眼镜蛇神经毒素多肽对鸦片类药品痛觉过敏和耐受的抑制及对其镇痛的协同增效作用的制作方法
技术领域:
本发明属于生物制药领域,涉及眼镜蛇神经毒素的新的临床应用,具体讲,涉及到抑制鸦片类药品的副作用及协同增效鸦片类药品的镇痛作用在临床上的应用。
背景技术:
阿片是从罂粟植物中提取的物质。它们与人体自身的类阿片受体结合,在合法处方时为急性或慢性疼痛患者提供止痛或镇痛;吗啡是阿片类镇痛剂的一个例子。短期使用这些药物被认为是一种安全的疼痛管理方法,然而,当它们被过度使用和/或经常使用时会产生一系列问题,包括疼痛过敏,耐受、药物依赖、便秘、呼吸抑制及致命的过量。根据美国疾病控制和预防中心的数据,2015年阿片类药物过量导致33000多人死亡。
吗啡等阿片类受体激动剂是临床上应用最为广泛的强效镇痛药。该类药物能有效缓解患者的疼痛,尤其是手术后及晚期癌症的疼痛。然而吗啡等鸦片类药物的使用极易导致患者出现耐受和痛觉过敏,而且往往在一周内即可形成。而一旦耐受和痛觉过敏形成就需要不断的提高剂量来继续维持其镇痛作用,即机体需要更大剂量的药物才能达到与最初相同的镇痛效应;而更大的剂量会导致更严重的痛觉过敏、耐受、便秘、成瘾和呼吸抑制。阿片类药品的痛觉过敏和耐受通过多种给药途径、不同剂量(从超低到高剂量)以及给药时间(如间歇性或持续)均可形成。所以要同时实现临床疗效以及对治疗方案的接受,阿片类药物痛觉过敏和耐受是一个亟待解决的问题。
药物所致的镇痛耐受(analgesictolerance)和痛觉过敏(hyperalgesia)是两个密切相关且易被混淆,但却彼此又不同的症状。前者指的是机体在长期应用阿片类受体激动剂后,该类药物的镇痛效应出现显著性下降,需要给予机体更大剂量的药物才能达到或维持与最初相同的镇痛效应。后者是指患者在长期或不正确应用吗啡等阿片类受体激动剂后,其对非伤害性刺激出现异常的痛觉反应,或是对相同的伤害性刺激出现高度敏感的痛觉反应。因此,镇痛耐受是药物缓解机体疼痛反应的能力下降,而痛觉过敏是机体对疼痛刺激的反应性增高。镇痛耐受和痛觉过敏虽然是由阿片类受体激动剂所诱发的两个不同的不良反应,但是它们均阻碍了吗啡类药物在临床上的长期使用,使需要长期使用此类药物的病人饱受痛苦,成为临床上一大未被满足的需求。
目前对吗啡耐受和疼痛过敏的治疗大多仍停留在实验阶段,如电针治疗,药物治疗包括二甲胺四环素、己酮可可碱、他汀类药物、白藜芦醇等。虽然阿片类药物和其它药物联合应用已经成为加强阿片类药物镇痛作用和抑制镇痛耐受和痛觉过敏的有效策略,但还没有一种疗效确切的药物能够使用,所以临床上一直期待有一款正真能让医生和病人满意的产品。
眼镜蛇神经毒素多肽是烟碱乙酰胆碱受体(nachr)的拮抗剂,它与肌型和神经元型烟碱乙酰胆碱受体(nachr)呈拮抗性和缓慢可逆性结合,此类眼镜蛇神经毒素多肽因其能阻断烟碱乙酰胆碱受体(nachr)的功能被称作为突触后神经毒素或α神经毒素。[1,2]烟碱乙酰胆碱受体(nachr)参与感觉、认知、疼痛和神经元的保护作用和神经递质传递,[3]故烟碱乙酰胆碱受体(nachr)被阻断后认知、疼痛、神经元保护及神经递质传递等作用就会受到不同程度的影响。
技术实现要素:
眼镜蛇神经毒素多肽的镇痛作用已有报道,但眼镜蛇神经毒素多肽既能抑制鸦片类药品的痛觉过敏和耐受的产生,又能增效鸦片类药品的镇痛疗效还是首次被发现。眼镜蛇神经毒素多肽在镇痛过程中不具有鸦片类药物的药物依赖性或需要增加剂量的问题,当他们和阿片类药物结合在一起时,这种组合能允许阿片类药物(如吗啡)在较低剂量下既能具有和高剂量吗啡同样甚至更好的镇痛作用,又能避免和抑制鸦片类药品的痛觉过敏和耐受的产生,可以长期使用,从而解决了阿片类药物耐受和痛觉过敏这一临床难题。
眼镜蛇神经毒素多肽与吗啡联用后具有协同镇痛的作用,并能显著延长吗啡镇痛作用的持续时间,在临床上治疗疼痛时,该复方制剂可以减少吗啡每次用药剂量和延长给药时间间隔。眼镜蛇神经毒素多肽,作为一种非吗啡类镇痛剂,因其拮抗吗啡镇痛耐受和痛觉过敏的优势,从而减少了吗啡等阿片类药物的严重不良反应而有望发展成一个辅助阿片类药物镇痛的有效佐剂。
本发明的意义在于可能能让患者用上一种能拮抗鸦片类药品所导致的镇痛耐受和痛觉过敏的药物;本发明的另一意义是可能能够开发一种和阿片类药物结合一起使用时,能使后者在较低剂量下仍能有效镇痛甚至更好镇痛的佐剂,因而避免了为了长期镇痛需不断增加吗啡剂量而导致的呼吸抑制,便秘,成瘾等严重副反应,同时因这一效果而满足了鸦片类药品长期使用的目的;本发明的最后一个意义可能是能够让患者在一定剂量鸦片类药品镇痛作用不理想的情况下能够联合应用这种佐剂而增强鸦片类药物的镇痛作用,这将可用于治疗临床上对较高剂量吗啡镇痛无效或疗效不好的病人,但对于二者的联合使用仍有良好的镇痛作用而无需再次提高吗啡的剂量。
实施方法:
本发明通过建立吗啡诱导的小鼠疼痛过敏和镇痛耐受模型,及眼镜蛇神经毒素多肽与吗啡合用后镇痛作用的时效关系和量效关系,包括它们之间的叠加增效、协同增效、延长吗啡药物作用时间等效应进行了系统性的实验,以模型观察眼镜蛇神经毒素多肽在上述各方面的作用。
以下结合具体实施例对上述方案做进一步说明,应理解,这些实施例是用于说明本发明而不在于限制本发明的范围。
实施步骤:
a.小鼠基础痛觉值的测量,从第一到第四天,将100个昆明小鼠进行4天的尾压力试验来测量小鼠机械痛觉值,作为基础痛觉值。
1.将小鼠轻轻地放入约束器中,并将其尾巴放在制痛仪的圆锥形尖端下,按下脚踏开关,均匀增加对尾部近端的压力,直到第一次痛觉反应(挣扎,吱吱声)发生;动物反应时记录引起痛觉的压力(单位:克)作为痛觉值,在没有任何反应的情况下,使用700g截止值来避免组织损伤;在同一只小鼠的尾巴的中部和远端重复这个测量,间隔至少30秒。
2.把测试完的小鼠放进笼子里,测试下一只直到该组所有小鼠都经过测试。三次测量的平均值(即每只小鼠尾巴的近端、中端和远端)作为每只小鼠的痛觉值(克),直到所有小鼠都经过小鼠尾压力试验。
3.在随后的3天,对所有小鼠重复尾压力痛觉值测试。
4.把通过以上尾压力试验为痛觉测量方式的小鼠中痛觉数值大于220克和小于180克的去除,然后将小鼠分随机分为“生理盐水组”、“吗啡组”、“眼镜蛇神经毒素多肽组”及“眼镜蛇神经毒素多肽+吗啡组”共4组,每组20个,用来对相应的药品进行疼痛过敏及镇痛耐受试验,多余的小鼠出组。
5.最后,再把每组20个小鼠一分为二,变成每组10个小鼠,分别用于眼镜蛇神经毒素多肽氨基酸序列seqidno.1,和眼镜蛇神经毒素多肽氨基酸序列seqidno.2的各项平行试验。
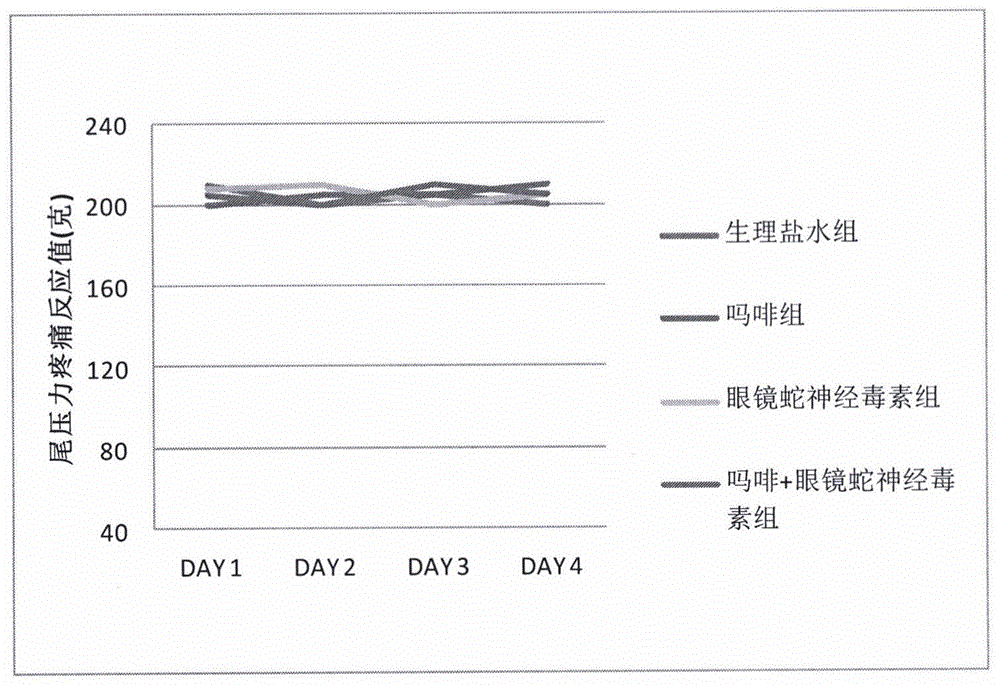
它们中的每组小鼠基础痛觉反应平均值(机械压力单位:克)的情况如图-1和图-2:
图-1是“生理盐水组”、“吗啡组”、“眼镜蛇神经毒素多肽组”及“眼镜蛇神经毒素多肽+吗啡组”四天基础痛觉反应平均值对应的折线图。所用眼镜蛇神经毒素多肽氨基酸序列是seqidno.1。
图-2是:“生理盐水组”、“吗啡组”、“眼镜蛇神经毒素多肽组”及“眼镜蛇神经毒素多肽+吗啡组”四天基础痛觉反应平均值对应的折线图。所用眼镜蛇神经毒素多肽氨基酸序列是seqidno.2。
每组小鼠的尾压力痛觉反应平均值将作为基础痛觉值为下一步实验中“生理盐水”,“吗啡”、眼镜蛇神经毒素多肽、眼镜蛇神经毒素多肽+吗啡这4种药物产生的痛觉值提供对比。
b.吗啡诱导的小鼠疼痛过敏和镇痛耐受模型的建立
吗啡诱导的小鼠疼痛过敏和镇痛耐受模型原理:
对小鼠进行连续7天的吗啡注射,使之产生疼痛过敏和镇痛耐。在第一次注射后开始,连续6天分别在注射前对小鼠痛觉进行测量,作为吗啡诱导的痛觉过敏指标;在注射后当天,第4天和第7天,对小鼠进行注射后的痛觉测量,即测量吗啡的镇痛效果,作为镇痛耐受指标,这些指标可以和对照组(“生理盐水组”,眼镜蛇神经毒素多肽组”、“眼镜蛇神经毒素多肽+吗啡组”)小鼠的指标及小鼠基础痛觉值作对比,以确认在连续吗啡注射后小鼠是否有疼痛过敏和镇痛耐受,眼镜蛇神经毒素多肽(氨基酸序列seqidno.1),和眼镜蛇神经毒素多肽(氨基酸序列seqidno.2)将分别平行的被用于以下的每一种试验。
具体步骤如下:
1.第五天对以上4组小鼠分别对应皮下注射无菌生理盐水(nacl0.9%1ml),吗啡(5mg/kg),眼镜蛇神经毒素多肽(50ug/kg),及眼镜蛇神经毒素多肽(50ug/kg)+吗啡(5mg/kg)。一个小时后(因吗啡的可能最大镇痛效应在45~90分钟)用尾压力试验测量“生理盐水组”,“吗啡组”、“眼镜蛇神经毒素多肽组”、“眼镜蛇神经毒素多肽+吗啡组”每只小鼠的尾压力疼痛反应值(上述a中的1-2步骤),以了解以上4种药品的镇痛作用。
2.从第六天到到第11天,小鼠在接受各自药物注射前先用尾压力试验测量“生理盐水组”,“吗啡组”、“眼镜蛇神经毒素多肽组”、“眼镜蛇神经毒素多肽+吗啡组”每只小鼠痛觉值(上述a中的1-2步骤)作为疼痛过敏指标;然后重复第五天的给药方法,对以上4组小鼠分别皮下注射无菌生理盐水(nacl0.9%1ml),吗啡(5mg/kg)、眼镜蛇神经毒素多肽(50ug/kg)、眼镜蛇神经毒素多肽(50ug/kg)+吗啡(5mg/kg)。
3.在第八天和第十一天,在注射一个小时后,再次用尾压试验来测量“生理盐水组”,“吗啡组”、“眼镜蛇神经毒素多肽组”、“眼镜蛇神经毒素多肽+吗啡组”每只小鼠疼痛反应值(上述a中的1-2步骤),以了解以上4种药品的的镇痛耐受情况。
第五天到第十一天(共7天),注射前尾压试验测量的4组小鼠平均疼痛反应值(即疼痛过敏指标,机械压力单位:克)情况如图-3和图-4。
图-3是“生理盐水组”、“吗啡组”、“眼镜蛇神经毒素多肽组”及“眼镜蛇神经毒素多肽+吗啡组”第五天到第十一天注射前尾压试验测量的痛觉反应平均值对应的折线图。所用眼镜蛇神经毒素多肽氨基酸序列是seqidno.1。
图-4是“生理盐水组”、“吗啡组”、“眼镜蛇神经毒素多肽组”及“眼镜蛇神经毒素多肽+吗啡组”第五天到第十一天注射前尾压试验测量的痛觉反应平均值对应的折线图。所用眼镜蛇神经毒素多肽氨基酸序列是seqidno.2。
##表示“吗啡组”和“眼镜蛇神经毒素多肽+吗啡组”之间的数值有统计学上显著性差异,p<0.05。
第五天,第八天和第十一天在药物注射后一个小时用尾压力试验测量的“生理盐水组”,“吗啡组”、“眼镜蛇神经毒素多肽组”、“眼镜蛇神经毒素多肽+吗啡组”每组小鼠的平均痛觉反应值(即镇痛耐受情况,机械压力单位:克)如图-5和图-6。
图-5是“生理盐水组”,“吗啡组”、“眼镜蛇神经毒素多肽组”及“眼镜蛇神经毒素多肽+吗啡组”的疼痛反应值对应的柱状图(镇痛耐受情况),所用眼镜蛇神经毒素多肽的氨基酸序列是seqidno.1。
“眼镜蛇神经毒素多肽+吗啡组”的痛觉反应值高于“吗啡组”和“眼镜蛇神经毒素多肽组,且在统计学上有显著性意义。试验数据不仅提示眼镜蛇神经毒素多肽能抑制吗啡诱导的镇痛耐受,而且证明眼镜蛇神经毒素多肽对吗啡的镇痛有叠加增效作用。
图-6是“生理盐水组”,“吗啡组”、“眼镜蛇神经毒素多肽组”及“眼镜蛇神经毒素多肽+吗啡组”的疼痛反应值对应的柱状图(镇痛耐受情况),所用眼镜蛇神经毒素多肽的氨基酸序列是seqidno.2。
“眼镜蛇神经毒素多肽+吗啡组”的痛觉反应值高于“吗啡组”和“眼镜蛇神经毒素多肽组,且在统计学上有显著性意义。试验数据不仅提示眼镜蛇神经毒素多肽能抑制吗啡诱导的镇痛耐受,而且证明眼镜蛇神经毒素多肽对吗啡的镇痛有叠加增效作用。
###表示“吗啡组”与“眼镜蛇神经毒素多肽+吗啡组”之间的数值有统计学上显著性差异,p<0.01;同时也表示吗啡组第五、八、十一天之间的数值有统计学上显著性差异,p<0.01。***表示“眼镜蛇神经毒素多肽组”与“眼镜蛇神经毒素多肽+吗啡组”之间的数值有统计学上显著性差异,p<0.01。
眼镜蛇神经毒素多肽和鸦片类药品的协同镇痛作用及延长其镇痛时间的具体实验步骤如下:
c.眼镜蛇神经毒素多肽和吗啡的协同镇痛试验
1.将40个sd大鼠随机分为“生理盐水组”,“吗啡组”、“眼镜蛇神经毒素多肽组”、“眼镜蛇神经毒素多肽+吗啡组”,每组10个。
2.对以上4组大鼠分别皮下注射无菌生理盐水(nacl0.9%1ml),吗啡(3mg/kg)、眼镜蛇神经毒素多肽(50ug/kg)及眼镜蛇神经毒素多肽(25ug/kg)+吗啡(1.5mg/kg)。
3.在注射给药后60分钟时给sd大鼠腹腔注射1.5%醋酸溶液1ml/只造模,用大鼠扭体试验测量得到的“生理盐水组”,“吗啡组”、“眼镜蛇神经毒素多肽组”、“眼镜蛇神经毒素多肽+吗啡组”每组大鼠1小时内平均扭体数作为衡量4种药品的可能最大镇痛效应。
图-7是4组大鼠分别皮下注射无菌生理盐水(nacl0.9%1ml),吗啡(3mg/kg)、眼镜蛇神经毒素多肽(50ug/kg)及眼镜蛇神经毒素多肽(25ug/kg)+吗啡(1.5mg/kg)60分钟后腹腔注射1ml(1.5%)醋酸溶液之后的1小时内平均扭体数,所用眼镜蛇神经毒素多肽为氨基酸序列seqidno.1。
图-8是4组大鼠分别皮下注射无菌生理盐水(nacl0.9%1ml),吗啡(3mg/kg)、眼镜蛇神经毒素多肽(50ug/kg)及眼镜蛇神经毒素多肽(25ug/kg)+吗啡(1.5mg/kg)60分钟后腹腔注射1ml(1.5%)醋酸溶液之后的1小时内平均扭体数,所用眼镜蛇神经毒素多肽为氨基酸序列seqidno.2。
可以看到半数剂量的眼镜蛇神经毒素多肽(25ug/kg)+半数剂量吗啡(1.5mg/kg)的镇痛效应要优于单独全剂量的吗啡(3mg/kg)及单独的全剂量的眼镜蛇神经毒素多肽(50ug/kg),且有显著性意义。以上数据显示眼镜蛇神经毒素多肽+吗啡能产生1+1大于2的镇痛效应,这是一种协同镇痛效应。
d.眼镜蛇神经毒素多肽延长吗啡镇痛时间的试验
继续以上试验c,上述4组sd大鼠按注射给药后150分钟后、210分钟后分别给sd大鼠腹腔注射1.5%醋酸溶液1ml/只造模,sd大鼠吗啡组在150分钟后、210分钟后已无镇痛作用;sd大鼠眼镜蛇神经毒素多肽组仍有镇痛作用,但疗效仍次于半数剂量的眼镜蛇神经毒素多肽(25ug/kg)+半数剂量的吗啡(1.5mg/kg)组,且差别有统计学上显著性意义。故显示出眼镜蛇神经毒素多肽+吗啡不仅有协同增效作用,而且在镇痛时间上半数剂量的眼镜蛇神经毒素多肽+半数剂量的吗啡和单独全剂量的吗啡相比能显著延长镇痛时间,和单独全剂量的眼镜蛇神经毒素多肽相比能有更强的镇痛作用,且并未随单独吗啡的镇痛作用递减而减弱这种协同增效的镇痛效应,由此证明眼镜蛇神经毒素多肽能延长吗啡的镇痛作用时间。
4组大鼠在分别注射4种药品后的60分钟腹腔注射1ml(1.5%)醋酸溶液,而后它们的60、150、210分钟后的一小时内扭体数如图-7和图-8。
图-7是4组大鼠分别皮下注射无菌生理盐水(nacl0.9%1ml),吗啡(3mg/kg)、眼镜蛇神经毒素多肽(50ug/kg)及眼镜蛇神经毒素多肽(25ug/kg)+吗啡(1.5mg/kg)的60分钟后腹腔注射1ml(1.5%)醋酸溶液之后的60、150、210分钟后一小时内扭体数对应的柱状图,所用眼镜蛇神经毒素多肽为氨基酸序列seqidno.1。
图-8是4组大鼠分别皮下注射无菌生理盐水(nacl0.9%1ml),吗啡(3mg/kg)、眼镜蛇神经毒素多肽(50ug/kg)及眼镜蛇神经毒素多肽(25ug/kg)+吗啡(1.5mg/kg)的60分钟后腹腔注射1ml(1.5%)醋酸溶液之后的60、150、210分钟后一小时内扭体数对应的柱状图,所用眼镜蛇神经毒素多肽为氨基酸序列seqidno.2。
在图-7和图-8中,###表示“吗啡组”与“眼镜蛇神经毒素多肽+吗啡组”之间的数值有统计学上显著性差异,p<0.01;同时也表示吗啡组第60、150、210分钟之间的数值有统计学上显著性差异,p<0.01。**和***分别表示“眼镜蛇神经毒素多肽组”与“眼镜蛇神经毒素多肽+吗啡组”之间的数值有统计学上显著性差异,分别为p<0.05和p<0.01。
机理研究:
我们也对眼镜蛇神经毒素多肽抑制鸦片类药品的痛觉过敏和耐受的机理作了进一步的研究,具体步骤如下:
il-1β,il-6及nos活性和no含量的测定
吗啡耐受和疼痛过敏试验完成后,参与眼镜蛇神经毒素多肽抑制鸦片类药品的痛觉过敏和耐受模型的小鼠在完成最后一天实验的2个小时后,取水合氯醛麻醉吗啡组及眼镜蛇神经毒素多肽+吗啡组的小鼠,脱臼处死。然后在含有冰块的板面上快速取出腰段脊髓,用冰水洗净后,待测定il-1β,il-6及nos活性和no含量的脊髓组织称重后放入预冷冻的生理盐水中,4000转/分离10分钟制备成10%的均浆待测,用elisa法测定腰段脊髓组织的以上指标的数值,按说明书步骤检测il-1β,il-6及nos和no的释放,考马斯亮蓝法测定各样本匀浆中的总蛋白含量,吗啡组及眼镜蛇神经毒素多肽+吗啡组的各项指标如下:
表-9
吗啡耐受组的il-1b,il-6、no及nos活性均分别显著高于眼镜蛇神经毒素多肽+吗啡组的数值,且有显著性意义。
根据公开报道的实验结果,促炎性细胞因子与病理性神经痛等多种疼痛有关。il-1b在神经病理性疼痛,人为皮下注射福尔马林、或蛛网膜下腔注射制疼剂所致疼痛时,脊髓胶质细胞分泌的il-1b明显增多,阻断il-1b受体可以减轻疼痛;[4]il-6可诱发机械性痛敏和痛觉过敏,il-6基因敲除可抑制坐骨神经结扎大鼠疼痛行为。[5]同时促炎性细胞因子确能通过多种途径而加重疼痛。神经元上表达有细胞因子受体时,促炎性细胞因子可能直接作用于中枢神经系统的神经元而增强疼痛;这类细胞因子还能通过调节初级传入神经纤维释放神经介质而增强疼痛。
促炎性细胞因子还能诱导星形胶质细胞和小胶质细胞使兴奋性氨基酸(excitatoryaminoacid,eaa)合成与释放增加,一氧化氮合成酶(nitricoxidesynthase,nos)被激活、一氧化氮(nitricoxide,no)合成增多,这些物质通过间接作用而使疼痛程度增加。[6-10]根据已经公开发表的实验数据来看,吗啡诱导的疼痛过敏和耐受也会伴随着il-1,il-6、nos活性及no含量的最高,[11-12]而我们在实验中发现眼镜蛇神经毒素多肽能够降低这些炎性因子和相关物质的指标。
另外,相当数量的实验数据已经证明烟碱乙酰胆碱受体(nachr)种类繁多,是调节以上促炎性细胞因子,nos及no的一个重要中间环节。烟碱乙酰胆碱受体拮抗剂通过拮抗烟碱乙酰胆碱受体(nachr)而调升了体内碱乙酰胆碱受体激动剂(如烟碱、gts21等)的含量,这些激动剂或是直接降低了促炎性细胞因子,nos活性或no含量,或是通过激活一些特殊的烟碱乙酰胆碱受体,如a7-nachr,a9-nachr等来调低促炎性细胞因子及nos活性,no的含量。[13-17]各种通道最后达到的结果是降低了以上这些相关物质的指标。最后,其他一些公开的实验结果还证实了烟碱乙酰胆碱受体(nachr)拮抗剂直接参与了降低神经性疼痛的过程。[18-23]
眼镜蛇神经毒素多肽,作为最重要的烟碱乙酰胆碱受体的拮抗剂,在我们的实验中也被证实能够降低促炎性细胞因子及nos活性,no的含量,和文献上其他烟碱乙酰胆碱受体拮抗剂对促炎性细胞因子il-1b,il-6及nos,no的调节效果一致。
眼镜蛇神经毒素多肽作为最主要的烟碱乙酰胆碱受体(nachr)拮抗剂,或是通过烟碱乙酰胆碱受体(nachr)来调控促炎性细胞因子,nos、no等与疼痛密切相关的因子;或是通过直接作用,这都和眼镜蛇神经毒素多肽是烟碱乙酰胆碱受体的拮抗剂这一共性有关,本发明用了2种不同的眼镜蛇神经毒素多肽来进行试验,达到了几乎一致的实验结果,也证实了眼镜蛇神经毒素多肽这一共性。
上述实例只为说明本发明的实施思路和技术特点,其目的在于让熟悉此技术的人员能够了解本发明的内容并据以实施,并不能以此而限制本发明的保护范围。凡根据本发明精神实质所做的任何等效改变或修饰,都应涵盖在本发明的保护范围之内。
references:
1.naguibmetal.advancesinneurobiologyoftheneuromuscularjunction:implicationsfortheanesthesiologist.jamsocanesthesiol,2002,96:202-31.
2.abbasm,rahmans.effectsofαα-7nicotinicacetylcholinereceptorpositiveallostericmodulatoronlipopolysaccharide-inducedneuroinflammatorypaininmice.eurjpharmacol,2016,783:85-91.
3.李江兵等.α神经毒素与烟碱型乙酰胆碱受体相互作用研究进展,生命科学2017年1月第29卷第1期.[
4.milliganetal.intrathecalhiv-1envelopglycoproteingp120enhancedstatesmediatedbyspinalcodeproinflammatorycytokins.jneuroscience2001(8)2808-2819.
5.murphyetal.endogenousinterleukin-6contributetohypersensitivitytocutanousstimuliandchangesinneuropeptidesassociatedwithchroninerveconstrictioninmice.eurjneurosci1999,11(7),2243-2253.
6.项红兵等.氯胺酮对吗啡耐受小鼠脊髓星形胶质细胞的影响.中华麻醉学杂志,2004,24(4)290-293.
7.rainerviktoretal.theroleofspinalneuroimmuneactivationinmorphineinducedtolerance/hyperplasiainneuropathicandshamoperatedrats.jneurosci2002,22(22)9980-9989.
8.haberbergeretal.nicotinicreceptoralpha7-subunitsarecoupledtothestimulationofnitricoxidesynthaseinratdorsalrootganglionneurons。histochemcellbiol(2003)120:173-181.
9.s.papadopolouetal.nicotinicreceptormediatedstimulationofno-generationinneuronsofratthoracicdorsalrootganglia.neuroscienceletters361(2004)32-35.
10.watkinsetal.spinalcordglianewplayerinpain,pain2001,93(3):201-205.
11.梁慧春.归元抗吗啡镇痛耐受和痛觉过敏的药效评价及其机制探讨.中国人民解放军军事科学院,2014.
12.简道林等.吗啡耐受对大鼠脊髓il-1b和il-6水平的影响,实用医学杂志,2005.第21卷第20期.
13.zakrzewicza,jetal.(2017)canonicalandnovelnoncanonicalcholinergicagonistsinhibitatp-inducedreleaseofmonocyticinterleukin-1betaviadifferentcombinationsofnicotinicacetylcholinereceptorsubunitsalpha7,alpha9andalpha10.frontcellneurosci11,189.
14.pateletal.anti-inflammatoryeffectsofastroglialα7nicotinicacetylcholinereceptorsaremediatedbyinhibitionofthenf-κbpathwayandactivationofthenrf2pathwayjournalofneuroinflammation(2017)14:192.
15.papadopolous,etal.nicotinicreceptormediatedstimulationofno-generationinneuronsofratthoracicdorsalrootganglia.neuroscilett361,32-35.77(2004).
16.thippeswamyt,etal.inhibitionofneuronalnitricoxidesynthaseresultsinneurodegenerativechangesintheaxotomiseddorsalrootganglionneurons:evidenceforaneuroprotectiveroleofnitricoxideinvivo.neuroscienceresearch2001.05.
17.richterk,etal.(2016)phosphocholine-anagonistofmetabotropicbutnotofionotropicfunctionsofalpha9-containingnicotinicacetylcholinereceptors.scirep6,28660.
18.paciniaetal.thealpha9alpha10nicotinicreceptorantagonistalpha-conotoxinrgiapreventsneuropathicpaininducedbyoxaliplatintreatment.expneurol2016.282,37-48.
19.romerohketal.inhibitionofa9a10nicotinicacetylcholinereceptorspreventschemotherapy-inducedneuropathicpain.procnatlacadsciusa2017.114,1825-1832.
20.vinclermetal.molecularmechanismforanalgesiainvolvingspecificantagonismofalpha9alpha10nicotinicacetylcholinereceptors.procnatlacadsciusa2006.103,17880-17884.
21.luos,etal.cloning,synthesis,andcharacterizationofalphaoconotoxingexiva,apotentalpha9alpha10nicotinicacetylcholinereceptorantagonist.procnatlacadsciusa2015.112,e4026-e4035.
22.holtmanjretal.thenovelsmallmoleculealpha9alpha10nicotinicacetylcholinereceptorantagonistzz-204gisanalgesic.eurjpharmacol2011.670,500-508.
23.walaepetal.novelsmallmoleculealpha9alpha10nicotinicreceptorantagonistpreventsandreverseschemotherapy-evokedneuropathicpaininrats.anesthanalg.2012.115,713-720.
- 还没有人留言评论。精彩留言会获得点赞!