一种拷贝数变异分析方法、系统及计算机可读存储介质与流程
本发明属于拷贝数变异(cnv)注释技术领域,尤其涉及一种拷贝数变异注释以及对于cnv的临床意义判断和解读的分析方法;用于拷贝数变异注释以及对于cnv的临床意义判断和解读的分析系统;以及用于实现该分析系统的计算机可读存储介质。
背景技术:
拷贝数变异(cnv,copynumbervariant)是一类染色体亚显微水平的结构变异,即染色体的某些区域或基因片段在基因组中发生了重复或缺失,有研究称基因组中有约12%的区域易于发生cnv。异常的拷贝数变化是许多人类疾病(如癌症、遗传性疾病、心血管疾病等)的一种重要分子机制。然而目前缺乏快速、全面注释包含这些数据库信息的软件及方法,只能手动依次去这些数据库网站上搜寻,非常不便。另一方面,目前的cnv主要是通过染色体芯片分析(cma芯片)和ngs方法进行检测。由于检测技术的限制,都很难准确的检测到cnv的断点。这也为判断两个cnv是否为同一cnv判定带来了难度。因此,一种能够便捷地为cnv注释并智能判断致病性的分析方法成为实际需求。
技术实现要素:
本发明的目的在于提供一种拷贝数变异分析方法、系统及计算机可读存储介质,旨在解决现有技术对cnv手动注释不便捷,致病性判断不智能的问题。
为了解决上述技术问题,本发明提供了一种拷贝数变异分析方法,其特征在于,该方法包括:
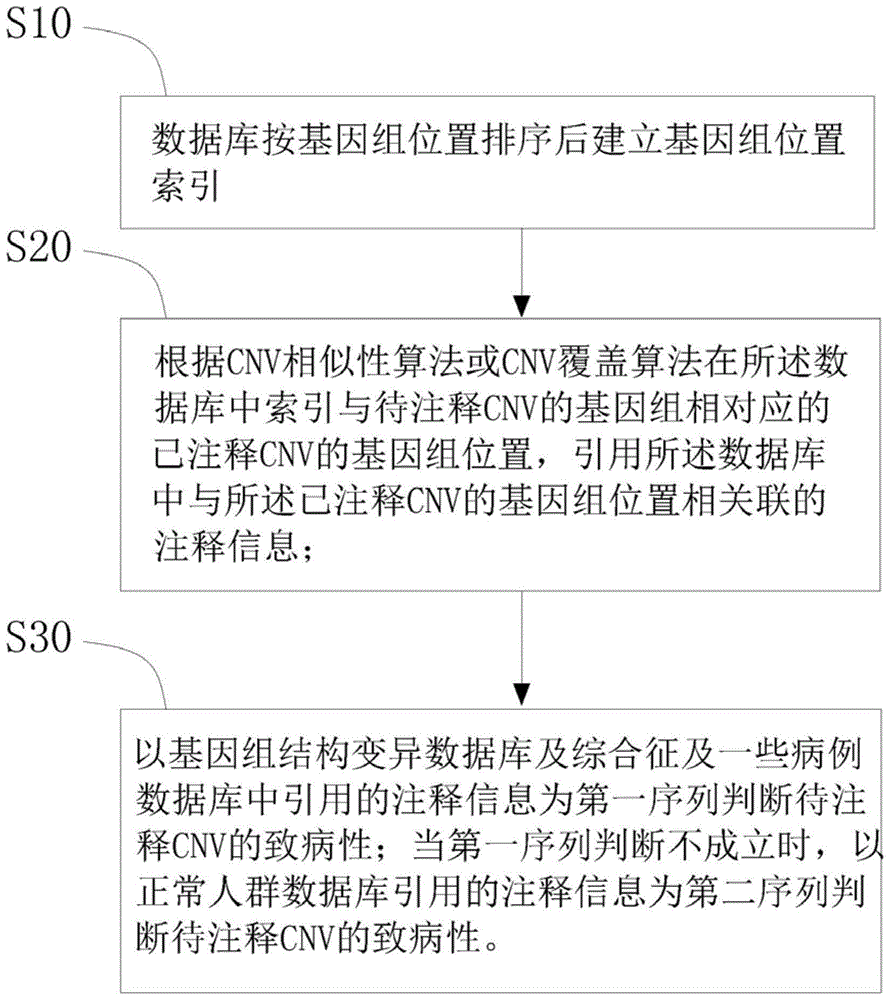
数据库按基因组位置排序后建立基因组位置索引,所述数据库包括正常人群cnv数据库、基因组结构变异数据库及综合征及一些病例数据库;
根据cnv相似性算法或cnv覆盖算法在所述数据库中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述数据库中与所述已注释cnv的基因组位置相关联的注释信息;
以基因组结构变异数据库及综合征及一些病例数据库中引用的注释信息为第一序列判断待注释cnv的致病性;当第一序列判断不成立时,
以正常人群数据库引用的注释信息为第二序列判断待注释cnv的致病性。
具体地,第一序列中:
根据cnv相似性算法在所述基因组结构变异数据库中匹配出与待注释cnv相似的若干个已注释cnv,
根据cnv覆盖算法在所述若干个已注释cnv中匹配出与待注释cnv相覆盖的已注释cnv,
索引与所述待注释cnv相覆盖的已注释cnv的基因组位置,引用所述基因组结构变异数据库中与所述已注释cnv的基因组位置相关联的临床意义和表型信息。
具体地,第一序列中:
根据cnv相似性算法在所述综合征及一些病例数据库的综合征子数据库中匹配出与待注释cnv相似的若干个已注释cnv,
根据cnv覆盖算法在所述若干个已注释cnv中匹配出与待注释cnv相覆盖的已注释cnv,
索引与所述待注释cnv相覆盖的已注释cnv的基因组位置,引用所述综合征子数据库中与所述已注释cnv的基因组位置相关联的综合征信息;
根据cnv相似性算法在所述综合征及一些病例数据库的人群子数据库中匹配出与待注释cnv相似的若干个已注释cnv,
索引与所述待注释cnv相似的若干个已注释cnv的基因组位置,引用所述综合征及一些病例数据库的人群子数据库中与所述若干个已注释cnv的基因组位置相关联的人群频率信息;
索引与所述待注释cnv相似的若干个已注释cnv的基因组位置,引用所述综合征及一些病例数据库的单倍不足预测子数据库中与所述若干个已注释cnv的基因组位置相关联的单倍不足基因信息。
具体地,所述数据库按基因组位置排序后建立基因组位置索引,所述数据库包括正常人群数据库、基因组结构变异数据库及综合征及一些病例数据库;根据cnv相似性算法或cnv覆盖算法在所述数据库中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述数据库中与所述已注释cnv的基因组位置相关联的注释信息的步骤中:
所述数据库按基因组位置排序后建立基因组位置索引,所述数据库还包括基因信息数据库及染色体区带信息数据库;
根据待注释cnv的染色体、基因组起始位置及基因组终止位置索引基因信息数据库中相对应的基因组位置,引用所述基因信息数据库中与所述基因组位置相关联的基因信息;
根据待注释cnv的染色体、基因组起始位置及基因组终止位置索引染色体区带信息数据库中相对应的基因组位置,引用所述染色体区带信息数据库中与所述基因组位置相关联的染色体区带信息。
具体地,所述数据库按基因组位置排序后建立基因组位置索引,所述数据库包括正常人群数据库、基因组结构变异数据库及综合征及一些病例数据库;根据cnv相似性算法或cnv覆盖算法在所述数据库中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述数据库中与所述已注释cnv的基因组位置相关联的注释信息的步骤中:
数据库按基因组位置排序后建立基因组位置索引,所述数据库还包括综合性肿瘤数据库;
根据cnv相似性算法在所述综合性肿瘤数据库中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述综合性肿瘤数据库中与所述已注释cnv的基因组位置相关联的注释信息。
一种拷贝数变异分析系统,所述系统包括:
数据库单元,按基因组位置排序后建立基因组位置索引,所述基因组位置与对应的注释信息相关联,所述数据库单元存储有正常人群数据库、基因组结构变异数据库及综合征及一些病例数据库;
算法单元,采用cnv相似性算法或cnv覆盖算法在所述数据库中单元中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置;
注释单元,引用所述数据库单元中与所述已注释cnv的基因组位置相关联的注释信息;
致病性判断单元,以基因组结构变异数据库及综合征及一些病例数据库中引用的注释信息为第一序列判断待注释cnv的致病性;当第一序列判断不成立时,以正常人群数据库引用的注释信息为第二序列判断待注释cnv的致病性;
所述数据库单元分别与算法单元和注释单元相连接,所述致病性判断单元与注释单元相连接。
具体地,所述综合征及一些病例数据库还存储有综合征子数据库、人群子数据库和单倍不足预测子数据库,所述注释信息包括临床意义、表型信息、人群频率信息和单倍不足基因信息,
所述算法单元根据cnv相似性算法在所述基因组结构变异数据库中匹配出与待注释cnv相似的若干个已注释cnv,
所述算法单元根据cnv覆盖算法在所述若干个已注释cnv中匹配出与待注释cnv相覆盖的已注释cnv,
所述数据库单元索引与所述待注释cnv相覆盖的已注释cnv的基因组位置,引用所述基因组结构变异数据库中与所述已注释cnv的基因组位置相关联的临床意义和表型信息;
所述算法单元根据cnv相似性算法在所述综合征及一些病例数据库的综合征子数据库中匹配出与待注释cnv相似的若干个已注释cnv,
所述算法单元根据cnv覆盖算法在所述若干个已注释cnv中匹配出与待注释cnv相覆盖的已注释cnv,
所述数据库索引与所述待注释cnv相覆盖的已注释cnv的基因组位置,引用所述综合征子数据库中与所述已注释cnv的基因组位置相关联的综合征信息;
所述算法单元根据cnv相似性算法在所述综合征及一些病例数据库的人群子数据库中匹配出与待注释cnv相似的若干个已注释cnv,
所述数据库索引与所述待注释cnv相似的若干个已注释cnv的基因组位置,引用所述综合征及一些病例数据库的人群子数据库中与所述若干个已注释cnv的基因组位置相关联的人群频率信息;
所述数据库索引与所述待注释cnv相似的若干个已注释cnv的基因组位置,引用所述综合征及一些病例数据库的单倍不足预测子数据库中与所述若干个已注释cnv的基因组位置相关联的单倍不足基因信息。
具体地,所述数据库单元还存储有基因信息数据库及染色体区带信息数据库,所述注释信息还包括基因信息和染色体区带信息;
所述算法单元根据待注释cnv的染色体、基因组起始位置及基因组终止位置索引基因信息数据库中相对应的基因组位置,引用所述基因信息数据库中与所述基因组位置相关联的基因信息;
所述算法单元根据待注释cnv的染色体、基因组起始位置及基因组终止位置索引染色体区带信息数据库中相对应的基因组位置,引用所述染色体区带信息数据库中与所述基因组位置相关联的染色体区带信息。
具体地,所述数据库单元还存储有综合性肿瘤数据库,所述注释信息还包括肿瘤类型信息;
所述算法单元根据cnv相似性算法在所述综合性肿瘤数据库中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述综合性肿瘤数据库中与所述已注释cnv的基因组位置相关联的肿瘤类型信息。
一种计算机可读存储介质,所述计算机可读存储介质存储有计算机程序,所述计算机程序被处理器执行时实现上述任一所述系统的功能。
本发明通过将在数据库中整合正常人群数据库、基因组结构变异数据库及综合征及一些病例数据库,按照基因组位置排序并建立索引,使得用户能够根据基因组位置引用相关联的注释信息,通过cnv相似性算法和cnv覆盖算法匹配到与待注释cnv相似的已注释cnv在数据库中的位置,使得用户能够根据待注释cnv的基因排序引用相关的注释信息,通过引用数据库的注释信息获知该待注释cnv的人群出现频率、临床意义、表型信息等,进而判断该待注释cnv的致病性。
附图说明
图1是本发明实施例一的整体方法流程图;
图2是本发明实施例一的步骤s10的方法流程图;
图3是本发明实施例一的步骤s20的方法流程图;
图4是本发明实施例一的步骤s23的方法流程图;
图5是本发明实施例一的步骤s24的方法流程图;
图6是本发明实施例一的步骤s25的方法流程图;
图7是本发明实施例二的步骤s10的方法流程图;
图8是本发明实施例二的步骤s20的方法流程图;
图9是本发明实施例三的步骤s10的方法流程图;
图10是本发明实施例三的步骤s20的方法流程图;
图11是本发明实施例四的系统结构框图;
数据库单元~10,基因信息数据库~11,染色体区带信息数据库~12,正常人群数据库~13、基因组结构变异数据库~14、综合征及一些病例数据库~15,综合性肿瘤数据库~16,算法单元~20,注释单元~30,致病性判断单元~40,
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
近年来ngs技术在生命科学的许多领域得到广泛应用,取得了很多突破性的进展,该技术也被广泛运用于cnv检测。ngs具有信息量大、通量高等特点,能快速准确的检测cnv,并且具有较高的分辨率。准确检测到cnv检测结果后还需要对cnv的片段中包含的基因信息、人群出现频率、临床意义、表型等进行注释及解读。目前公共的cnv数据库如正常人群数据库13dgv,ncbi的基因组结构变异数据库14dbvar,综合征及一些病例数据库15decipher以及肿瘤相关数据库cosmic等。这些公共数据库为cnv注释及解读提供了重要的依据。然而目前缺乏快速、全面注释包含这些数据库信息的软件及方法,只能手动依次去这些数据库网站上搜寻,非常不便捷和自动化。另一方面,目前的cnv主要是通过染色体芯片分析(cma芯片)和ngs方法进行检测。由于检测技术的限制,都很难准确的检测到cnv的断点。这也为两个cnv是否为同一cnv判定带来了难度。本研究通过一种cnv相似性算法能够有效的搜索出数据库中存在的和该cnv相似性高(可能为同一cnv)的cnv,以及全面注释目前权威的公共数据库refgene,dgv,dbvar,decipher,cosmic等数据库中的基因信息、相关人群分布、临床意义、表型等相关信息,并自动根据数据库信息判断该cnv是否致病,为cnv的后续解读提供全面、准确的依据。
实施例一
参见图1,本实施例提供的一种拷贝数变异分析方法,该方法包括:
步骤s10:数据库按基因组位置排序后建立基因组位置索引,
具体地,通过ngs技术准确检测到cnv检测结果后还需要对cnv的片段中包含的基因信息、人群出现频率、临床意义、表型等进行注释及解读,通过上述解读进一步分析该cnv样本是否为致病性,本实施主要通过引用正常人群数据库13、基因组结构变异数据库14及综合征及一些病例数据库15对cnv的人群出现频率、临床意义、表型信息等进行注释和解读以判断该样本cnv的致病性。
具体地,所述数据库包括正常人群数据库13、基因组结构变异数据库14及综合征及一些病例数据库15;数据库从外部导入正常人群数据库13、基因组结构变异数据库14及综合征及一些病例数据库15数据,由于是外部数据,且外部导入正常人群数据库13、基因组结构变异数据库14及综合征及一些病例数据库15数据分别有不同的来源,需要数据库对三者建立统一索引,该索引必须建立在统一规则下以便用户直接通过数据库获取正常人群数据库13、基因组结构变异数据库14及综合征及一些病例数据库15数据的数据资源,所述正常人群数据库13、基因组结构变异数据库14及综合征及一些病例数据库15数据的数据资源为现有技术中的外部数据库,该外部数据库至少包含有基因组信息,与该基因组相对应的注释信息,所述基因组信息是本领域通过大量医学临床积累记录的cnv基因组,以及临床上记录的与该cnv基因组相关联的人群出现频率、临床意义、表型信息等有助于医生判断病人病症、病理的注释信息。这类注释信息种类繁多,不同的医生往往有自己的经验,却没有一个统一合理的方法优化这一分析过程。本实施例中,仅采用正常人群数据库13、基因组结构变异数据库14及综合征及一些病例数据库15数据的相关注释信息获得样本cnv的致病性结论。
具体地,数据库按所述正常人群数据库13、基因组结构变异数据库14及综合征及一些病例数据库15中已注释cnv的基因组位置建立统一的排序,并建立基因组位置索引,使得用户能够根据样本cnv的基因组在数据库便捷地锁定相应已注释cnv的基因组位置,以便于调用与该已注释cnv的基因组相关联的注释信息。
具体地,参见图2,所述步骤s10可以具体分解为:
步骤s13:数据库获取正常人群数据库13的数据文件,并按照数据文件中基因组位置排序,再按照基因组位置建立索引。
具体地,数据库从ucsc(universityofcaliforniasantacruzgenomebrowser)数据库中下载合并后的dgv(正常人群cnv数据库13)数据库文件,按基因组位置排序后使用bgzip(一种现有技术中的文件压缩工具)进行压缩,再使用tabix(一种现有技术中的建立索引工具)对其基因组位置建立索引。
步骤s14:数据库获取基因组结构变异数据库14的数据文件,并按照数据文件中基因组位置排序,再按照基因组位置建立索引。
具体地,从ncbi中下载dbvar(基因组结构变异数据库14)数据库文件,按基因组位置排序后使用bgzip进行压缩,再使用tabix对其基因组位置建立索引。
步骤s15:数据库获取综合征及一些病例数据库15的数据文件,并按照数据文件中基因组位置排序,再按照基因组位置建立索引。
具体地,从https://decipher.sanger.ac.uk下载decipher(综合征及一些病例数据库15)单倍不足预测数据库、拷贝数人群频率数据库以及综合征数据库,按基因组排序后使用bgzip进行压缩,再使用tabix对其基因组位置建立索引。
步骤s20:根据cnv相似性算法或cnv覆盖算法在所述数据库中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述数据库中与所述已注释cnv的基因组位置相关联的注释信息;
具体地,所述cnv相似性算法或cnv覆盖算法为现有技术中通过计算机程序比对两基因组的基因序列以判断其相似的执行算法,
进一步地,所述相似cnv算法具体为:
两个cnv类型一致(同为dup或同为del),他们包含的基因相同,并且两个cnv重叠部分占两个cnv长度比例都达到70%以上。
进一步地,所述cnv覆盖算法为:
两个cnv类型一致(同为dup或同为del),其中一个cnv90%以上的区间在另一个cnv中,并且其所有的gene都在另一个cnv中包含。
具体地,所述数据库根据cnv相似性算法或cnv覆盖算法在所述数据库中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,所述数据库根据cnv相似性算法或cnv覆盖算法指的是仅采用cnv相似性算法、或仅采用cnv覆盖算法、或先后采用cnv相似性算法和cnv覆盖算法三种情况中的一种。
具体地,所述待注释cnv为通过ngs技术准确检测到cnv检测结果后还需要对cnv的片段中包含的基因信息、人群出现频率、临床意义、表型信息等进行注释及解读的待注释cnv。该待注释cnv需要通过与现有记录在数据库中已知的已注释cnv相比较,以确定该待注释cnv的致病类型,致病类型至少包括该cnv致病、该cnv可能致病、该cnv为良性三种情况,还可以根据具体情况直接给出相应的临床意义及表型信息,或标明临床意义注释为空。
具体地,引用所述数据库中与所述已注释cnv的基因组位置相关联的注释信息,本实施例中,引用的所述数据库包括正常人群数据库13、基因组结构变异数据库14及综合征及一些病例数据库15,所述注释信息相对应的为:正常人群数据库13存储的该基因组的cnv案例数量,基因组结构变异数据库14存储的该基因组的cnv临床意义和表型信息,综合征及一些病例数据库15存储的人群频率信息、单倍不足基因信息和综合征信息。
具体地,参见图3,步骤s20可以具体分解为:
步骤s23:根据cnv相似性算法和cnv覆盖算法在所述正常人群数据库13中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述正常人群数据库13中与所述已注释cnv的基因组位置相关联的案例数量;
步骤s24:根据cnv相似性算法和cnv覆盖算法在所述基因组结构变异数据库14中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述基因组结构变异数据库14中与所述已注释cnv的基因组位置相关联的临床意义和表型信息;
步骤s25:根据cnv相似性算法和cnv覆盖算法在所述综合征及一些病例数据库15中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述综合征及一些病例数据库15中与所述已注释cnv的基因组位置相关联的人群频率信息、单倍不足基因信息和综合征信息;
进一步地,参见图4,步骤s23具体包括:
步骤s23a:根据cnv相似性算法在所述正常人群数据库13中匹配出与待注释cnv相似的若干个已注释cnv,
步骤s23b:根据cnv覆盖算法在所述若干个已注释cnv中匹配出与待注释cnv相覆盖的已注释cnv,统计和待注释cnv相似的cnv的案例数量。
步骤s23c:索引与所述待注释cnv相覆盖的已注释cnv的基因组位置,引用所述正常人群数据库13中与所述已注释cnv的基因组位置相关联的案例数量。
具体地,使用pysam包对dgv数据库(正常人群数据库13)快速搜索与cnv的基因组位置有交集的已知人群cnv,使用cnv相似性算法判断待注释的cnv在dgv中是否存在相似cnv,统计和待注释cnv相似的cnv的案例数量;使用cnv覆盖算法判断待注释的cnv在dgv中是否有已知cnv覆盖,覆盖待注释cnv的cnv案例数量。
进一步地,参见图5,步骤s24具体包括:
步骤s24a:根据cnv相似性算法在所述基因组结构变异数据库14中匹配出与待注释cnv相似的若干个已注释cnv,
步骤s24b:根据cnv覆盖算法在所述若干个已注释cnv中匹配出与待注释cnv相覆盖的已注释cnv,统计和待注释cnv相似的cnv的案例数量。
步骤s24c:索引与所述待注释cnv相覆盖的已注释cnv的基因组位置,引用所述基因组结构变异数据库14中与所述已注释cnv的基因组位置相关联的临床意义和表型信息。
具体地,使用pysam包对dbvar数据库快速搜索与待注释cnv的基因组位置有交集的已知cnv;使用cnv相似性算法找出和待注释cnv相似的cnv,使用cnv覆盖算法找出待注释cnv覆盖的dbvar中的cnv,并获得对应的临床意义和表型信息。
进一步地,参见图6,步骤s25具体包括:
s25a:根据cnv相似性算法在所述综合征及一些病例数据库15的综合征子数据库中匹配出与待注释cnv相似的若干个已注释cnv,
s25b:根据cnv覆盖算法在所述若干个已注释cnv中匹配出与待注释cnv相覆盖的已注释cnv,
s25c:索引与所述待注释cnv相覆盖的已注释cnv的基因组位置,引用所述综合征子数据库中与所述已注释cnv的基因组位置相关联的综合征信息;
s25d:根据cnv相似性算法在所述综合征及一些病例数据库15的人群子数据库中匹配出与待注释cnv相似的若干个已注释cnv,
s25e:索引与所述待注释cnv相似的若干个已注释cnv的基因组位置,引用所述综合征及一些病例数据库15的人群子数据库中与所述若干个已注释cnv的基因组位置相关联的人群频率信息;
s25f:索引与所述待注释cnv相似的若干个已注释cnv的基因组位置,引用所述综合征及一些病例数据库15的单倍不足预测子数据库中与所述若干个已注释cnv的基因组位置相关联的单倍不足基因信息。
具体地,使用pysam对decipher综合征数据库快速搜索与待注释cnv的基因组位置有交集的综合征相关cnv;使用cnv相似算法找出步骤s25a中与待注释cnv相似的cnv,使用cnv覆盖算法找出步骤s25a中被待注释cnv覆盖的cnv;提取步骤s25b中cnv对应的综合征信息;使用pysam对decipher人群数据库快速搜索与待注释cnv的基因组位置有交集的cnv;使用cnv相似算法找出步骤s25a中与待注释cnv相似的cnv,并获取这些cnv对应的人群频率信息;使用pysam对decipherhi_predictionsdatabase(单倍不足预测数据库)快速搜索与cnv的基因组位置有交集的单倍不足基因信息,并合并得到cnv中包含的所有单倍不足基因信息。
步骤s30:以基因组结构变异数据库14及综合征及一些病例数据库15中引用的注释信息为第一序列判断待注释cnv的致病性;当第一序列判断不成立时,以正常人群数据库13引用的注释信息为第二序列判断待注释cnv的致病性。
具体地,若基因组结构变异数据库14及综合征及一些病例数据库15存在该待注释cnv,直接给出相应的临床意义及表型信息。
具体地,当基因组结构变异数据库14及综合征及一些病例数据库15不存在该待注释cnv时,若待注释cnv覆盖的已注释cnv中(基因组结构变异数据库14及综合征及一些病例数据库15)有明确致病,则注释该待注释cnv致病。
具体地,当基因组结构变异数据库14及综合征及一些病例数据库15不存在该待注释cnv,且待注释cnv覆盖的已注释cnv中(基因组结构变异数据库14及综合征及一些病例数据库15)无明确致病时,若待注释cnv覆盖的已注释cnv中(基因组结构变异数据库14及综合征及一些病例数据库15),有可能致病,则注释该待注释cnv可能致病。
具体地,当基因组结构变异数据库14及综合征及一些病例数据库15不存在该待注释cnv,且待注释cnv覆盖的已注释cnv中(基因组结构变异数据库14及综合征及一些病例数据库15)无明确致病和可能致病时,若正常人群数据库13中存在该待注释cnv,或者待注释的cnv被正常人群数据库13中的已注释cnv覆盖,且研究案例>=3,则认为该待注释cnv为良性。
实施例二
本实施例二与实施例一的区别在于,步骤s10:所述数据库按基因组位置排序后建立基因组位置索引,所述数据库包括正常人群数据库13、基因组结构变异数据库14及综合征及一些病例数据库15;步骤s20根据cnv相似性算法或cnv覆盖算法在所述数据库中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述数据库中与所述已注释cnv的基因组位置相关联的注释信息的步骤中:
所述数据库按基因组位置排序后建立基因组位置索引,所述数据库还包括基因信息数据库11及染色体区带信息数据库12;
进一步地,参见图7,则步骤s10具体包括:
步骤s11:数据库获取基因信息数据库11的数据文件,并按照数据文件中基因组位置排序,再按照基因组位置建立索引。
具体地,从universityofcaliforniasantacruz(ucsc)genomebrowser数据库中下载refgene.txt文件,按基因组位置进行排序后使用bgzip软件进行压缩,再使用tabix对其基因组位置建立索引。
步骤s12:数据库获取染色体区带信息数据库12的数据文件,并按照数据文件中基因组位置排序,再按照基因组位置建立索引。
具体地,从ucsc数据库中下载cytoband区间信息文件按基因组位置排序后使用bgzip进行压缩,再使用tabix对其基因组位置建立索引。
步骤s13:数据库获取正常人群cnv数据库13的数据文件,并按照数据文件中基因组位置排序,再按照基因组位置建立索引。
步骤s14:数据库获取基因组结构变异数据库14的数据文件,并按照数据文件中基因组位置排序,再按照基因组位置建立索引。
步骤s15:数据库获取综合征及一些病例数据库15的数据文件,并按照数据文件中基因组位置排序,再按照基因组位置建立索引。
进一步地,参见图8,步骤s20具体包括:
步骤s21:根据待注释cnv的染色体、基因组起始位置及基因组终止位置索引基因信息数据库11中相对应的基因组位置,引用所述基因信息数据库11中与所述基因组位置相关联的基因信息;
具体地,需要注释的待注释cnv至少需要包括染色体、基因组起始位置、基因组终止位置信息,使用pysam包对refgene快速搜索与cnv的基因组位置(染色体,起始位置,终止位置)有交集的基因信息,并提取转录本信息及cnv区间所覆盖的各基因的外显子信息。
步骤s22:根据待注释cnv的染色体、基因组起始位置及基因组终止位置索引染色体区带信息数据库12中相对应的基因组位置,引用所述染色体区带信息数据库12中与所述基因组位置相关联的染色体区带信息。
具体地,使用pysam包对cytoband数据库(染色体区带信息数据库12)快速搜索与待注释cnv的基因组位置(染色体,起始位置,终止位置)有交集的染色体区带信息,合并得到cnv所跨的染色体区带信息。
步骤s23:根据cnv相似性算法和cnv覆盖算法在所述正常人群数据库13中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述正常人群数据库13中与所述已注释cnv的基因组位置相关联的案例数量;
步骤s24:根据cnv相似性算法和cnv覆盖算法在所述基因组结构变异数据库14中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述基因组结构变异数据库14中与所述已注释cnv的基因组位置相关联的临床意义和表型信息;
步骤s25:根据cnv相似性算法和cnv覆盖算法在所述综合征及一些病例数据库15中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述综合征及一些病例数据库15中与所述已注释cnv的基因组位置相关联的人群频率信息、单倍不足基因信息和综合征信息;
步骤s30中,用户可以在第一序列和第二序列,即通过正常人群数据库13、基因组结构变异数据库14和综合征及一些病例数据库15引用的注释信息无法判断待注释cnv致病性时,可以通过基因信息和染色体区带信息为用户提供一定参考。例如,当基因组结构变异数据库14及综合征及一些病例数据库15不存在该待注释cnv,且待注释cnv覆盖的已注释cnv中(基因组结构变异数据库14及综合征及一些病例数据库15)无明确致病和可能致病时,且正常人群数据库13中不存在该待注释cnv,并且该待注释的cnv未被正常人群数据库13中的已注释cnv覆盖,抑或研究案例<3时,若待注释的cnv不包含任何基因信息,则认为其为良性。又例如,当上述正常人群cnv数据库13、基因组结构变异数据库14和综合征及一些病例数据库15、基因信息数据库11及染色体区带信息数据库12给出的注释信息均无法判断cnv致病性时,则临床意义注释为空。
实施例三
本实施例三与实施例一的区别在于,所述数据库按基因组位置排序后建立基因组位置索引,所述数据库包括正常人群cnv数据库13、基因组结构变异数据库14及综合征及一些病例数据库15;根据cnv相似性算法或cnv覆盖算法在所述数据库中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述数据库中与所述已注释cnv的基因组位置相关联的注释信息的步骤中:
数据库按基因组位置排序后建立基因组位置索引,所述数据库还包括综合性肿瘤数据库16;
根据cnv相似性算法在所述综合性肿瘤数据库16中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述综合性肿瘤数据库16中与所述已注释cnv的基因组位置相关联的注释信息。
进一步地,参见图9,所述步骤s10具体包括:
步骤s11:数据库获取基因信息数据库11的数据文件,并按照数据文件中基因组位置排序,再按照基因组位置建立索引。
步骤s12:数据库获取染色体区带信息数据库12的数据文件,并按照数据文件中基因组位置排序,再按照基因组位置建立索引。
步骤s13:数据库获取正常人群数据库13的数据文件,并按照数据文件中基因组位置排序,再按照基因组位置建立索引。
步骤s14:数据库获取基因组结构变异数据库14的数据文件,并按照数据文件中基因组位置排序,再按照基因组位置建立索引。
步骤s15:数据库获取综合征及一些病例数据库15的数据文件,并按照数据文件中基因组位置排序,再按照基因组位置建立索引。
步骤s16:数据库获取综合性肿瘤数据库16的数据文件,并按照数据文件中基因组位置排序,再按照基因组位置建立索引。
具体地,从https://cancer.sanger.ac.uk/cosmic下载cosmiccnv数据库,按基因组排序后使用bgzip进行压缩,再使用tabix对其基因组位置建立索引。
进一步地,参见图10,所述步骤s20具体包括:
步骤s21:根据待注释cnv的染色体、基因组起始位置及基因组终止位置索引基因信息数据库11中相对应的基因组位置,引用所述基因信息数据库11中与所述基因组位置相关联的基因信息;
步骤s22:根据待注释cnv的染色体、基因组起始位置及基因组终止位置索引染色体区带信息数据库12中相对应的基因组位置,引用所述染色体区带信息数据库12中与所述基因组位置相关联的染色体区带信息。
具体地,使用pysam包对cytoband数据库(染色体区带信息数据库12)快速搜索与待注释cnv的基因组位置(染色体,起始位置,终止位置)有交集的染色体区带信息,合并得到cnv所跨的染色体区带信息。
步骤s23:根据cnv相似性算法和cnv覆盖算法在所述正常人群数据库13中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述正常人群数据库13中与所述已注释cnv的基因组位置相关联的案例数量;
步骤s24:根据cnv相似性算法和cnv覆盖算法在所述基因组结构变异数据库14中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述基因组结构变异数据库14中与所述已注释cnv的基因组位置相关联的临床意义和表型信息;
步骤s25:根据cnv相似性算法和cnv覆盖算法在所述综合征及一些病例数据库15中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述综合征及一些病例数据库15中与所述已注释cnv的基因组位置相关联的人群频率信息、单倍不足基因信息和综合征信息;
步骤s26:根据cnv相似性算法在所述综合性肿瘤数据库16中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述综合性肿瘤数据库16中与所述已注释cnv的基因组位置相关联的肿瘤类型信息;
具体地,使用pysam对cosmic人群数据库(综合性肿瘤数据库16)快速搜索与待注释cnv的基因组位置有交集的cnv。使用cnv相似算法找出与待注释cnv相似的已注释cnv,并获取对应的肿瘤类型信息。
实施例四
本实施例提供一种拷贝数变异分析系统,参见图11,所述系统包括:
数据库单元10,按基因组位置排序后建立基因组位置索引,所述基因组位置与对应的注释信息相关联,所述数据库单元10存储有基因信息数据库11、染色体区带信息数据库12、正常人群数据库13、基因组结构变异数据库14、综合征及一些病例数据库15和综合性肿瘤数据库16;
算法单元20,采用cnv相似性算法或cnv覆盖算法在所述数据库中单元中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置;
注释单元30,引用所述数据库单元10中与所述已注释cnv的基因组位置相关联的注释信息;
致病性判断单元40,以基因组结构变异数据库14及综合征及一些病例数据库15中引用的注释信息为第一序列判断待注释cnv的致病性;当第一序列判断不成立时,以正常人群数据库13引用的注释信息为第二序列判断待注释cnv的致病性;
具体地,所述数据库单元10分别与算法单元20和注释单元30相连接,所述致病性判断单元40与注释单元30相连接。
具体地,所述综合征及一些病例数据库15还存储有综合征子数据库、人群子数据库和单倍不足预测子数据库,所述注释信息包括所述注释信息还包括基因信息和染色体区带信息、临床意义、表型信息、人群频率信息、单倍不足基因信息和肿瘤类型信息,
具体地,所述算法单元20根据cnv相似性算法在所述基因组结构变异数据库14中匹配出与待注释cnv相似的若干个已注释cnv,
进一步地,所述算法单元20根据cnv覆盖算法在所述若干个已注释cnv中匹配出与待注释cnv相覆盖的已注释cnv,
进一步地,所述数据库单元10索引与所述待注释cnv相覆盖的已注释cnv的基因组位置,引用所述基因组结构变异数据库14中与所述已注释cnv的基因组位置相关联的临床意义和表型信息;
具体地,所述算法单元20根据cnv相似性算法在所述综合征及一些病例数据库15的综合征子数据库中匹配出与待注释cnv相似的若干个已注释cnv,
进一步地,所述算法单元20根据cnv覆盖算法在所述若干个已注释cnv中匹配出与待注释cnv相覆盖的已注释cnv,
进一步地,所述数据库索引与所述待注释cnv相覆盖的已注释cnv的基因组位置,引用所述综合征子数据库中与所述已注释cnv的基因组位置相关联的综合征信息;
进一步地,所述算法单元20根据cnv相似性算法在所述综合征及一些病例数据库15的人群子数据库中匹配出与待注释cnv相似的若干个已注释cnv,
进一步地,所述数据库索引与所述待注释cnv相似的若干个已注释cnv的基因组位置,引用所述综合征及一些病例数据库15的人群子数据库中与所述若干个已注释cnv的基因组位置相关联的人群频率信息;
具体地,所述数据库索引与所述待注释cnv相似的若干个已注释cnv的基因组位置,引用所述综合征及一些病例数据库15的单倍不足预测子数据库中与所述若干个已注释cnv的基因组位置相关联的单倍不足基因信息。
进一步地,所述算法单元20根据待注释cnv的染色体、基因组起始位置及基因组终止位置索引基因信息数据库11中相对应的基因组位置,引用所述基因信息数据库11中与所述基因组位置相关联的基因信息;
进一步地,所述算法单元20根据待注释cnv的染色体、基因组起始位置及基因组终止位置索引染色体区带信息数据库12中相对应的基因组位置,引用所述染色体区带信息数据库12中与所述基因组位置相关联的染色体区带信息。
进一步地,所述算法单元20根据cnv相似性算法在所述综合性肿瘤数据库16中索引与待注释cnv的基因组相对应的已注释cnv的基因组位置,引用所述综合性肿瘤数据库16中与所述已注释cnv的基因组位置相关联的肿瘤类型信息。
实施例五
本实施例提供一种控制终端,以及应用于该终端的计算机可读存储介质,所述计算机可读存储介质存储有计算机程序,所述计算机程序被处理器执行时实现上述实施例四所述系统的功能。
所述终端包括存储器、处理器以及存储在所述存储器中并可在所述处理器上运行的计算机程序,
示例性的,计算机程序可以被分割成一个或多个模块,一个或者多个模块被存储在存储器中,并由处理器执行,以完成本发明。一个或多个模块可以是能够完成特定功能的一系列计算机程序指令段,该指令段用于描述计算机程序在用户终端中的执行过程。
本领域技术人员可以理解,上述中控单元的描述仅仅是示例,并不构成对中控单元的限定,可以包括比上述描述更多或更少的部件,或者组合某些部件,或者不同的部件,例如可以包括输入输出设备、网络接入设备、总线等。
具体地,所述处理器可以是中央处理单元(centralprocessingunit,cpu),还可以是其他通用处理器、数字信号处理器(digitalsignalprocessor,dsp)、专用集成电路(applicationspecificintegratedcircuit,asic)、现成可编程门阵列fieldprogrammablegatearray,fpga)或者其他可编程逻辑器件、分立门或者晶体管逻辑器件、分立硬件组件等。
进一步地,通用处理器可以是微处理器或者该处理器也可以是任何常规的处理器等,所述处理器是所述中控单元的控制中心,利用各种接口和线路连接整个中控单元的各个部分。
具体地,所述存储器可用于存储所述计算机程序和/或模块,所述处理器通过运行或执行存储在所述存储器内的计算机程序和/或模块,以及调用存储在存储器内的数据,实现所述中控单元的各种功能。
进一步地,所述存储器可主要包括存储程序区和存储数据区。
其中,存储程序区可存储操作系统、至少一个功能所需的应用程序(比如声音播放功能、图像播放功能等)等;存储数据区可存储根据终端的使用所创建的数据(比如音频数据、电话本等)等。此外,存储器可以包括高速随机存取存储器,还可以包括非易失性存储器,例如硬盘、内存、插接式硬盘,智能存储卡(smartmediacard,smc),安全数字(securedigital,sd)卡,闪存卡(flashcard)、至少一个磁盘存储器件、闪存器件、或其他易失性固态存储器件。
具体地,所述中控单元集成的模块/单元如果以软件功能单元的形式实现并作为独立的产品销售或使用时,可以存储在一个计算机可读取存储介质中。
基于这样的理解,本发明实现上述实施例方法中的全部或部分流程,也可以通过计算机程序来指令相关的硬件来完成,所述的计算机程序可存储于一计算机可读存储介质中,该计算机程序在被处理器执行时,可实现上述方法实施例的步骤。
其中,所述计算机程序包括计算机程序代码,所述计算机程序代码可以为源代码形式、对象代码形式、可执行文件或某些中间形式等。所述计算机可读介质可以包括:能够携带所述计算机程序代码的任何实体或装置、记录介质、u盘、移动硬盘、磁碟、光盘、计算机存储器、只读存储器(rom,read-onlymemory)、随机存取存储器(ram,randomaccessmemory)、电载波信号、电信信号以及软件分发介质等。
藉此,本发明通过将在数据库中整合正常人群cnv数据库、基因组结构变异数据库及综合征及一些病例数据库,按照基因组位置排序并建立索引,使得用户能够根据基因组位置引用相关联的注释信息,通过cnv相似性算法和cnv覆盖算法匹配到与待注释cnv相似的已注释cnv在数据库中的位置,使得用户能够根据待注释cnv的基因排序引用相关的注释信息,通过引用数据库的注释信息获知该待注释cnv的人群出现频率、临床意义、表型信息等,进而判断该待注释cnv的致病性。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!