一种含乌苯美司的前药衍生物的固体剂型及其制备方法与流程
本发明涉及药物化学领域,具体涉及一种含乌苯美司的前药衍生物的固体剂型及其制备方法。
背景技术:
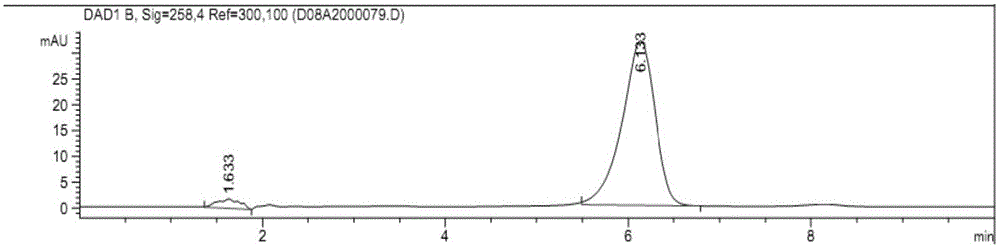
:乌苯美司(ubenimex,bestatin)是从网状橄榄链霉菌(streptomycesolivoreticuli)培养液中发现的一种二肽结构化合物,于1987年在日本上市作为免疫增强剂用于白血病的治疗;乌苯美司于1998年在国内上市。乌苯美司能够抑制apn活性(ic50在2.5-16.9μm),能够阻断肿瘤干细胞的作用,但本身几乎没有抑制肿瘤细胞增殖的能力,因而乌苯美司与其它细胞类抗肿瘤药物联合应用会大大提高其疗效。有文献研究表明,在小鼠肝癌移植瘤实验中,联合使用乌苯美司与五氟尿嘧啶明显优于单独分别使用抑瘤活性(jclininvest2010,120,3326-3339),这一研究为乌苯美司与其它化疗药物的联合应用提供很好实验依据。在临床上,乌苯美司可配合化疗、放疗及联合应用于白血病、多发性骨髓瘤、骨髓增生异常综合症,以及其它实体瘤等疾病。5-氟尿嘧啶(5-fluorouracil,5fu)是一类广泛应用于临床的抗代谢抗肿瘤药物,其抗肿瘤作用疗效确切且一直作为胃癌、直肠癌、乳腺癌等治疗的首选药物。但5-氟尿嘧啶使用中出现的较严重的骨髓抑制和胃肠道反应、半衰期短、选择性较差等不足限制了其广泛应用。近年来,研究者通过各种途径来减少或避免5fu不足,已合成许多5fu的前体药物并有一些已应用于临床,如fto、hcfu、cap、5'dfurd、bof-a2、atfu等。另外,5fu的n3的脲的衍生物通过消除原理形成原药5fu,其原理如下:来那度胺(lenalidomide)由美国gelgene公司开发,是沙利度胺的衍生物,临床上用于治疗骨髓增生异常综合症及与地塞米松联合用药治疗多发性骨髓瘤。来那度胺对细胞内多种生物途径都有影响。在临床i、ii期研究中发现其对多种肿瘤都有抑制作用,包括多发性骨髓瘤、前列腺癌、甲状腺癌、肾癌、黑色素瘤、肝癌、慢性淋巴细胞白血病。表阿霉素(epirubicin)是一类抗生素类抗肿瘤药,其结构为阿霉素的同分异构体。表阿霉素主要的抗肿瘤作用机制是能直接嵌入dna核碱对之间,干扰转录过程,从而阻止mrna的形成,起到抑制dna和rna的合成。此外,经研究发现,表阿霉素对拓扑异构酶ⅱ也有抑制作用,是一类细胞周期非特异性药物。其在临床上主要用于急性白血病、肾母细胞瘤、软组织肉瘤、膀胱癌、睾丸癌、前列腺癌、胃癌、恶性淋巴瘤、乳腺癌、支气管肺癌、卵巢癌、肝癌(包括原发性肝细胞癌和转移性细胞癌)等多种实体瘤。目前,关于表阿霉素的联合应用比较多,如与紫杉醇联合用、与索拉菲尼联合用于晚期乳腺癌等。阿糖胞苷(cytarabine)是胞嘧啶与阿拉伯糖形成的糖苷化合物,是dna聚合酶的竞争性抑制剂,能够抑制体内dna的生物合成,临床上主要用于白血病的治疗。但临床研究发现,阿糖胞苷存在一系列的缺点:脂溶性差、生物利用度低、毒副作用大等。因此,科学家研究阿糖胞苷与其它药物联合治疗相关疾病,从而改善其药物活性及化学稳定性,较大提高阿糖胞苷临床疗效(阿糖胞苷联合氟达拉滨、米托蒽醌等其它药物治疗各种白血病、淋巴瘤等疾病)。乌苯美司(bestatin)、5-氟尿嘧啶(5-fluorouracil,5fu)、来那度胺(lenalidomide,le)、阿糖胞苷(cytarabine,cy)、表阿霉素(epirubicin,ep)的化学结构式如下。目前,协同前药已成为针对恶性肿瘤的多重发病机制、多个病例环节设计药物的重要策略。药物在体内代谢降解,释放出多个药效结构后再分别结合不同作用部位而实现协同调节作用,可以产生更好的药效与降低毒副作用等。协同前药比联合用药具有单一的理化特性和均一的药代动力学特性,可以避免药物混合后组分间相互作用以及各组分吸收分布代谢不同步等问题。技术实现要素:本发明的目的是为了克服现有的抗肿瘤活性化合物各自存在的上述缺点,提供一种含乌苯美司的前药衍生物的固体剂型及其制备方法。本发明提供了一种含乌苯美司的前药衍生物的固体剂型,其含有药物活性组分、填充剂、崩解剂和润滑剂,以所述固体剂型的总重量为基准,所述药物活性组分的含量为20-80重量%,所述填充剂的含量为10-70重量%,所述崩解剂的含量为0.5-5重量%,所述润滑剂的含量为0.5-5重量%,其中,所述药物活性组分为式(i)所示的乌苯美司的前药衍生物或其光学异构体、非对映异构体、消旋体混合物及其药学上可接受的盐,其中,式(i)中r为具有抗肿瘤活性的化合物残基。优选地,r为优选地,以所述固体剂型的总重量为基准,所述活性组分的含量为50-80重量%,所述填充剂的含量为16-46重量%,所述崩解剂的含量为0.5-2重量%,所述润滑剂的含量为0.5-2重量%。优选地,所述填充剂为无水乳糖、乳糖一水合物、蔗糖、淀粉、糊精、微晶纤维素和预胶化淀粉中的至少一种。优选地,所述崩解剂为交联羧甲基纤维素钠、预胶化淀粉、羧甲基纤维素钠、聚乙烯吡咯烷酮和交联聚乙烯吡咯烷酮中的至少一种。优选地,所述润滑剂为硬脂酸镁、硬脂酸钙、硬脂酸锌、滑石粉、二氧化硅和聚乙二醇粉中的至少一种。优选地,所述固体制剂为片剂或胶囊,且每片剂或每胶囊包含20mg、50mg、100mg、200mg或400mg所述药物活性组分。本发明还提供了一种制备上述含乌苯美司的前药衍生物的固体剂型的方法,该方法包括以下步骤:(1)按比例量取所述药物活性组分、填充剂、崩解剂和润滑剂,过60-100目筛;(2)将过筛物料放在三维混合机中进行总混,三维混合机的转速为5~20rpm,时间为5~30分钟;(3)将步骤(2)得到的混合物料直接压成片剂或填装胶囊。在本发明中,采用药效团拼合原理将乌苯美司(bestatin)通过脲键与其它具有抗肿瘤活性的化合物(如5-氟尿嘧啶、来那度胺、表阿霉素、阿糖胞苷)融合,设计合成一系列新的协同前药。在体内酶作用下可代谢释放出乌苯美司和另一抗癌药物片段,乌苯美司发挥其抑制干细胞作用,具有抗肿瘤活性的化合物发挥其特定的药效作用,从而实现两个药物的协同作用。因此,本发明所述的含乌苯美司的前药衍生物的固体剂型可以延长药物在体内的滞留时间,改善药物的药代动力学性质,提高药物的生物利用度,从而达到更好的抗肿瘤效果。附图说明图1是制备例1制备的化合物5fu-bestatin的液相色谱图;图2是制备例1制备的化合物5fu-bestatin在人工胃液的稳定性图;图3是制备例1制备的化合物5fu-bestatin在人工肠液的稳定性图;图4是制备例1制备的化合物5fu-bestatin在人血浆的稳定性图。具体实施方式以下对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。本发明所述的含乌苯美司的前药衍生物的固体剂型含有药物活性组分、填充剂、崩解剂和润滑剂。在所述含乌苯美司的前药衍生物的固体剂型中,以所述固体剂型的总重量为基准,所述药物活性组分的含量为20-80重量%,优选为50-80重量%;所述填充剂的含量为10-70重量%,优选为16-46重量%;所述崩解剂的含量为0.5-5重量%,优选为0.5-2重量%;所述润滑剂的含量为0.5-5重量%,优选为0.5-2重量%。在本发明中,所述药物活性组分为式(i)所示的乌苯美司的前药衍生物或其光学异构体、非对映异构体、消旋体混合物及其药学上可接受的盐,其中,式(i)中r为具有抗肿瘤活性的化合物残基。在优选的实施方式中,在式(i)中,r为5-氟尿嘧啶残基、来那度胺残基、阿糖胞苷残基或表阿霉素残基,其结构分别如下:在上述优选实施方式中,作为药物活性组分的所述乌苯美司的前药衍生物的结构式如下:对于化合物五氟尿嘧啶-乌苯美司(5fu-bestatin),其制备方法可以包括:使五氟尿嘧啶在活性炭催化下与三光气反应,再与乌苯美司反应得到目标化合物5fu-bestatin。其具体合成路线如下:反应条件a:btc,活性炭,py,0℃,其中btc为三光气,py为吡啶具体制备步骤如下:将0.65g五氟尿嘧啶(5mmol)于反应瓶中,加入20ml吡啶使其全部溶解,后加入0.65g的活性炭,于冰浴下,缓慢加入0.5g三光气,继续在冰浴下反应1小时,抽出光气,冲入n2,进行三次。冰浴下,缓慢加入1.7g乌苯美司(5mmol),之后室温反应12小时。过滤活性炭,用乙酸乙酯萃取,6mol/l盐酸溶液洗涤3次,再用饱和食盐水洗涤3次,无水硫酸钠干燥。蒸出乙酸乙酯待有固体析出停止,放入冰箱进行结晶,过滤得白色产物。对于化合物来那度胺-乌苯美司(le-bestatin),其制备方法可以包括:使来那度胺在活性炭催化下与三光气反应,再与乌苯美司反应得到目标化合物le-bestatin。合成路线如下:反应条件a:btc,活性炭,py,0℃,其中btc为三光气,py为吡啶对于化合物阿糖胞苷-乌苯美司(cy-bestatin),其制备方法可以包括:使阿糖胞苷在活性炭催化下与三光气反应,再与乌苯美司反应得到目标化合物cy-bestatin。其合成路线如下:反应条件a:btc,活性炭,py,0℃,其中btc为三光气,py为吡啶对于化合物表阿霉素-乌苯美司(ep-bestatin),其制备方法可以包括:使表阿霉素在活性炭催化下与三光气反应,再与乌苯美司反应得到目标化合物ep-bestatin。其合成路线如下:反应条件a:btc,活性炭,py,0℃,其中btc为三光气,py为吡啶本发明的“药学上可接受的盐”是指那些在合理的医学判断的范围内适用于与人类和低等动物的组织接触而没有不适当的毒性、刺激性、过敏反应等并且与合理的效益/风险比相称的盐。在本发明中,所述填充剂可以为本领域的常规选择。在具体的实施方式中,所述填充剂可以包括但不限于无水乳糖、乳糖一水合物、蔗糖、淀粉、糊精、微晶纤维素和预胶化淀粉中的至少一种。在本发明中,所述崩解剂可以为本领域的常规选择。在具体的实施方式中,所述崩解剂可以包括但不限于交联羧甲基纤维素钠、预胶化淀粉、羧甲基纤维素钠、聚乙烯吡咯烷酮和交联聚乙烯吡咯烷酮中的至少一种。在本发明中,所述润滑剂可以为本领域的常规选择。在具体的实施方式中,所述润滑剂可以包括但不限于硬脂酸镁、硬脂酸钙、硬脂酸锌、滑石粉、二氧化硅和聚乙二醇粉中的至少一种。在一种优选实施方式中,在所述含乌苯美司的前药衍生物的固体剂型中,所述填充剂为乳糖一水合物和微晶纤维素的混合物,所述崩解剂为羧甲基纤维素钠,所述润滑剂为硬脂酸镁。在本发明中,所述含乌苯美司的前药衍生物的固体剂型可以为片剂或胶囊。在优选情况下,每片剂或每胶囊包含20mg、50mg、100mg、200mg或400mg所述药物活性组分。本发明还提供了一种制备上述含乌苯美司的前药衍生物的固体剂型的方法,该方法包括以下步骤:(1)按比例量取所述药物活性组分、填充剂、崩解剂和润滑剂,过60-100目筛(优选100目);(2)将过筛物料放在三维混合机中进行总混,三维混合机的转速为5~20rpm(优选10rpm),时间为5~30分钟(优选15分钟);(3)将步骤(2)得到的混合物料直接压成片剂或填装胶囊。以下将通过实施例对本发明进行详细描述,但本发明的保护范围并不仅限于此。制备例1将0.65g五氟尿嘧啶(5mmol)于反应瓶中,加入20ml吡啶使其全部溶解,后加入0.65g的活性炭,于冰浴下,缓慢加入0.5g三光气,继续在冰浴下反应1小时,抽出光气,冲入n2,进行三次。冰浴下,缓慢加入1.7g乌苯美司(5mmol),之后室温反应12小时。过滤活性炭,用乙酸乙酯萃取,6mol/l盐酸溶液洗涤3次,再用饱和食盐水洗涤3次,无水硫酸钠干燥。蒸出乙酸乙酯待有固体析出停止,放入冰箱进行结晶,过滤得白色产物,其为化合物5fu-bestatin,白色固体,产率:36.7%,熔点=165-167℃。ms-esi:[m-1]=463.6;[m-1+na]=485.8。其核磁共振氢谱数据如下:300mhz1hnmr1(d6-dmso)δ=0.65(d,j=5.7hz,3h),0.74(d,j=5.7hz,2h)1.49(m,1h),2.76-2.82(m,1h),2.88-2.96(m,1h)4.01(m,1h),4.21-4.28(m,1h),4.34-4.21(m,1h),6.69-6.71(d,1h),7.20-7.34(m,5h),7.72(d,1h),8.33(d,1h)9.44(d,1h)12.44(d,1h)12.66(s,1h)。按照与制备例1类似的方法,可以制备化合物le-bestatin、化合物cy-bestatin和化合物ep-bestatin。测试例1(1)体外抑酶活性1、材料和方法氨肽酶n、底物l-亮氨酸-p-硝基苯胺(购自sigma公司)缓冲液的配制,12.89gnah2po4·2h2o,溶解于1000ml容量瓶中,加新煮沸放冷的蒸馏水定容,得ph为7.2的50mm磷酸盐缓冲溶液,室温放置备用;氨肽酶n溶解在缓冲液中配成0.1iu/ml的溶液;底物l-亮氨酸-p-硝基苯胺溶解在dmso中配成16mmol/ml溶液,放置冰箱备用。2、实验步骤编号氨肽酶n溶液(μl)底物溶液(μl)缓冲液(μl)抑制剂100%组1051850空白组051950抑制剂组1051454096孔板中分别加入上述氨肽酶n溶液10μl,底物l-亮氨酸-p-硝基苯胺溶液5μl,不同梯度浓度的化合物40μl,以50mm磷酸盐缓冲溶液(ph7.2)补足200μl,100%组不含抑制剂。空白组用5μl底物以缓冲液补足200μl。37℃孵育0.5小时,于405nm波长处测定吸收值。按照如下公式计算抑制率:根据化合物的浓度与相应的抑制率,利用origin7.5软件拟合曲线,得到各化合物的ic50,结果如下表1所示。表1体外抑酶活性实验结果显示,本发明所述的目标化合物5fu-bestatin表现出对氨肽酶n的抑制作用,且与阳性药乌苯美司抑制活性相当。(2)体外药效实验(mtt法)以mtt法检测化合物对卵巢透明细胞癌es-2细胞株、人白血病k562细胞株、人前列腺癌pc-3细胞株、人结肠癌hct-116细胞株的生长抑制率。取指数生长期细胞,经hank's液洗涤后,以0.02%胰蛋白酶+0.05%edata消化分解单个细胞,台盼蓝(trypanblue)染色计数活细胞在95%以上。取细胞接种于96孔培养板中,细胞数5000/孔,加入不同浓度化合物(不加细胞的孔读数时作空白,加细胞不加化合物的孔作100%组,每组均设三个复孔,5fu-bestatin作为化合物阳性对照)。于37℃,5%co2中孵育48h,每孔加入100μl5%的mtt染色液,继续孵育4h后,2500rpm离心12min,然后抛板弃孔中培养基,加入dmso,100μl/孔。在酶标仪上于570nm(吸收波长),630nm(辅助波长)处测定每孔的吸光度od值,以od570-od630的差值进行计算。细胞生长抑制率按下式计算(公式中的od值即是od570-od630的差值):结果如表2所示。表2体外药效实验显示,本发明所述的目标化合物5fu-bestatin对上述细胞都有一定的增值抑制作用。与阳性对照药乌苯美司(bestatin)、五氟尿嘧啶(5fu)相比,对卵巢透明细胞癌es-2、人白血病k562、人前列腺癌pc-3、人结肠癌hct-116都表现了比乌苯美司、五氟尿嘧啶更好的增殖抑制活性。(3)体外稳定实验(hplc法)对化合物5fu-bestatin进行人工胃液、人工肠液、人血浆进行研究,为进一步动物实验提供依据。本实验中人工胃液、人工肠液的配制参考《中国药典》二部。色谱条件:venusilxbpc-18柱(4.6mm×150mm,5μm);流动相:乙腈-甲醇-0.2%甲酸水溶液(8-47-45,v/v/v);进样量:20μl;流速:1.0ml/min;检测波长:258nm;柱温:室温;化合物5fu-bestatin保留时间t=6.133min(如图1所示)。化合物5fu-bestatin的稳定性结果如图2、3、4所示:从图中可以看出,化合物5fu-bestatin在人工胃液稳定,在人工肠液和人血浆中缓慢降解。由此说明化合物5fu-bestatin如预期设计那样进入体内后能缓慢降解释放出原药。实施例1将化合物5fu-bestatin、乳糖一水合物、微晶纤维素、羧甲基纤维素钠和硬脂酸镁过100目筛。将过筛后物料放在三维混合机中进行总混,三维混合机的转速为10rpm,时间15分钟。将混合后物料直接压片或填装胶囊制备成10万个单位,每个单位含20mg化合物5fu-bestatin。实施例2将化合物le-bestatin、乳糖一水合物、微晶纤维素、羧甲基纤维素钠和硬脂酸镁过100目筛。将过筛后物料放在三维混合机中进行总混,三维混合机的转速为10rpm,时间15分钟。将混合后物料直接压片或填装胶囊制备成10万个单位,每个单位含50mg化合物le-bestatin。实施例3将化合物cy-bestatin、乳糖一水合物、微晶纤维素、羧甲基纤维素钠和硬脂酸镁过100目筛。将过筛后物料放在三维混合机中进行总混,三维混合机的转速为10rpm,时间15分钟。将混合后物料直接压片或填装胶囊制备成10万个单位,每个单位含100mg化合物cy-bestatin。实施例4将化合物ep-bestatin、乳糖一水合物、微晶纤维素、羧甲基纤维素钠和硬脂酸镁过100目筛。将过筛后物料放在三维混合机中进行总混,三维混合机的转速为10rpm,时间15分钟。将混合后物料直接压片或填装胶囊制备成10万个单位,每个单位含200mg化合物ep-bestatin。测试例2分别取10片实施例1-6中制备的固体剂型,在水介质中进行溶出曲线测量,检测并计算不同的时间的溶出度平均值,结果如下表2所示。表2由表2的数据可以看出,按照本发明所述的方法制备的固体剂型在10分钟内90%以上的药物活性化合物溶解于水中,符合固体剂型的溶出标准。以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1