基于吲哚美辛薄荷酯的药物组合物及其作为抗炎剂的用途的制作方法
本发明涉及药物和抗炎药,特别地,涉及基于吲哚美辛酯的药物组合物,其可用于治疗急性和慢性炎性疾病。
背景技术:
与所公开的化合物最接近的化合物(即本发明的原型)是吲哚美辛(1-(4-氯苯甲酰基)-5-甲氧基-2-甲基-1h-吲哚-3-乙酸),其是一种众所周知的非甾体抗炎药(nsaid),可发挥有效的抗炎、镇痛和解热作用。吲哚美辛以胶囊、片剂、栓剂、注射剂和其他全身作用剂型的形式广泛用于治疗关节炎和其他肌肉骨骼系统的炎性和退行性疾病、用于治疗不同原因的疼痛、用于一些感染性-炎性和肿瘤性疾病的联合治疗。
然而,吲哚美辛具有高毒性(口服给药于大鼠的ld50小于15mg/kg)并且在全身使用中诱导严重的不良反应。特别地,吲哚美辛发挥所谓的致溃疡作用,严重刺激胃肠道(gi道)器官的粘膜,引起溃疡和危险出血。这种致溃疡作用与吲哚美辛和其他nsaid的作用机制直接相关。吲哚美辛是两种形式的环氧合酶(cox-1和cox-2)的抑制剂,所述环氧合酶负责前列腺素的合成。当吲哚美辛在血液中达到一定浓度时,前列腺素的合成在炎症部位和胃粘膜中均被破坏,导致糜烂和溃疡。
研究了不同的吲哚美辛衍生物以降低吲哚美辛的胃肠毒性。合成了不同的酯、酰胺、曼尼希碱(mannichbase)和一些其它可水解的化合物。在一些情况下,这种修饰降低了致溃疡作用或改变了环氧合酶抑制的选择性[1;2]。专利[3]公开了含有吲哚美辛薄荷酯(mei)的药物组合物作为局部使用的眼科药物以用于治疗炎性眼病的用途。文章[4]还描述了几种吲哚美辛衍生物(包括其薄荷酯)的合成,以及它们作为nsaid的用途。mei的功效与其在生物体内的水解和具有有效抗炎作用的游离吲哚美辛的产生有关。然而,目前还没有基于吲哚美辛衍生物(包括mei)的具有全身作用的药物,所述药物将高抗炎活性和使用安全性(特别是低胃肠毒性)结合起来。其原因之一是抗炎作用的效力与不良致溃疡作用的严重程度之间的关系。在mei的情况下,存在第二个问题-该化合物的生物利用度低:与吲哚美辛不同,口服给药时mei实际上不会在胃肠道中吸收。因此,为了实现全身抗炎效果,必须开发特定的药物组合物,其将确保mei的生物利用度,达到吲哚美辛在血液中的有效浓度。
技术实现要素:
本发明的目的是创建基于mei的药物组合物,该药物组合物将在没有(或低严重程度)不良致溃疡作用的情况下确保吲哚美辛的生物利用度并达到其有效血液浓度,提供抗炎活性,这将允许使用基于mei的药物组合物作为具有全身作用的有效且安全的nsaid。
该目的通过创建基于mei的药物组合物来实现,该药物组合物确保其在没有不良致溃疡作用的情况下的高生物利用度和抗炎活性。这些药物组合物包含mei(5-甲基-2-(丙-2-基)环己烷-1-基]{[2-甲基-5-甲氧基-7-(4-氯苯甲酰基)-1h-吲哚-3-基]乙酸酯})和不同的药学上可接受的赋形剂。
在一个实施方案中,药物组合物包含脂质体包封的mei(负载有mei的脂质体)。脂质(优选磷脂,如卵磷脂,例如氢化大豆卵磷脂粉末或液体卵磷脂)用作脂质体的结构形成组分以便产生脂质体包封的mei。当使用卵磷脂时,需要加入增溶剂,例如甘油、乙醇或其他药学上可接受的增溶剂。药学上可接受的非极性溶剂(例如甘油三酯,包括辛酸和癸酸的中链甘油三酯)用作mei的溶剂(分子载体)。为了制备负载mei的脂质体,加入疏水性抗氧化剂(如抗坏血酸棕榈酸酯),渗透剂(如麦芽糖糊精)和其他药学上可接受的成分(如生育酚乙酸酯、棕榈酸视黄酯、红棕榈油、辅酶q)和基于脂质体包封的mei产生稳定的药物组合物所需的其他成分。
在其他实施方案中,药物组合物是基于mei和赋形剂的溶液或分散体系:非极性溶剂,如脂肪酸的甘油单酯、甘油二酯和甘油三酯和其他药学上可接受的非极性溶剂;醇、水和其他极性溶剂;乳化剂,表面活性剂,如卵磷脂、聚山梨酸酯和产生稳定的分散体系或溶液所需的其他药学上可接受的赋形剂,该溶液在水溶液和生物介质中会转变成稳定的分散体系。
基于mei的药物组合物也可以是栓剂,例如直肠栓剂,使用聚乙二醇(如peg400、peg(macrogol)1500、4000或6000),或固体脂肪(脂肪酸的甘油三酯)(如witepsol或supsuosyl)作为栓剂形成的基质。
基于mei的药物组合物可以包括在用于肠内使用(包括用于口服和直肠给药)的剂型中,或用于胃肠外使用(包括给药到生物体的皮肤和粘膜上,或通过注射(例如静脉内、皮下、肌内、关节内注射)或确保全身作用的其他方法)的剂型中,以及还包括用于局部使用的剂型中,除了滴眼剂和其他眼科组合物。基于mei的药物组合物可以包括在液体剂型(包括溶液、悬浮液和乳液);软剂型(包括软膏);栓剂和贴剂;固体剂型(包括片剂和胶囊),以及其他剂型中,这些剂型确保mei及其代谢产物吲哚美辛递送至炎性部位以用于治疗急性和慢性炎性疾病。
使用体外和体内模型研究所产生的基于mei的药物组合物,以评价它们在血液中的生物利用度和药代动力学概况,并且还评价抗炎活性和不良致溃疡作用。
实施例1
以下组分(按1个治疗剂量的mei计算)用于产生药物组合物,基于脂质体包封的mei的乳液:
1)mei物质(粉末)-10至100mg;
2)卵磷脂60%-10至100mg;
产生稳定的脂质体乳液所需量的其他成分,包括:
3)乙醇96%;
4)药用甘油;
5)麦芽糖糊精粉末;
6)角叉菜胶gs;
7)山梨酸钾;
8)苯甲酸钠;
9)grindox抗氧化剂;
10)纯净水。
由于mei是亲脂性的,通过将其与0.5/1-10/1比例的卵磷脂/mei(优选2/1比例的卵磷脂/mei)融合而将mei与磷脂结合。以该比例,当存储在凝胶中时,所产生的脂质体保持稳定,并且在脂质体膜中和在最终的脂质体凝胶剂型中实现了高浓度的活性剂。基于mei的物理性质选择卵磷脂与mei物质的融合温度且为75±5℃。在该温度下,该混合物具有所需的流动性和粘度以产生前体脂质体双层,随后它们自身闭合,产生球形囊泡。在该过程中,mei以其分子形式存在于熔体中,从而保证其在脂质体双层中的均匀分布。通过将熔融的卵磷脂/mei混合物与乙醇-甘油混合物以2/1的比例在25-55℃(优选40±5℃)的温度下混合来制备前体脂质体。通过以1000-10000rpm(优选2000-3000rpm)的速率均质化1-10分钟(优选1-2分钟),在与前体脂质体相同的温度下(优选在40±5℃下)制备脂质体乳液。脂质体乳液由角叉菜胶混合物(角叉菜胶gs)在75℃的温度下稳定。
实施例2
以下组分(按1个治疗剂量的mei计算)用于产生基于脂质体包封的mei的药物组合物粉末:
1)mei物质(粉末)-10至100mg;
2)氢化卵磷脂(粉末)-10至100mg;
3)麦芽糖糊精-20至100mg;
4)辛酸和癸酸的中链甘油三酯(mctg)-50至200mg;
产生稳定的脂质体乳液所需量的其他成分,包括:
5)grindox抗氧化剂;
6)纯净水。
将氢化卵磷脂和麦芽糖糊精以5/1至1/5的比例(优选1/1的比例)混合,并溶解于纯净水中,产生含有磷脂双层的粘性物质,然后在至少50℃的温度下加入脂溶性组分(分子载体mctg中的熔融mei)和剩余量的纯净水。以0.5/1至10/1的比例(优选1/1的比例)mctg/mei产生mctg中的mei熔体。使用的温度为75±5℃,在该温度下,mei以其分子形式存在于熔体中,确保其在脂质体双层中的均匀分布,而熔体具有向前体脂质体中给药所需的流动性。加入含有丁基羟基甲苯、丁基羟基茴香醚、抗坏血酸棕榈酸酯、卵磷脂和菜籽油的grindox抗氧化剂至浓度为脂肪质量的0.02-0.1%。为了生产脂质体乳液,将水中含有20%浓度干燥组分的混合物在35-70℃(优选55±5℃)的温度下均质1-10分钟(优选1-2分钟)。将脂质体乳液通过0.16-0.2mm的尼龙过滤器过滤,并使用喷雾干燥器在60-80℃(优选70℃)的温度下干燥,产生水分含量不超过5%的细分散粉末。
实施例3
以下组分用于产生基于mei的药物组合物,在水性介质中产生乳液的甘油三酯溶液:
1)mei物质(粉末)-10至100mg;
2)脂质(lipoid)c80-20至100mg;
3)聚氧乙烯脱水山梨糖醇单油酸酯(吐温80)-1至20mg;
4)中链甘油三酯(miglyol812n)-200至1000mg。
将预称重的miglyol812n放入100ml小瓶中,放入水浴中,打开搅拌器,并将miglyol加热至40-50℃。达到40-50℃的温度时,在搅拌的同时加入细研磨和预称重的脂质(lipoid)c80,将混合物搅拌30分钟,直到脂质(lipoid)c80完全溶解。然后,在以1/2至2/1(优选1/2至1/1)比例的脂质/mei持续搅拌的同时,将40-50℃下预称重的mei物质分成小份,并搅拌15分钟直到该物质完全溶解。产生均匀溶液后,在40-50℃下持续搅拌的同时加入预称量的吐温80,并搅拌5分钟。搅拌后,在40-50℃下加入剩余的miglyol812n,并搅拌10分钟,直到该溶液变得澄清和均匀。mei在miglyol812n中的良好溶解性以及两种表面活性剂(脂质(lipoid)c80和聚氧乙烯脱水山梨糖醇单油酸酯)的组合添加可确保当组合物进入胃中时形成稳定的乳液,从而提高mei的生物利用度。
实施例4
以下组分用于产生基于mei的药物组合物,在水性介质中产生乳液的脂肪酸的甘油单酯和甘油二酯的溶液:
1)mei物质(粉末)-10至100mg;
2)聚山梨酸酯20nf-5至15mg;
3)食品级脂肪酸的甘油单酯和甘油二酯(e471)-200至1000mg。
将预称重的e471放入100ml小瓶中,放入水浴中,打开搅拌器,并将e471加热至40-50℃。然后,在以1/1至20/1(优选5/1至10/1)比例的e471/mei持续搅拌的同时,将40-50℃下预称重的mei物质分成小份,并搅拌20分钟直到该物质完全溶解。产生均匀溶液后,在40℃下持续搅拌的同时,加入预称重的聚山梨酸酯20nf的量为混合物的2-15%,并搅拌10-15分钟,直到该溶液变得澄清和均匀。当与水或胃内容物混合后,所得的药物组合物可制得稳定的乳液。
实施例5
以下组分(按1个栓剂含有1个治疗剂量的mei计算)用于产生基于mei的药物组合物,即直肠栓剂:
1)mei物质(粉末)-10至100mg;
2)二甲基亚砜(dmso)-200至400mg;
3)吐温80-5至20mg;
4)苯甲醇-5至20mg;
5)纯净水-3至10mg;
6)栓剂基质-1.3至2.36g。
该栓剂具有以下物理化学特征:颜色-浅黄色,熔点-不高于35.5℃。
实施例6
以下组分(按1g软膏计算)用于产生基于mei的药物组合物,即软膏:
1)mei物质(粉末)-25至100mg;
2)二甲基亚砜(dmso)-150至300mg;
3)聚乙二醇(peg400)-20至100mg;
软膏基质:
4)聚乙二醇(peg4000)-200至300mg;
5)丙二醇-100至200mg。
将软膏基质放入研钵中,并在加热的同时在超声浴中熔融。将该物质放入研钵中,加入dmso和peg400,在研钵中混合直到该物质完全溶解,然后加入软膏基质并混合直到软膏变得均匀。所得的含有2.5-10%mei含量的软膏在60℃下30分钟不分离。
mei在生物体内代谢,产生其活性代谢物吲哚美辛,其具有有效的抗炎作用,同时也具有有效的不良致溃疡作用。然而,如果没有特定的赋形剂(如淀粉溶液或羧甲基纤维素中的悬浮液),mei几乎是无法生物利用的,它不会吸收到血液中并且不具有全身作用。因此,重要的是确定基于mei的组合物的药代动力学性质,特别是确定口服给药基于mei的组合物后血液中的吲哚美辛浓度。
实施例7
两种基于mei的组合物的药代动力学性质:
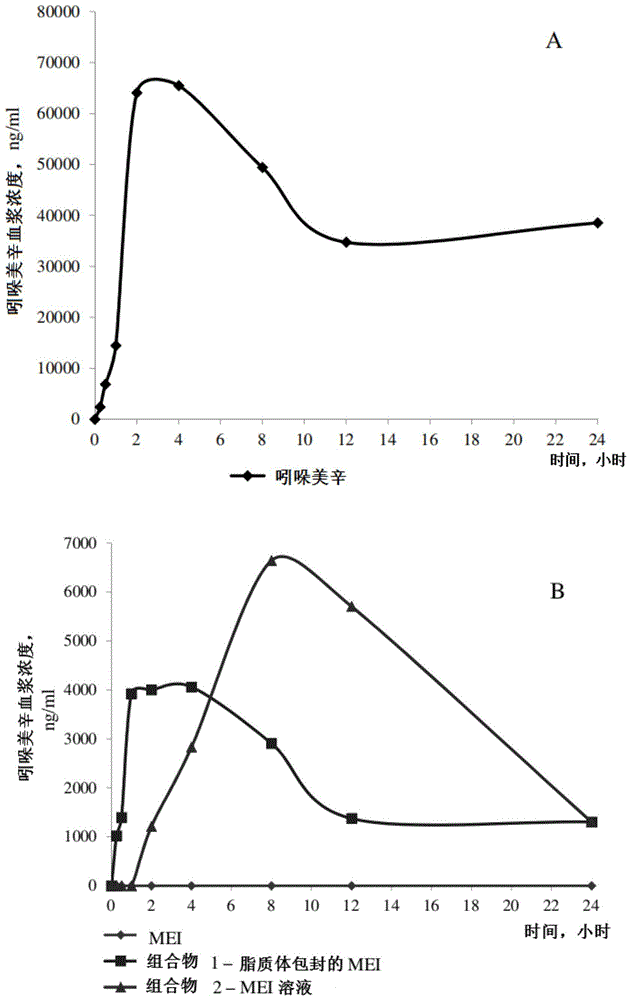
如实施例1所述产生的组合物1(脂质体包封的mei)和如实施例3所述产生的组合物3(mei溶液)。mei物质(在淀粉溶液中的悬浮液)和吲哚美辛用作参考药物。使用了斯普拉格-道利(spraguedawley)库存的大鼠。将所有测试化合物以25mg/kg的剂量口服给药于大鼠。在某些时间点(0;0.25;0.5;1,2,4,8,16和24小时)对动物血液进行采样以供分析。使用高效液相色谱(hplc)方法测定动物血浆中吲哚美辛的含量。给药测试化合物后吲哚美辛在血浆中的药代动力学概况如图1所示。
数据示出了口服给药吲哚美辛25mg/kg剂量后,其在大鼠血浆中的最大浓度(сmax)为65496ng/ml,达到最大浓度的时间(tmax)为4小时(图1a)。在以25mg/kg的剂量给药mei物质(作为淀粉溶液中的悬浮液)后,在任何时间点在血浆中均未检测到吲哚美辛,其浓度低于检测限,即这种形式的mei实际上是非生物利用的,并不能发挥其抗炎作用。在以25mg/kg剂量给药基于mei的药物组合物1和3后,吲哚美辛的cmax分别为6647和4061ng/ml,tmax分别为8小时和4小时(图1b)。因此,当以特定组合物(在脂质体或溶液中)给药时,mei是可生物利用的。同时,给药基于mei的组合物后血浆中吲哚美辛的最大浓度比给药吲哚美辛后血浆中吲哚美辛的最大浓度低约10-15,允许降低了不良全身毒性作用,例如致溃疡作用。
实施例8
在急性炎症模型(由炎性介质5-羟色胺诱导的爪肿胀)中研究了口服给药后与mei物质(作为淀粉溶液中的悬浮液)和吲哚美辛相比的基于mei的药物组合物的抗炎活性。使用了斯普拉格-道利(spraguedawley)库存的大鼠。通过在一只后爪中足底给药5-羟色胺(0.1mg/ml),在另一只后爪(对照爪)中足底给药盐水来诱导炎症。在给药5-羟色胺前2小时口服给药测试化合物。在给药5-羟色胺后30分钟使用肢体肿大容积测量仪(plethysmometer,意大利ugobasile)测量炎症的严重程度。通过肢体肿大容积测量术来测量炎症的严重程度,以对照与测试爪的质量之差表示,单位为ml。通过抑制指数评价测试化合物的抗炎活性,该抑制指数使用下式计算:
ii=((sct-st)/sct)×100%,
其中ii-炎症抑制指数,单位为%;
sct-对照组炎症的严重程度,ml;
st-测试组炎症的严重程度,ml。
研究结果如表1所示。
表1-基于mei的药物组合物在5-羟色胺诱导的肿胀模型中的抗炎活性(炎症抑制指数,%,n=8)
结果表明,口服给药后的mei(作为物质在淀粉溶液中的悬浮液)在急性炎症模型中没有明显的抗炎作用,而以25mg/kg剂量口服给药后基于mei的药物组合物具有有效的抗炎作用,仅比参考药物稍弱。因此,所开发的组合物确保了mei的生物利用度及其全身抗炎作用。
实施例9
在慢性炎症模型(佐剂诱导的类风湿关节炎大鼠)中研究了口服给药后基于mei的药物组合物的抗炎和镇痛活性。为了诱导关节炎,向大鼠右后爪的足底表面给药由sigmaaldrich提供的100ml完全弗氏佐剂(cfa)。对照组(完整)1接受了100ml盐水。注射cfa后,几个小时后发生局部炎症,以及随后掌指关节(metacarpophalangealjoint)和远端指间关节(distalinterphalangealjoint)的病理变化。从病理诱导后的第7天开始,每天口服给药一次测试化合物(组合物3-mei溶液)和参考药物(吲哚美辛),持续22天。以6.25、12.5和25mg/kg的剂量给药测试化合物。以1mg/kg的剂量给药参考药物吲哚美辛(以更高的剂量(5mg/kg)观察到动物死亡)。为了通过肢体肿大容积测量术评价mei在动物中的抗炎作用,将炎症严重程度测量为对照组与测试爪的质量之差,单位为ml。使用chaplan等人的方法[5]通过触觉反应性阈值(tactilereactivitythreshold)使用8个标准触觉测量套件(vonfreyhair,美国斯托尔廷(stoelting))来评价mei在动物中的镇痛活性。
实验结果如表2和表3所示。
表2-基于mei的药物组合物在佐剂诱导的类风湿关节炎模型中的抗炎活性(炎症严重程度,ml,x±se,n=8)
备注:
1-*-在检查日,与对照组2的差异在统计学上具有显著性,fisher准则(lsd测试),p<0.05;
2-**-在给药测试化合物之前在第7天进行测试。
表3-基于mei的药物组合物在佐剂诱导的类风湿关节炎模型中的镇痛活性(注射肢体中位有效触觉反应性阈值,g,x±se,n=8)
备注:
1-*-在检查日,与对照组2的差异在统计学上具有显著性,fisher准则(lsd测试),p<0.05;
2-**-在给药测试化合物之前在第7天进行测试。
结果表明,所有测试剂量的基于mei的药物组合物(6.25至25mg/kg)均发挥有效的抗炎作用,而在12.5和25mg/kg的剂量中,其可在慢性炎症模型(佐剂诱导的类风湿关节炎)中发挥有效的镇痛作用,其疗效不低于参考药物吲哚美辛。
实施例10
在角叉菜胶诱导的肿胀模型中研究了胃肠外给药后基于mei的药物组合物的抗炎活性[6]。使用了斯普拉格-道利(spraguedawley)库存的大鼠。将50ml盐水(“用于输注的0.9%氯化钠溶液”,俄罗斯npk)足底内注射到一只后爪(对照爪)中,而将50ml1%λ角叉菜胶溶液注射到另一只爪中。
4小时后使用肢体肿大容积测量仪(iitclifescience,美国)测量炎症严重程度。通过肢体肿大容积测量术将炎症严重程度测量为对照爪与测试爪的质量之差,单位为ml。在给药λ角叉菜胶前1小时肠胃外(腹膜内)给药测试化合物(药物组合物№4(mei溶液)),将接受相同量载体的动物用作对照。在研究前24小时,动物被剥夺了食物,但水的获取不受限制。
实验结果如表4所示。
表4-在角叉菜胶诱导的大鼠肿胀模型中腹膜内给药后基于mei的药物组合物的抗炎活性(x±se,n=7)
备注-*与对照差异在统计学上具有显著性,р<0.05。
结果表明,在急性炎症模型中以0.5至50mg/kg的剂量经肠胃外(腹膜内)给药基于mei的药物组合物可具有有效的抗炎作用。
实施例11
在角叉菜胶诱导的肿胀模型中研究了局部给药后基于mei的药物组合物(软膏)的局部抗炎活性。使用了斯普拉格-道利(spraguedawley)库存的大鼠。如实施例10所述产生角叉菜胶诱导的肿胀。在给药角叉菜胶后立即在大鼠后爪表面施用测试化合物(作为10%软膏的基于mei的药物组合物),然后再施用3次,每次施用之间间隔1小时。在施用测试化合物之后,将动物固定4-5分钟,然后返回笼子。将不含mei的软膏基质施用于对照组动物。在研究前24小时,动物被剥夺了食物,但水的获取不受限制。
实验结果如表5所示。
表5-在角叉菜胶诱导的大鼠肿胀模型中基于mei的药物组合物(10%软膏)的抗炎活性(x±se,n=8)
备注-*与对照差异在统计学上具有显著性,р<0.05。
结果表明,在急性炎症模型中局部给药基于mei的药物组合物(作为含有10%mei的软膏)可具有有效的局部抗炎作用。
实施例12
在酵母诱发的大鼠发烧模型中研究了基于mei的药物组合物的解热活性。使用了斯普拉格-道利(spraguedawley)库存的大鼠。给药测试化合物前18小时,通过以2ml体积皮下给药2000mg/kg剂量(以干酵母为基础计算)的贝克酵母(baker’syeast)悬浮液到动物体内,从而诱导大鼠发烧。使用电子直肠温度计(mlt1403直肠温度探针和powerlab4/354通道配准系统模块,adinstrumentsptyltd,澳大利亚)测量动物体温。在给药酵母前和给药酵母后18小时测量温度。之后,以25、50和100mg/kg的剂量向动物给药测试化合物(基于mei的药物组合物)(组合物3-如实施例3所述产生的mei溶液)。以10mg/kg的剂量的吲哚美辛用作参考药物。对照组中的动物已接收载体。在给药测试化合物后2和4小时测量温度。
实验结果如表6所示。
表6-在酵母诱导的大鼠发热模型中基于mei的药物组合物的解热活性(n=6)
结果表明,在酵母诱导的发热模型中口服给药基于mei的药物组合物(溶液)可具有有效的解热作用。
实施例13
为了评价不良致溃疡作用的严重程度,将测试化合物(组合物1-mei溶液)和参考药物(吲哚美辛)口服给药给大鼠4次。最后一次给药后3小时,处死动物,摘除它们的胃,在最小曲线处解剖并用盐水洗涤以除去内容物。使用以下4分制来测量致溃疡作用:
0-无损坏;
0.5-胃粘膜轻度充血;
1-胃粘膜的弱局部损坏(单点出血,严重充血);
2-胃粘膜的弱分布性损坏(小溃疡,糜烂,多发性出血);
3-胃粘膜的严重且分布性损坏(糜烂,溃疡,出血);
4-整个胃严重损坏(大量出血,糜烂,溃疡,穿孔)。
实验结果如表7所示。
表7-基于mei的药物组合物的致溃疡作用的严重程度
结果表明,接受四次5mg/kg剂量吲哚美辛的动物有多个溃疡和糜烂,而接受25和50mg/kg剂量的基于mei的药物组合物的动物没有胃粘膜疾病,而接受最大剂量为100mg/kg的基于mei的药物组合物的动物只有弱点损坏。因此,与吲哚美辛不同,以研究剂量四次给药的经测试的基于mei的药物组合物不具有致溃疡作用。
上述实施例表明,所公开的基于吲哚美辛薄荷酯的药物组合物的肠内、肠胃外和局部给药可以允许吲哚美辛达到有效浓度,具有足够的有效抗炎活性,而没有负面不良致溃疡作用。因此,所公开的基于mei的药物组合物可以用作有效和安全的nsaid。
参考文献
1.uspatentno.6,306,890.
2.uspatentno.6,762,182.
3.uspatentno.8,097,646.
4.sawrajsh.,bhardawajt.r.,sharmap.ddesign,synthesis,andevaluationofnovelindomethacin–antioxidantcodrugsasgastrosparingnsaids.medchemres(2012)21:834–843doi10.1007/s00044-011-9589-1.
5.chaplan,s.r.,etal.quantitativeassessmentoftactileallodyniaintheratpaw.//j.neurosci.methods.1994.vol.53.p.55-63.
6.methodicalrecommendationsforpreclinicalresearchofnon-steroidalanti-inflammatorydrugs./guidelineonpreclinicalresearchofdrugs.moscow:grifandco.part1.2012.p.748-760.
- 还没有人留言评论。精彩留言会获得点赞!