一种利用纳米孔测序的转录组分析方法与流程
本发明属于分子生物学技术领域,具体涉及一种利用纳米孔测序的转录组分析方法。
背景技术:
转录组学是从整体转录水平系统研究基因转录图谱并揭示复杂生物学通路和性状调控网络分子机制的学科。在高通量测序技术发展以前,基于cdna杂交荧光检测的高通量基因表达芯片(expressionarray)和基因表达系列分析技术(serialanalysisofgeneexpression,sage)是从整体水平研究动植物组织中基因表达信息的主要手段。在2008年前后,高通量测序技术开始应用于细胞和组织中转录本(主要是mrna)的种类和表达量的研究,转录组测序(rnasequencing,rnaseq)这样的名词开始出现并被广泛应用。与基因表达芯片方法不同,rna-seq不仅能够检测与现有基因组序列相对应的转录本,并能发现和定量新的转录本,对选择性剪接事件、新基因和转录本以及融合转录本的研究更具优势,从而能更加系统地研究转录组学。2010年前后,三代测序技术(单分子测序技术)兴起,因其具有测序读长较长的优点,在研究全长转录本上具有二代测序短reads所不能达到的优势。
随着测序技术的发展和成本的降低,使得核酸的检测与定量更加便捷和准确,高通量测序在转录组学的研究上越来越普遍,大有替代基因表达芯片的趋势。运用现有的转录组研究手段系统、准确地探究从dna向rna转录这一复杂而精细的调控层次,是揭示生物学过程中的复杂性状和解析转录调控网络的重要方面。
三代测序技术也叫单分子测序技术(singlemoleculesequencing),具有超长读长(平均读长10-15kb,最长读长可达60kb、无pcr扩增偏向性及gc偏好性的特点,被认为是进行全基因组denovo组装、全长转录本测序及表观遗传学测序的理想测序平台。pacbio公司的单分子实时测序技术(singlemoleculerealtimesequencing,smrt-seq)和oxfordnanoporetechnologies的纳米孔单分子测序平台是目前主流的三代测序平台。
纳米孔测序技术(oxfordnanoporetechnologies)是最近几年兴起的新一代测序技术。与其他技术不同,nanopore是基于电信号而非光信号。该技术的关键是设计一种特殊纳米孔,在dna分子通过纳米孔的过程中实现测序。样本加到flowcell上,当dna分子通过纳米孔时,电荷发生变化,从而短暂影响通过纳米孔的电流强度,每种碱基所影响的电流变化幅度不同,通过检测电流变化鉴定碱基。目前市场上广泛接受的纳米孔测序平台是oxfordnanoporetechnologies(ont)公司的minion,gridionx5和promethion三款不同类型测序仪。ont测序的特点是单分子测序,测序读长长,测序速度快,测序数据实时监控,机器方便携带等。ont测序的读长很长,约几十甚至上百kb;测序通量高,起始dna在测序过程中不被破坏;ont测序样本制备简单便宜;数据可实时读取,也可直接读取甲基化的胞嘧啶,理论上也可检测rna。ont测序单碱基错误率高,为随机错误,可通过提高测序深度进而提高碱基准确率。
技术实现要素:
为了克服三代nanopore测序数据错误率高的问题,本发明提供一种利用纳米孔测序的转录组分析方法,通过对nanopore转录组测序数据进行网络形聚类并进行纠错,能够较大程度提高后续分析中nanopore数据的正确率。其具体技术方案如下:
一种利用纳米孔测序的转录组分析方法,包含如下步骤:
步骤1,比对获得序列之间的关系:
利用mimimap2软件对三代nanopore全长转录组reads之间进行比对,找出每条序列间的一一对应关系;通过比对果paf文件中的cigar值获得序列间比对的具体情况;
步骤2,计算序列之间比对的相似性、覆盖度、最大缺失、最大插入:
通过minimap2比对的结果,进一步挖掘序列之间的比对特征,同时通过设置similarity>=0.85、deletionall<=5.0、insertionall<=5.0过滤掉比对质量比较差的结果,剩下为质量较高的序列比对结果,将质量较高的序列比对结果用于后续聚类;
步骤3,利用carnac-lr软件进行网络状聚类,获得序列间网络状关系:
将质量较高的序列比对结果输入到carnac-lr软件中,同时设置coverage>=0.98作为权重进行网络状聚类,获得每条序列与所有序列间的网络关系,通过计算序列之间的网络状关系,将相似的序列聚集到一个cluster中,获得高度相似的序列集;
步骤4,纠错获得ccs:
过滤掉包含3条以下的cluster,以保证纠错后ccs的准确性;利用聚类后的cluster中的fasta文件,经过格式转换后使用pbccs软件进行纠错处理,利用pbccs纠错软件将subreads序列转换成ccs序列,获得高质量的ccs序列;经过纠错后获得的高质量ccs序列作为后续转录组分析的参考序列,用于计算基因的表达量;
所述步骤1中,minimap2软件参数为-xava-ont--cs=long-c,其它参数为默认;
所述步骤2中,比对特征包含match、coverage、similarity、deletionall、deletionmax、insertionall、insertionmax;
所述步骤4中,ccs为circularconsensussequence。
本发明的一种利用纳米孔测序的转录组分析方法,与现有技术相比,有益效果为:
一、本发明是基于三代nanopore测序平台,相对于二代illumina测序平台,其读长更长,无碱基偏好性,不用组装直接获取全长转录本序列,从而避免短reads组装导致的错误。
二、本发明使用三代nanopore测序平台,相对于三代pacbio测序平台,其建库过程中几乎没有经过pcr扩增,没有对序列进行多次重复测序,从而使其能够用于后续的定量分析。
三、本发明采用的是网络状聚类方式,可以获得每条序列与所有序列的网络状关系。从而可以将序列按照转录本水平进行区分,获得转录本水平的定量结果。
四、目前基于nanopore测序的三代转录组分析,大多数是通过二代数据对三代数据进行纠错从而获得较为准确的序列,用于后续作为参考序列、可变剪切体分析。本发明中巧妙的通过网络状聚类方式,利用三代nanopore序列进行自我纠错,从而克服nanopore平台测序错误率高的缺点。同时,在网络状聚类过程中获得了序列之间的网络状关系,为后续基因定量提供基础。
五、本发明方法基于网络状聚类并纠错三代nanopore转录组数据。目前,nanopore转录组数据主要是用来辅助二代转录组分析,从而获得更加准确的组装和可变剪切体结果。本发明通过纯三代nanopore转录组数据,先进行三代自我纠错获得高质量的ccs序列,然后进行转录组定量、可变剪接体等流程分析。
六、本发明通过多次网络状方式聚类,进行纠错。将纠错后的序列与原始参考基因组进行比对,其准确性最高可以达到99%,中位数为93%,可以满足后续分析的需要。
附图说明
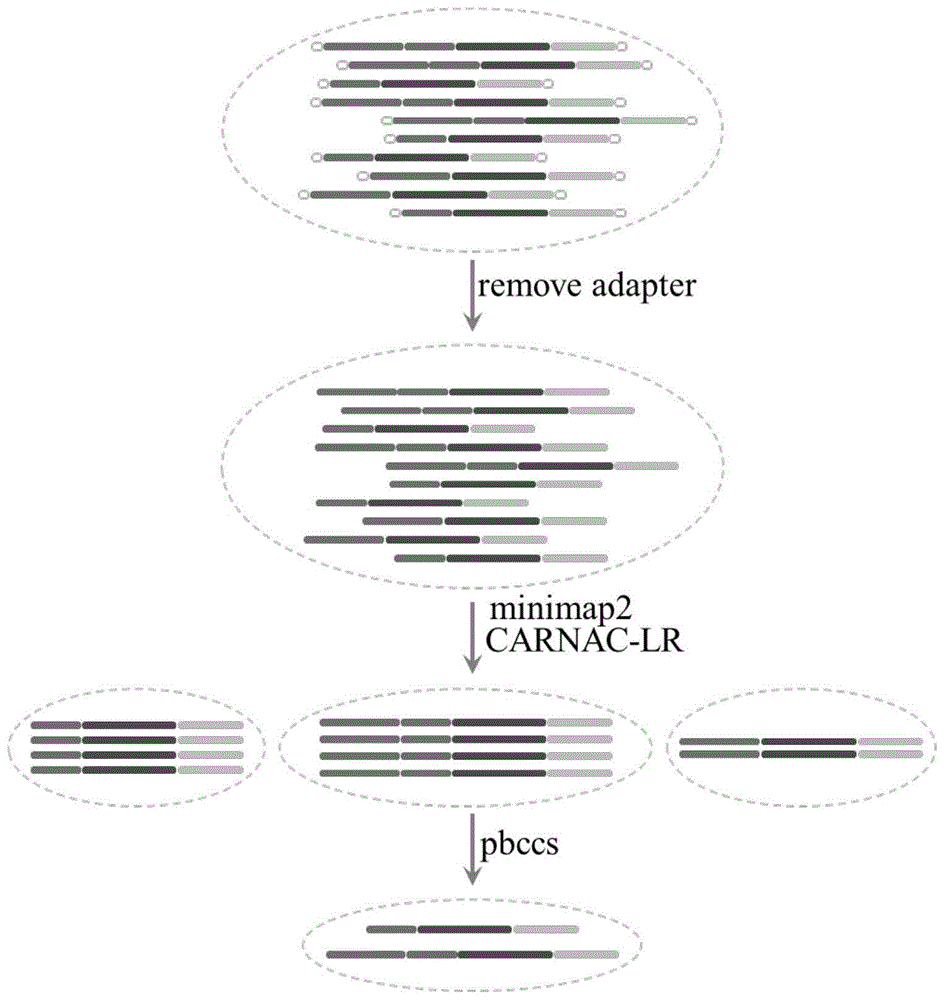
图1为本发明实施例1的nanopore转录组测序数据处理示意图。
具体实施方试
下面结合具体实施案例和附图1对本发明作进一步说明,但本发明并不局限于这些实施例。
一种利用纳米孔测序的转录组分析方法,包含如下步骤:
1)利用ont测序仪进行转录组测序
根据文库制备试剂盒操作说明,构建牛组织样品nanopore测序文库。使用oxfordnanoporetechnologies(ont)公司的minion测序平台进行高通量测序。本次测序一共测了6个牛样本,数据量约为5g。
2)数据去接头
通过脚本程序去除nanopore三代转录组数据的接头序列,以及统计测序数据的相关指标,如rawdata大小、reads总数、碱基总数、全长率、接头剪切率等。同时,将全长的nanopore三代全长转录组的全长reads挑选出来进行后续分析。
3)minimap2比对和比对特征计算
使用minimap2软件将挑选出来的全长转录组序列自我比对,minimap2软件参数为-xava-ont--cs=long-c,其它参数为默认;根据minimap2软件比对后生成的paf文件中的cigar值计算序列之间比对的特征,即readslength、match、deletionall、insertionall、coverage、similarity;同时通过similarity>=0.85、deletionall<=5.0、insertionall<=5.0过滤掉一部分质量低的比对结果,剩下为质量较高的序列比对结果,将质量较高的序列比对结果用于后续聚类;
4)reads聚类
将minimap2比对后的质量较高的序列比对结果输入到carnac-lr软件中,同时设置coverage>=0.98作为聚类的权重;通过计算序列之间的网络状关系,将相似的序列聚集到一个cluster中;由于将coverage作为聚类的权重,cluster中的序列两两间序列相似性和覆盖度高度相似,因此,聚集到一起的序列可以认为是转录本水平的聚类结果,该cluster中的序列也可以近似认为是该转录本的表达量;
5)纠错
获得nanopore转录组水平的聚类后,借鉴pacbio三代转录组的提高reads准确性的方法,将nanopore三代转录组数据聚类后的cluster作为subreads,利用pbccs纠错软件将subreads序列转换成ccs序列;经过纠错后获得的高质量ccs序列可以作为后续转录组分析的参考序列,用于计算基因的表达量等。
如图1所示,本发明实施例通过多次网络状方式聚类,进行纠错。一方面,将纠错后的序列与原始参考基因组进行比对,其准确性最高可以达到99%,中位数为93%,可以满足后续分析的需要。另一方面,我们可以通过序列间的网络状关系,进行基因定量分析。
- 还没有人留言评论。精彩留言会获得点赞!