randomwalk来代替dfs(深度优先搜索算法)或者bfs(广度优先搜索算法),
在基因网络上通过随机游走的方式,产生一系列基因节点序列;该随机游走方式是一种有偏的游走策略,即从一个节点向下一个节点的游走概率是不同的,这个概率由回归参数p和输入输出参数q来控制,最终产生一系列的节点序列;记开始基因节点为c0=u,通过上述随机游走的方式,
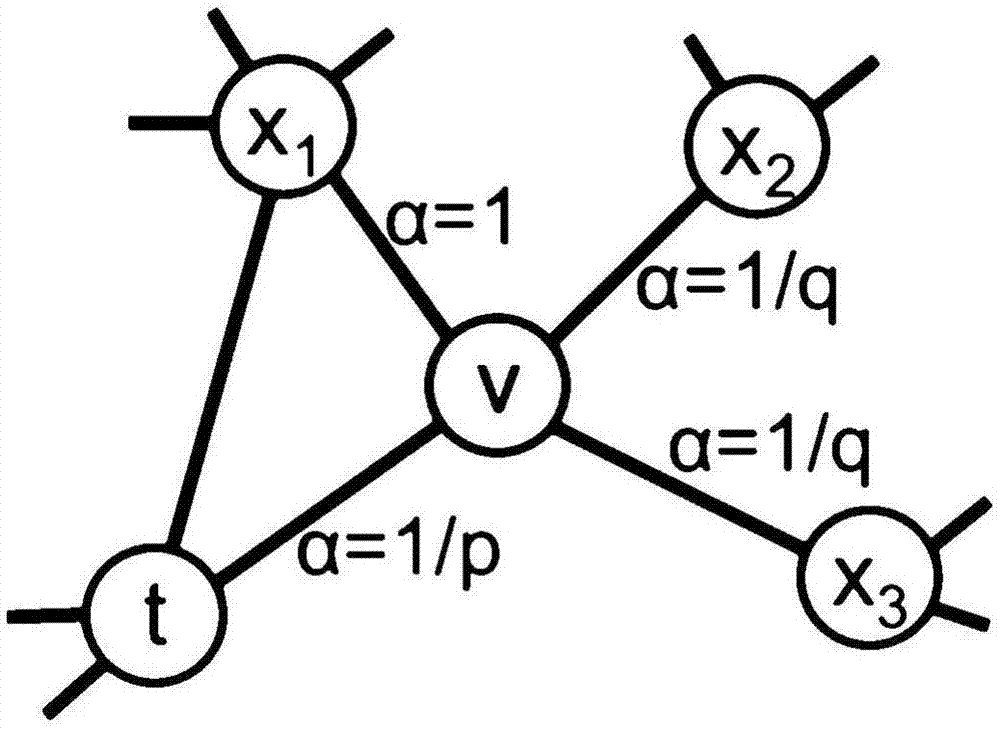
有偏的随机游走选择下一个基因节点的公式为:
即若图e中存在边(v,x),则以
即当下一个节点x与前一个节点t和当前节点v等距时,α=1;当下一个节点x是上一个节点时,
2-1-2.通过word2vec计算基因节点向量表征
将randomwalk得到每个基因节点的游走序列,通过用word2vec工具为每个基因节点生成一个特征向量,即实现基因节点向量化;
2-1-3.节点间距离的计算
通过欧式距离函数计算两个基因节点s和t之间的相似度值d(s,t)。
欧氏距离(euclideandistance),也称欧几里得度量、欧几里得度量,是一个通常采用的距离定义,它是在n维空间中两个点之间的真实距离。
n维空间的公式:
其中,d(x,y)为点(x1,x2,…,xn)与点(y1,y2,…,yn)之间的欧氏距离。
步骤3所述的基于网络的药物与疾病之间的接近度计算,具体实现如下:
要将药物与疾病的接近程度计算出来,就要考虑药物的靶标基因和疾病的致病基因之间的路径长度,通过各种距离测量来评估疾病和药物之间的接近度;设致病基因集合s,药物的靶标基因集合t;网络中基因节点s和t之间的最短路径长度d(s,t),其中基因节点s为致病基因集合s中的元素,基因节点t为药物的靶标基因集合t中的元素,使用以下公式计算出药物与疾病的接近度:
其中,公式中的d(s,t)指代两个基因节点s和t之间的相似度值,具体指疾病的蛋白质致病基因与药物的靶标基因之间的距离;公式(3)中dc(s,t)计算的是药物的每一个靶标基因与所有致病基因的距离取最小值,再对所有靶标基因求平均得到的是最近距离;
公式(4)中ds(s,t)计算的是药物的每一个靶标基因与所有致病基因的距离取平均值,再对所有靶标基因求平均得到的是平均距离,其中dc(s,t)或ds(s,t)越小,表示该药物的治疗效果越好。
步骤4所述的结果展示与评测具体实现如下:
针对疾病a,根据步骤3计算得出多种药物与疾病a的接近度表格,同时调用原有数据库中治疗疾病a的药物表格,将两个表格进行对比;使用roc曲线与x轴和y轴所包围的面积即auc值,评估接近度表格的正确率;
所述的roc曲线的横坐标的分母是所有药物数量n,n取值为2833;纵坐标的分母是药物表格中药物的个数k,分子是这些药物与前top个药物交集的个数;例如:roc曲线的横坐标的分母是所有药物数为2833,依次进行遍历得到1/2833、2/2833、3/2833……top/2833……,如图2所示。
本发明有益效果如下:
首先,本发明结合非监督学习计算出基于网络的靶标基因与致病基因之间的相似度方法,再依据最短距离和平均距离公式计算出药物对疾病的作用程度,最后基于roc曲线对药物效果进行评估。
综上所述,本发明由于采用了基因网络分析与机器学习相结合的方法,不同于传统方法中对药物疗效筛选的思路,进而较为准确地得出了基于网络的药物与疾病之间的接近程度。在一定程度上促进解决对罕见疾病的药物预测问题,对于临床决策具有重要的指导意义。
附图说明
图1是本发明在node2vec上的随机漫步过程说明图
图2是本发明得出的针对某一疾病的roc曲线图
图3是本发明方法流程
具体实施方式
以下结合附图和具体实施例来详细描述本发明。
如图1-3所示,一种基于复杂网络的药物疗效筛选方法。将收集到的近36万对基因关系数据,采用node2vec机器学习方法进行网络结构特征提取,进而准确高效地计算出节点间的相似度。利用得出的相似度,再计算出所有药物治疗每一种疾病的可能性。这种方法在技术上实现了所有药物对某种疾病的一个疗效预测,解决了现有方法的数据不够充分,结果准确率低,计算效率低下等问题,具体实现如下:
步骤1、数据采集与数据处理
1-1.从omim基因库和gwas基因库中采集疾病—基因关联数据;
1-2.对疾病—基因关联数据作预处理:对于输入的疾病a,在有基因库中查询该疾病a的所有致病基因;
1-3.每一种药物都有许多的靶标基因组成。在所有的药物与靶标基因的关系数据中,将药物与靶标基因进行分类和统计,统计出每一种药物对应的所有靶标基因。首先将不同的药物放入数组drug[]中,然后将同一药物对应的多个靶标基因以元素的形式存为一个数组,即代表同一种药物的所有靶标基因。然后再连续地放在数组target[]中,即代表所有药物的靶标基因;
疾病基因,药物基因和基因间相互作用数据:疾病种类大概一百多种,其中每种疾病对应的致病基因主要来源于两个库,分别是omim基因库和gwas基因库。因此,疾病—基因关联数据是在这两个库中检索而来。寻找的信息还包括一些在drugbank数据库中经过fda批准了的药物以及它们相对应的靶标基因。若要计算某种疾病与所有药物的关联度,这就必须要借助于基因—基因相互作用网络。为此,我们采集了大约三十五万对基因。该模块主要是对数据进行一些初步的处理。首先是对一种疾病,要知道它所有基因库中的致病基因。接着,将药物与靶标基因进行分类,即每一种药物有哪些基因。要将药物一一列出,为下一模块做准备。
步骤2、药物疗效计算
药物疗效的计算主要实现的功能是构建基因网络,并利用步骤1-2以及1-3中分类和统计后的疾病—基因和药物—基因关联数据,进行基因间的欧氏距离计算。最后得出每种经过fda批准的药物与罕见疾病之间的接近程度。具体内容详述如下:
2-1.基于网络的基因与基因之间的相似性计算
使用node2vec方法(一种对于图中的节点使用向量建模的方法)将基因网络中的基因节点向量化,引入了“randomwalks”的邻居节点选择方式。通过调用gensim库中的word2vec进行模型训练,具体步骤如下:
2-1-1.采用randomwalk来代替dfs(深度优先搜索算法)或者bfs(广度优先搜索算法),在基因网络上通过随机游走的方式,产生一系列基因节点序列。与普通随机游走不同的是,本发明采用一种有偏的游走策略,即从一个节点向下一个节点的游走概率是不同的,这个概率由回归参数p(returnparameter)和输入输出参数q(in-outparameter)来控制。记开始基因节点为c0=u,有偏的随机游走选择下一个基因节点的公式为:
即若图e中存在边(v,x),则以
对于回归参数p(returnparameter)和输入输出参数q(in-outparameter),如图1所示。即当下一个节点x与前一个节点t和当前节点v等距时,α=1;当下一个节点x是上一个节点时,
通过上述随机游走的方式,最终产生一系列的节点序列。
2-1-2.通过word2vec计算基因节点向量表征
word2vec是google于2013年开源的一款将词表征为实数值向量的高效工具,其基本思想是通过训练将每个词映射成k维实数向量。
将randomwalk得到每个基因节点的游走序列,然后使用word2vec工具为每个基因节点生成一个特征向量,即实现基因节点向量化。
2-1-3.节点间距离的计算
通过欧式距离函数计算两个基因节点s和t之间的相似度值d(s,t)。
欧氏距离(euclideandistance),也称欧几里得度量、欧几里得度量,是一个通常采用的距离定义,它是在m维空间中两个点之间的真实距离。
n维空间的公式:
其中,d(x,y)为点(x1,x2,…,xn)与点(y1,y2,…,yn)之间的欧氏距离。
步骤3、基于网络的药物与疾病之间的接近度计算
要将药物与疾病的接近程度计算出来,就要考虑药物的靶标基因和疾病的致病基因之间的路径长度,通过各种距离测量来评估疾病和药物之间的接近度。给致病基因集合s,靶标基因集合t;网络中基因节点s和t之间的最短路径长度d(s,t),其中基因节点s为致病基因集合s中的元素,基因节点t为靶标基因集合t中的元素,使用以下公式计算出药物与疾病的接近度:
其中,公式中的d(s,t)指疾病的蛋白质致病基因与药物的靶标基因之间的距离。公式(3)中dc(s,t)计算的是药物的每一个靶标基因与所有致病基因的距离取最小值,再对所有靶标基因求平均得到的是最近距离;公式(4)中ds(s,t)计算的是药物的每一个靶标基因与所有致病基因的距离取平均值,再对所有靶标基因求平均得到的是平均距离。
其中dc(s,t)或ds(s,t)越小,表示该药物的治疗效果越好。
步骤4、结果展示与评测
针对疾病b,根据其多种药物与疾病b的接近度表格,同时调用原有数据库中治疗疾病b的药物表格,将两个表格进行对比;
使用roc曲线与x轴和y轴所包围的面积即auc值,评估接近度表格的正确率。所述的roc曲线的横坐标的分母是所有药物数量n,n取值为2833;纵坐标的分母是药物表格中药物的个数k,分子是这些药物与前top个药物交集的个数;例如:roc曲线的横坐标的分母是所有药物数为2833,依次进行遍历得到1/2833、2/2833、3/2833……top/2833……,如图2所示。
- 还没有人留言评论。精彩留言会获得点赞!