一种单细胞转录组数据中分析双细胞的方法与流程
本发明属于单细胞转录组测序技术领域,具体地说本发明涉及一种单细胞转录组数据中分析双细胞的方法。
背景技术:
单细胞转录组测序技术(scrna-seq)在近年来发展迅速,相比于常规转录组测序而言,该技术可以将基因表达的分辨率从群体水平的平均值精确到单个细胞水平(具体可参见文献1、2)。尤其是从2015年以来刚刚兴起的基于微流控技术的各类海量单细胞测序技术(massivelyparallelsinglecellsequencing)(具体可参见文献3-5),可以将大量细胞分离成单细胞并给每个单细胞加上分子标签后联合扩增建库测序,避免了人工分离单细胞的繁琐实验流程。
具体而言,海量单细胞测序技术通过液滴或微孔阵列的微流控系统分离单细胞。根据泊松分布的原理,当细胞数远少于液滴数或微孔数时,大部分液滴或微孔为空,一部分液滴或微孔含有一个细胞,只有极少比例的的液滴或微孔含有2个或2个以上的细胞。由于总液滴数或微孔数一定,当细胞数增多时,双细胞(doublet)比例也随之提高。当分析10000个细胞时,双细胞比例通常情况下约为5%。该技术可以在双细胞率较低的基础上实现同时分析成千上万个单细胞。
然而,即使如上所述双细胞率相对较低,但在通量较高的实验中,双细胞个数也会达到几百个。这些双细胞基因表达谱在后续的数据分析中也可能会影响细胞亚型聚类、细胞分化状态分析、细胞亚型标志物分析等的结果,造成对生物学意义理解上的偏差或误导。因此,在数据中鉴别出双细胞可以进一步提高单细胞转录组实验的精度,降低实验噪音,减少后续分析错误的可能性。
在现有的实践之中,通常会采用两种方法鉴定并去除单细胞表达谱中的双细胞:i).去除总表达分子数偏高的细胞;ii).通过已知细胞类型的标志物,去除表达两种或两种以上细胞亚群标志物的细胞(如文献6、7)。但是第一种方法假设任意两个细胞表达的分子总数大于任意单个细胞的表达分子数。而事实上,单细胞表达谱的动态范围很大,且不同细胞类型的mrna总表达水平可相差几倍。因此,采用这种方法去除双细胞所基于的假设并不成立。而第二种方法依赖于已有的生物学知识积累,通过手动注释的方法,去除已知标志物的细胞类型混合而成的双细胞,但有可能错误的去除了细胞类型转化的中间状态。因此,第二种方法需要极高的知识背景,步骤繁琐,而且假阳性和假阴性都很高。
上述文献1-7具体为:
1.kolodziejczyk,a.a.,etal.,thetechnologyandbiologyofsingle-cellrnasequencing.molcell,2015.58(4):p.610-20.
2.shapiro,e.,t.biezuner,ands.linnarsson,single-cellsequencing-basedtechnologieswillrevolutionizewhole-organismscience.natrevgenet,2013.14(9):p.618-30.
3.fan,h.c.,g.k.fu,ands.p.fodor,expressionprofiling.combinatoriallabelingofsinglecellsforgeneexpressioncytometry.science,2015.347(6222):p.1258367.
4.klein,a.m.,etal.,dropletbarcodingforsingle-celltranscriptomicsappliedtoembryonicstemcells.cell,2015.161(5):p.1187-1201.
5.macosko,e.z.,etal.,highlyparallelgenome-wideexpressionprofilingofindividualcellsusingnanoliterdroplets.cell,2015.161(5):p.1202-1214.
6.griffithsja,scialdonea,marionijc:usingsingle-cellgenomicstounderstanddevelopmentalprocessesandcellfatedecisions.molsystbiol2018,14:e8046.
7.stoeckius,m.etal.cell"hashing"withbarcodedantibodiesenablesmultiplexinganddoubletdetectionforsinglecellgenomics.biorxiv(2017)。
技术实现要素:
本发明目的是:为了克服现有技术鉴定并去除单细胞表达谱中的双细胞存在的问题,提供一种自动化鉴定双细胞并拆分双细胞中细胞表达谱的方法。
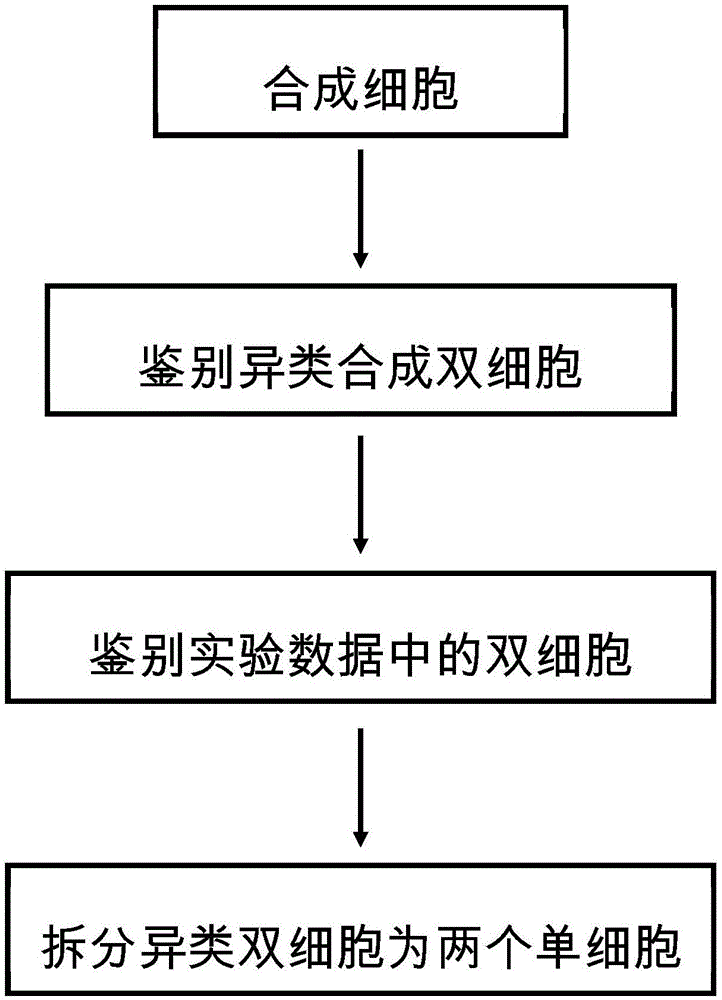
具体地说,本发明是采用以下技术方案实现的,包括以下步骤:
合成细胞的步骤:通过计算机模拟,基于实验数据合成细胞表达谱,得到合成细胞,所述实验数据为检测到的细胞数据;
鉴别合成异类双细胞的步骤:将合成细胞与实验数据合并,采用k近邻算法计算合并的数据集中每个合成细胞的近邻及近邻中合成双细胞的比例,并基于该比例采用聚类算法将合成细胞分成两类,其中高于分类阈值的那类为合成异类双细胞;
鉴别实验数据中的双细胞的步骤:将合成异类双细胞和实验数据合并,采用k近邻算法计算合并的数据集中实验数据中每个检测到的细胞周围近邻及近邻中合成异类双细胞的比例,并基于该比例采用聚类算法将检测到的细胞分为两类,其中高于分类阈值的那类为实验数据中的异类双细胞。
进一步而言,上述方案还包括拆分鉴别出的异类双细胞为两个单细胞表达谱的步骤:对每个鉴别出的实验数据中的异类双细胞,选取该异类双细胞所有近邻中的合成异类双细胞及各合成异类双细胞的来源单细胞,使用聚类算法把来源单细胞分为两类,得到两个单细胞亚型表达谱的分布,通过在两个单细胞亚群表达谱分布中多轮次随机抽样,采用最大似然估计寻找两个叠加之后和该异类双细胞表达谱距离最小的单细胞,作为该异类双细胞的两个替换对应单细胞。
进一步而言,所述鉴别合成异类双细胞的步骤或者鉴别实验数据中的双细胞的步骤中,所述聚类算法为kmean、dbscan、层次聚类或其他聚类算法。
本发明的有益效果如下:本发明既不依赖于对细胞类型的现有知识,也不依赖于对细胞总表达量的假设,而是在计算机模拟的基础上就可实现全自动化双细胞的鉴别。在此基础上,在双细胞鉴别出之后,通过拆分双细胞表达谱,可以将检测到的双细胞表达谱拆分成为2个单细胞表达谱。因此,采用本发明,可以无需去除鉴别出的双细胞,在最大程度上利用数据,提高实验中检测到细胞数的效率。
附图说明
图1:本发明流程示意图。
图2:本发明数据示例的同类双细胞与个异类双细胞的分类结果图。
图3:本发明数据示例的待检测细胞的近邻中合成双细胞的比例计算结果图。
具体实施方式
下面结合实施例并参照附图对本发明作进一步详细描述。
实施例1:
本实施例公开了一种自动化鉴定双细胞并拆分双细胞中细胞表达谱的方法,其原理是基于计算机模拟进行双细胞的鉴定分析,主要包括以下四个过程:
合成细胞:通过计算机模拟,基于实验数据,合成细胞表达谱。实验数据为检测到的细胞数据,由于实验数据中双细胞的比例很低,绝大多数为单细胞,所以绝大多数合成细胞将由检测到的单细胞数据中的两个随机来源单细胞叠加而成,即形成合成双细胞。
双细胞分为两种,同类双细胞由2个属于同种细胞类型的细胞混合而成,而异类双细胞由2个不同种类的细胞类型混合而成。故形成的合成双细胞也分为合成同类双细胞和合成异类双细胞两类。
而同类双细胞的表达谱和其来源细胞类型的单细胞表达谱高度相似,并不会对下游分析解读造成很大的影响;而异类双细胞具有两种细胞类型的表达特征,因此是需要专注的对象。
鉴别合成异类双细胞:合并合成细胞和实验数据,采用k近邻算法计算每个合成细胞的近邻及近邻中合成双细胞的比例,并以此给每个双细胞打分。而同类双细胞的近邻中将既有合成的同类双细胞,也有检测到的单细胞。而异类双细胞的近邻中将有合成异类双细胞和实验数据中待鉴定的异类双细胞。由于待鉴定的双细胞比例很小,异类双细胞的近邻将绝大多数为合成异类双细胞。可以基于这一原理,基于近邻中合成双细胞的比例采用聚类算法将合成细胞分成两类,其中高于分类阈值的那类为合成异类双细胞。
鉴别实验数据中的双细胞:通过合并合成异类双细胞和实验数据,采用k近邻算法计算实验数据中每个检测到的细胞周围近邻及近邻中合成异类双细胞的比例,实验数据中每个检测到的细胞即为每个待鉴定的细胞,并以此给每个待鉴定的细胞打分。和上面同样的原理,真实的单细胞的近邻大部分为其他单细胞,而检测到的异类双细胞的近邻中应含有较多的合成异类双细胞。再次采用聚类算法将检测到的细胞分为两类,其中高于分类阈值的那类为实验数据中的异类双细胞。
拆分鉴别出的异类双细胞为两个单细胞表达谱:对每个鉴别出的异类双细胞,选取该鉴别出的异类双细胞所有近邻中的合成异类双细胞,并找到每个近邻中的合成异类双细胞的两个来源单细胞。采用聚类算法将所有近邻的来源单细胞分为两类,并得到两个单细胞亚型表达谱的分布。这里的假设是表达谱相似的合成双细胞的来源细胞是来自于相同的两个细胞亚型。通过在两个细胞亚群表达谱分布中多轮次随机抽样,采用最大似然估计寻找两个叠加之后和该鉴别出的异类双细胞表达谱距离最小的单细胞,作为该鉴别出的异类双细胞的两个替换对应单细胞。这两个单细胞可在原检测到的实验数据中替换对应的异类双细胞。
上述的聚类算法可以为kmean、dbscan或层次聚类等聚类算法,其中对于kmean算法,采用k=2的k-mean聚类。
以下给出一个具体的计算实例:
1)设检测到的细胞数据可以表述成一个n*m的表达矩阵a,其中n为总细胞数,m为总基因数。矩阵a的元素为基因表达量,为整数值。从a中随机按行抽取2个1*m的向量(有放回),将2个抽取的向量按列求和,得到一个合成细胞表达向量。重复s次后得到一个s*m的合成细胞矩阵b,即得到s个合成细胞。
2)将矩阵a和b按列合并,并计算合并后的矩阵中所有s个合成细胞的k近邻。这里采用pearson相关系数或其他类似指标作为衡量两点之间距离远近的指标。k为用户可设参数。
3)计算s个合成细胞的近邻中合成双细胞所占比例{f1,f2,…,fs}。
4)通过k=2的k-mean聚类的方法计算最佳的分类阈值c将{f1,f2,…,fs}分为两部分,从而将合成细胞表达矩阵b分成两个矩阵b1、b2,其中b1包含近邻中合成双细胞比例低于阈值c的合成细胞,b2包含近邻中合成双细胞比例高于阈值c的双细胞(即合成异类双细胞)。
5)将矩阵a和b2按列合并,并计算合并后的矩阵中所有n个细胞的k近邻,计算采用的参数与上述步骤2)中参数相同。
6)计算n个细胞的近邻中合成异类双细胞所占比例{g1,g2,…,gn}。
7)通过k=2的k-mean聚类计算最佳的分类阈值c’将{g1,g2,…,gn}分为两部分。其中近邻中合成异类双细胞比例高于阈值c’的细胞注释为异类双细胞。
8)对于每一个表达矩阵a中的异类双细胞x,提取其所有近邻合成异类双细胞{n1,n2,…,nk}和各近邻合成异类双细胞的来源单细胞对{(p1,q1),(p2,q2),….(pk,qk)}。使用k=2的k-mean聚类算法将单细胞{p1,p2,…,pk,q1,q2,…,qk}分成2类d1、d2。从d1和d2两个分布中随机抽样合成双细胞y,并计算其与x的距离。采用最大似然估计,在1000次抽样中选择yi使得yi-x=min{y1-x,y2-x,…,y1000-x}。此时可以认为yi的两个来源细胞pyi,qyi的表达谱可近似为双细胞x拆分后的单细胞表达谱。
数据示例如下:
实验数据采用10xgenomics平台公开数据:50%:50%jurkat:293tcellmixture,其中总细胞数为3388,总基因数为32738。各步骤的相关情况如下:
1、通过1000次抽样,每次随机抽取2个细胞并求和,得到1000个合成双细胞。
2、合并实验检测到的细胞和合成双细胞后得到总共4388个细胞。
3、通过k=10的k近邻算法计算1000个合成双细胞的近邻。
4、计算1000个合成双细胞的近邻中,其他合成双细胞的比例。通过kmean聚类将1000个合成双细胞分成2类,得到220个同类双细胞,780个异类双细胞。如图2所示,图中最左侧两个立柱的上部(浅灰色部分)为3388个待检测细胞的近邻中的合成双细胞比例,其余部分为1000个合成双细胞的近邻中的合成双细胞比例。近邻中合成双细胞比例的阈值为0.4,左侧为检测到的单细胞和合成同类双细胞,右侧为合成异类双细胞。
5、合并实验检测到的3388个细胞,和步骤4中鉴定出的780个异类双细胞,得到共4168个细胞。通过k=10的k近邻算法计算3388个待检测细胞的近邻。计算3388个待检测细胞的近邻中合成双细胞的比例。如图3所示,为共4168个细胞的近邻中的合成双细胞比例。通过kmean聚类将待检测细胞分为2类,得到3349个单细胞,39个双细胞。近邻中合成双细胞比例的阈值为0.5,左侧为鉴定出的真实单细胞,右侧为鉴定出的双细胞和合成异类双细胞。
经过单细胞拆分步骤,将39个已鉴定出的的双细胞拆分为78个单细胞。将原始实验数据的3388个待检测细胞替换为3349个真实单细胞和78个拆分单细胞的集合。
虽然本发明已以较佳实施例公开如上,但实施例并不是用来限定本发明的。在不脱离本发明之精神和范围内,所做的任何等效变化或润饰,同样属于本发明之保护范围。因此本发明的保护范围应当以本申请的权利要求所界定的内容为标准。
- 还没有人留言评论。精彩留言会获得点赞!