一种快速导出末端限制性片段长度多态数据的方法与流程
本发明涉及一种快速导出末端限制性片段长度多态数据的方法,具体涉及到功能微生物末端限制性片段长度多态数据的整合及快速导出;本发明属于生物统计学领域。
背景技术:
末端限制性片段长度多态分析(t-rflp)是研究微生物生态的重要工具,其原理是根据基因序列保守区设计引物,其中一个引物5’端用荧光标记,通过pcr进行扩增,pcr产物经过限制性内切酶的消化后产生酶切片段,酶切片段通过测序仪进行基因扫描,扫描得到的信号强弱通过峰进行定量分析。不同长度的酶切片段代表不同种类的微生物,通过检测和定量得到微生物群落组成情况和相对丰度。
末端限制性片段长度多态是分析功能微生物群落组成和相对丰度的工具,酶切后的数据在abi测序仪产生的数据通常需要过程繁琐的提取、鉴定和分类等过程,目前主要通过人工操作完成,但是人工操作过于繁琐而且容易出错,特别是对于大通量的数据样品几乎无法靠人工来完成,如图1所示。本发明是基于genemapper软件导出数据繁琐,谱图校正困难等问题,利用色谱中常用的高斯函数对谱图进行拟合,提高数据导出的质量和效率。该技术的创新点在于基于高斯函数用r统计对t-rflp谱图进行批量校正、拟合、导出并实现数据处理可视化,易于操作。
技术实现要素:
本发明所要解决的技术问题是,克服现有技术的缺点,提供一种快速导出末端限制性片段长度多态数据的方法。
为了解决以上技术问题,本发明提供一种快速导出末端限制性片段长度多态数据的方法,其特征在于包含有以下几个步骤:
第一步、统计分析原始t-rflp数据
第1.1步骤、去除噪音峰图
利用标准物的峰高的最高值和最低值选择待测样品范围,将小于标准品最低值的峰高和大于标准品峰高的值去除;
第1.2步骤、校正与拟合重叠峰面积
(s1)、对重叠峰进行高斯函数校正可取得较好的校正效果,该数学模型为:
(s2)、上式简化为:
h(t)=s×z(σ,τ,tg,t);
其中,s为峰面积,tg、σ为高斯峰的中心位置与标准偏差,τ为指数衰减时间常数,t′为积分变量;
(s3)、保留时间为tr1和tr2的两个单峰发生部分重叠,构成峰顶时间分别为tp1和tp2,峰高为h1和h2的复合峰,假设两个峰拟合后面积分别为s1和s2,根据高斯函数得:
简化为:
从而峰面积可以计算得出:
通过归一化校正后的峰面积s拟合得到真实峰面积s′:
第二步、数据的导出和保存;
经过第一步对t-rflp图谱的杂峰进行去噪处理后,得到高质量的谱图,使数据更加准确。
第三步、数据的r统计语言进行数据合并和计算。
本发明的有益效果是:与常规的利用genemapper软件处理数据相比较,本方法更加快速,可以实现批量处理与导出数据,减少人工挑选图谱的误差。
附图说明
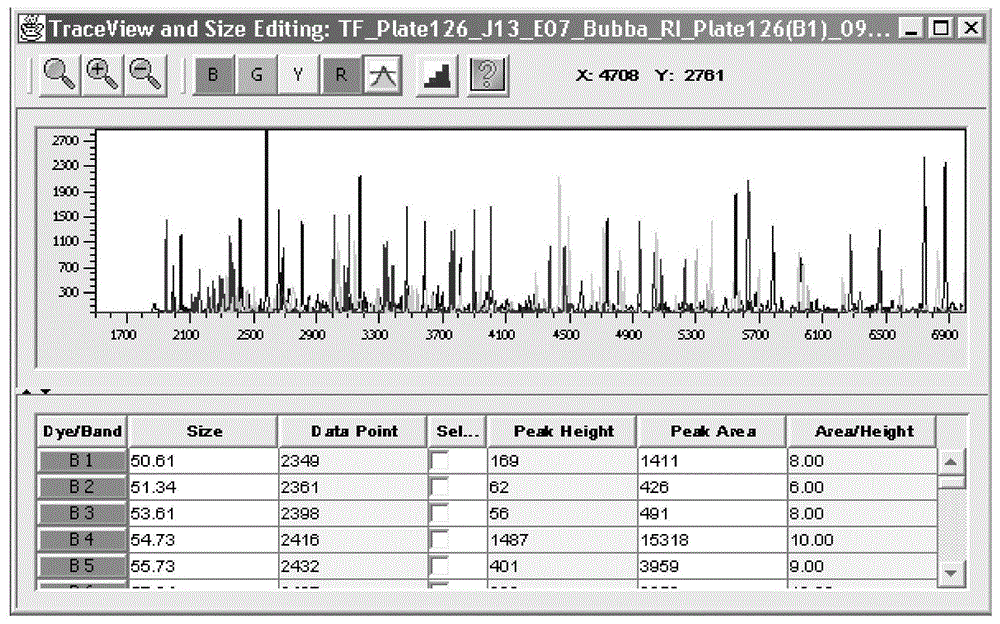
图1为abi测序仪原始数据在genemapper上的界面。
图2为产甲烷关键基因mcra扩增通过bstni酶切后的图谱。
图3为酶切位点起始点以及荧光染色的选择。
图4为文件保存路径。
图5为利用本方法导出产甲烷古菌酶切数据表。
图6为厌氧消化过程中产甲烷古菌群落nmds(非度量多维尺度)分析。
图7为原始数据导出后需要r进行数据合并和计算步骤。
具体实施方式
下面结合附图和具体实施方式对本发明做进一步的说明。
实施例1
本实施例提供的一种快速导出末端限制性片段长度多态数据的方法,包含有以下几个步骤:
第一步、统计分析原始t-rflp数据
第1.1步骤、去除噪音峰图
利用标准物的峰高的最高值和最低值选择待测样品范围,将小于标准品最低值的峰高<20和大于标准品峰高的值>5000去除;
第1.2步骤、校正与拟合重叠峰面积
(s1)、对重叠峰进行高斯函数校正可取得较好的校正效果,该数学模型为:
(s2)、上式简化为:
h(t)=s×z(σ,τ,tg,t);
其中,s为峰面积,tg、σ为高斯峰的中心位置与标准偏差,τ为指数衰减时间常数,t′为积分变量;
(s3)、保留时间为tr1和tr2的两个单峰发生部分重叠,构成峰顶时间分别为tp1和tp2,峰高为h1和h2的复合峰,假设两个峰拟合后面积分别为s1和s2,根据高斯函数得:
简化为:
从而峰面积可以计算得出:
通过归一化校正后的峰面积s拟合得到真实峰面积s′:
第二步、数据的导出和保存;
数据导出:
其中:
dyc/samplcpcak为引物用到的荧光标记物类型;
samplcfilcnamc为数据在λbi测序仪中的名称;
markcr为空白;
λllcl为空白;
sizc为序列读长,通常是空白;
hcight为峰高;
λrca为峰面积;
datapoint为λbi测序仪内部标记码;
第三步、原始数据导出后需要r进行数据合并和计算
如图7所示,以mcra扩增的产甲烷古菌为例,厌氧消化过程中产甲烷古菌利用t-rflp数据,并与环境因子结合分析得到图6。清晰地展示了厌氧发酵运行参数对产甲烷古菌群落组成的影响,表现在在nmds1&nmds2尺度上。
其主要步骤为:①厌氧发酵罐中采集到底物样品并进行总dna的提取;②利用产甲烷古菌功能基因mcra进行pcr扩增并对pcr产物进行纯化;③纯化后的pcr产物进行限制性内切酶的消化;④通过abi仪器对内切酶消化后的产物进行测序和检测荧光强度;⑤原始谱图如图1通过高斯函数进行校正和拟合后如图2。为了方便操作,代码进行了可视化编辑,使数据处理进一步简单化,通过对话框就能选择dna片段范围,如图3,并且所有的计算都在后台运行。图4为本方法文件保存路径;图5为利用本方法导出产甲烷古菌酶切数据表。
除上述实施例外,本发明还可以有其他实施方式。凡采用等同替换或等效变换形成的技术方案,均落在本发明要求的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!