光敏剂及其制备方法与流程
本发明涉及一种光敏剂,具体涉及一种纳米粒子上转换光敏剂。
背景技术:
肿瘤治疗技术主要包括手术切除、化疗、放射性治疗及光学治疗等。其中,光学治疗主要有光热治疗和光动力治疗。光热治疗是利用具有光热转换的材料,在外部光源的激发下能用产生的热能来杀死癌细胞的方法。而光动力治疗是在外部光源的作用下激发光敏剂产生活性氧进而导致细胞损伤及坏死的治疗方法。虽然光热治疗与光动力治疗得到了广泛的研究与报道,但是肿瘤具有很强异质性及其复杂的微环境,一般的光热治疗及光动力学治疗很难将其完全消除。
光热与光动力学联合治疗方法能有效克服肿瘤细胞的异质性,已被科学家们证实。但是,现今的联合光学治疗大多都是用较短波长光激发或多种波长激发光源,降低了治疗的效果以及操作性。而光热与光动力治疗的核心就是光敏剂。现今的光敏剂只能用于光动力治疗,而无法兼具光热与光动力治疗两方面。
因此,设计一种光热与光动力治疗光敏剂具有非常重要的研究意义与应用价值。
技术实现要素:
本发明的目的是提供一种光敏剂载体及其制备方法,以解决现有的光敏剂载体不能支持光热治疗的技术问题。
本发明的另一目的是提供一种光敏剂及其制备方法,以解决现有的光敏剂不具备光热治疗的功能的技术问题。
为了实现上述发明目的,本发明一方面提供了一种光敏剂载体,所述光敏剂载体为纳米级的核壳结构,所述核壳结构包括核体和包覆于所述核体的壳体,所述核体为聚多巴胺纳米颗粒,所述壳体包括gd离子化合物、yb离子化合物和er离子化合物。
优选地所述gd元素、yb元素和er元素的摩尔比例为70-80%:15-20%:1-5%。
优选地,所述光敏剂载体的粒径为120-160nm;
所述核体粒径为50-70nm;
所述壳体的厚度为70-90nm。
本发明另一方面提供了一种光敏剂,包括载体和结合于所述载体上的光敏剂功能成分,所述载体为所述的光敏剂载体,所述光敏剂功能成分为上转换光敏剂。
优选地,所述光敏剂功能成分与载体比值10%-15%。
优选地,所述上转换光敏剂功能成分包括光敏剂ce6分子。
本发明又一方面提供了一种光敏剂载体的制备方法,包括如下步骤:
在聚多巴胺纳米颗粒上包裹稀土碳酸氢氧物层;
将稀土碳酸氢氧物层转换成上转换发光层。
本发明又一方面提供了一种光敏剂的制备方法,其特征在于,包括如下步骤:
在聚多巴胺纳米颗粒上包裹稀土碳酸氢氧物层;
将稀土碳酸氢氧物层转换成上转换发光层。
本发明还一方面提供了一种光敏剂的制备方法,其特征在于,包括如下步骤:
在聚多巴胺纳米颗粒上包裹稀土碳酸氢氧物层;
将稀土碳酸氢氧物层转换成上转换发光层
在所述的上转换发光层上修饰活性基团;
在修饰有活性基团的上转换发光层上装载光敏剂。
优选地,所述活性基团包括羟基、羧基和氨基中的任意一种或多种。
光敏剂在制备光热光动力治疗药物方面的应用。
与现有产品相比,本发明的光敏剂载体结构简单,但却能兼具光热和光动力治疗两种功能。一方面选取的聚多巴胺纳米核心光热效应十分显著,使得所述光敏剂载体的光热性能突出;另一方面选取上转换纳米粒子作为壳体可以使长波长低能量,穿透性强的近红外光转变为能量高的短波长光从而激活光动力治疗。
所述光敏剂由于采用了所述光敏剂载体,因此也具备光热和光动力治疗两种功能,充分利用光能对需要治疗的部位双重破坏,效果更佳。并且还具有突出的光热性能,优秀的生物组织穿透性,并且相比短波长光照组织伤害更低。
所述光敏剂载体的制备方法,步骤精简,操作简单,也没有实用毒性高的溶剂,也不产生污染高的副产物,所选取的反应转化简单,产率高,具有很高的实用性。
所述光敏剂的制备方法由于采用了所述光敏剂载体,一方面具备步骤精简,操作简单,实验过程绿色;另一方面装载光敏剂功能成分的手段也十分简单有效,总体制备方法是简洁高效,绿色无害。
因此本发明所述的光敏剂可以被很好地应用于光热和光动力治疗中。
附图说明
图1本发明实施例所述光敏剂载体的合成步骤示意图;
图2为本发明实施例聚多巴胺纳米粒子a和光敏剂载体b的扫描电镜图;
图3本发明实施例提供的pda@ucnp的光热效果评价图;
图4是本发明实施例提供的pda@ucnp在980nm近红外光激发下的发射光谱及光敏分子的吸收光谱图。
具体实施方式
为了使本发明要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
本发明实施例一方面提供了一种光敏剂载体,所述光敏剂载体为纳米级的核壳结构,所述核壳结构包括核体和包覆于所述核体的壳体,所述核体为聚多巴胺纳米颗粒,所述壳体包括gd离子化合物、yb离子化合物和er离子化合物。本发明的光敏剂载体采用的是光热纳米材料,具有很强的光热性能,而不是像其他的光敏剂材料核体也是采用的上转换纳米材料。因此多具备一种光热性能。而采用的稀土元素的吸收波长是近红外区,并且通过上转化将两份长波长的近红外光转化为波长为一半的五百纳米以下的光,高能激发单线态氧,同时产生较强的光热效应。两者相互配合,达到消灭病灶的目的。
所述gd元素、yb元素和er元素的摩尔比例为70-80%:15-20%:1-5%。一方面多元素的掺杂可以使的吸收范围变广;二来由于各种元素的吸收强度发射强度都不尽相同,因此调整用量来平衡其总体强度。
所述光敏剂载体的粒径为120-160nm;
所述核体粒径为50-70nm;
所述壳体的厚度为70-90nm。
较小的粒径可以更充分的吸收能量传递能量,限于工艺条件,取条件所述的粒径范围。
本发明实施例另一方面提供了一种光敏剂载体的制备方法,包括如下步骤:
s01:在聚多巴胺纳米颗粒上包裹稀土碳酸氢氧物层;
s02:将稀土碳酸氢氧物层转换成上转换发光层。
在本发明实施例s01中,实现稀土碳酸氢氧物层的包裹是将包含稀土元素的盐溶解,加入聚多巴胺纳米颗粒并与碱性容易挥发的物质共热回流,得到稀土碳酸氢氧物层包裹的聚多巴胺颗粒。
在本发明实施例s02中,具体的所述稀土碳酸氢氧物层加入氟化物和氢氟酸,形成稀土元素的氟化物。
本发明实施例又一方面提供了一种光敏剂,包括载体和结合于所述载体上的光敏剂功能成分,载体为上述光敏剂载体,所述光敏剂功能成分为上转换光敏剂。
所述光敏剂功能成分与载体比值为10%-15%。需要足够的光敏剂载体才能让光敏剂功能发挥完全,过多的量则会造成浪费,因此选取此范围比例。
所述上转换光敏剂功能成分包括光敏剂ce6分子。这几种光敏剂的吸收波长与稀土元素上转换后的发射波长重合度较高,因此选取这几种光敏剂。
本发明还一方面提供了一种光敏剂的制备方法,其特征在于,包括如下步骤:
s01:在聚多巴胺纳米颗粒上包裹稀土碳酸氢氧物层;
s02:将稀土碳酸氢氧物层转换成上转换发光层。
s03:在所述的上转换发光层上修饰活性基团;
s04:在修饰有活性基团的上转换发光层上装载光敏剂。
本发明实施例步骤s03和s04中所述活性基团包括羟基、羧基和氨基中的任意一种或多种。具体的选取带有两种活性基团的连接化合物,先选择性的连接上稀土元素,然后通过另一个基团与光敏剂连接完成装载。
所述的光敏剂可被用在制备光热光动力治疗药物。具体的可以添加一些辅料,使之效果更好。
光敏剂及其制备方法(其中前面的步骤就是光敏剂载体及其制备方法,就不再单独列出)。
实施例1
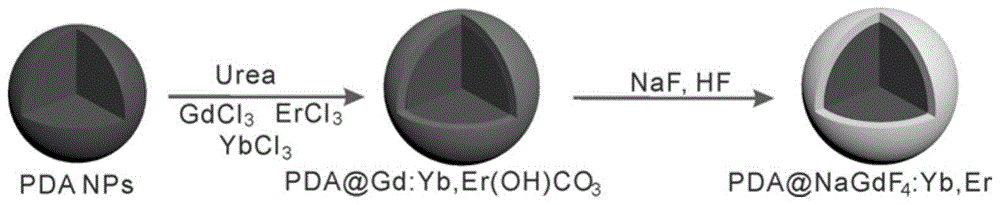
光敏剂的合成步骤如图1所示。
在20ml的乙醇-水(1:1)的混合溶液中,加入1ml稀土元素比例为78%:20%:2%(gdcl3:ybcl3:ercl3)的溶液,磁力搅拌10-30min形成透明溶液。加入步骤(1)中合成的3mg聚多巴胺纳米颗粒粉末,超声10-30min。随后将溶液在90℃下回流6h,即得到pda@gd,yb,er(oh)co3纳米颗粒。
将pda@gd,yb,er(oh)co3纳米颗粒分散于5ml水中,加入5ml乙二醇及20mgnaf,搅拌10min,转移至反应釜中,在180℃条件下处理8h。
将得到的纳米颗粒离心洗涤后分散于5-15ml乙醇-水溶液(1:1)中,加入0.1-0.5ml1mol/lnaf及0.1-0.5ml3mol/lhf溶液,搅拌10-30min,在100-180℃条件下处理4-12h,离心得到pda@ucnp纳米颗粒。
图2b所示为所得到的上转换发光层包覆的聚多巴胺纳米颗粒(pda@ucnp)的形貌图。图3为上转换发光层包覆的聚多巴胺纳米颗粒(pda@ucnp)在980nm近红外光以1w/cm2功率激发下的光热升温图。
将(2)中合成的pda@ucnp与多爪聚乙二醇(mpeg-pmhc18)按质量比1:1混合搅拌24h,洗涤,得到pda@ucnp-peg纳米颗粒。
加入光敏分子ce6乙醇溶液,搅拌12h,离心得到pda@ucnp-peg/ce6纳米探针。
图4为上转换发光层包覆的聚多巴胺纳米颗粒(pda@ucnp)在980nm近红外光激发下的发射光谱及光敏分子(ce6)的吸收光谱图。
实施例2
在20ml的乙醇-水(1:1)的混合溶液中,加入1ml稀土元素比例为60%:20%:20%(gdcl3:ybcl3:ercl3)的溶液,磁力搅拌10-30min形成透明溶液。加入步骤(1)中合成的3mg聚多巴胺纳米颗粒粉末,超声10-30min。随后将溶液在90℃下回流6h,即得到pda@gd,yb,er(oh)co3纳米颗粒。
将pda@gd,yb,er(oh)co3纳米颗粒分散于5ml水中,加入5ml乙二醇及20mgnaf,搅拌10min,转移至反应釜中,在180℃条件下处理8h。
将得到的纳米颗粒离心洗涤后分散于5-15ml乙醇-水溶液(1:1)中,加入0.1-0.5ml1mol/lnaf及0.1-0.5ml3mol/lhf溶液,搅拌10-30min,在100-180℃条件下处理4-12h,离心得到pda@ucnp纳米颗粒。
图2b所示为所得到的上转换发光层包覆的聚多巴胺纳米颗粒(pda@ucnp)的形貌图。图3为上转换发光层包覆的聚多巴胺纳米颗粒(pda@ucnp)在980nm近红外光以1w/cm2功率激发下的光热升温图。
将(2)中合成的pda@ucnp与多爪聚乙二醇(mpeg-pmhc18)按质量比1:1混合搅拌24h,洗涤,得到pda@ucnp-peg纳米颗粒。
加入光敏分子ce6乙醇溶液,搅拌12h,离心得到pda@ucnp-peg/ce6纳米探针。
实施例3
在20ml的乙醇-水(1:1)的混合溶液中,加入1ml稀土元素比例为78%:12%:10%(gdcl3:ybcl3:ercl3)的溶液,磁力搅拌10-30min形成透明溶液。加入步骤(1)中合成的3mg聚多巴胺纳米颗粒粉末,超声10-30min。随后将溶液在90℃下回流6h,即得到pda@gd,yb,er(oh)co3纳米颗粒。
将pda@gd,yb,er(oh)co3纳米颗粒分散于5ml水中,加入5ml乙二醇及20mgnaf,搅拌10min,转移至反应釜中,在180℃条件下处理8h。
将得到的纳米颗粒离心洗涤后分散于5-15ml乙醇-水溶液(1:1)中,加入0.1-0.5ml1mol/lnaf及0.1-0.5ml3mol/lhf溶液,搅拌10-30min,在100-180℃条件下处理4-12h,离心得到pda@ucnp纳米颗粒。
图2b所示为所得到的上转换发光层包覆的聚多巴胺纳米颗粒(pda@ucnp)的形貌图。图3为上转换发光层包覆的聚多巴胺纳米颗粒(pda@ucnp)在980nm近红外光以1w/cm2功率激发下的光热升温图。
将(2)中合成的pda@ucnp与多爪聚乙二醇(mpeg-pmhc18)按质量比1:1混合搅拌24h,洗涤,得到pda@ucnp-peg纳米颗粒。
加入光敏分子ce6乙醇溶液,搅拌12h,离心得到pda@ucnp-peg/ce6纳米探针。
- 还没有人留言评论。精彩留言会获得点赞!