PLAP-CAR-效应细胞的制作方法
序列表、表格或计算机程序的参考
序列表以ascii格式的文本文件通过efs-web与说明书同时提交,文件名为sequencelisting.txt,创建日期为2019年5月23日,并且大小为66.1千字节。通过efs-web提交的序列表是说明书的一部分,并在此通过引用全文并入本文。
本发明涉及plap(胎盘碱性磷酸酶)-car。本发明还涉及通过向患者施用plap-car-t细胞、plap-car-自然杀伤细胞,或plap-car-巨噬细胞来治疗plap阳性癌细胞的方法。
背景技术:
免疫疗法正在成为一种非常有前途的癌症治疗方法。t细胞或t淋巴细胞是我们免疫系统的武装力量,不断寻找外来抗原并将异常(癌症或经感染细胞)和正常细胞区分开来。利用car对t细胞进行基因修饰已成为设计肿瘤特异性t细胞最常见的方法。将靶向肿瘤相关抗原(taa)的car-t细胞输入患者体内(称为过继细胞转移或act)是一种有效的免疫治疗方法。与化疗或抗体相比,car-t技术的优点在于经重新编程的工程化t细胞能在患者体内实现增殖和持续存在(“一种活的药物”)。
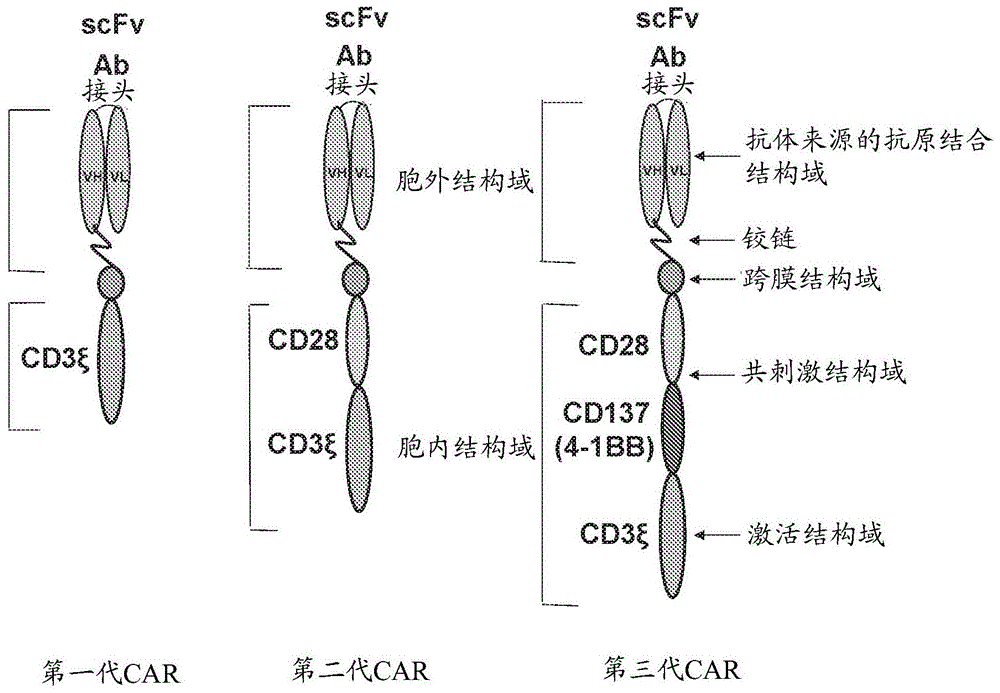
car(嵌合抗原受体)通常由源自单克隆抗体的单链可变片段(scfv)组成,该单链可变片段通过铰链和跨膜结构域与可变数量的胞内信号共刺激结构域相连:(i)cd28、ox-40、cd137(4-1bb)、gitr,或其他共刺激结构域;和(ii)共刺激结构域后的单一细胞激活cd3-ζ结构域(图1)。car的演变经历了从第一代(无共刺激域)到第二代(有一个共刺激域)再到第三代car(有若干个共刺激域)。具有多个共刺激结构域的car(所谓的第三代car)可增强细胞溶解活性,并显著改善car-t细胞的持久性,从而增强其抗肿瘤活性。
自然杀伤(nk)细胞是cd56+cd3-大颗粒淋巴细胞,可以杀死经病毒感染和转化的细胞,是构成先天免疫系统的关键细胞亚群。与具细胞毒性的cd8+t淋巴细胞不同,nk细胞对肿瘤细胞产生细胞毒性不需要预先致敏,并且还可以根除mhc-i阴性细胞。
car-t细胞疗法在治疗血液系统肿瘤患者的临床中取得了成功[1-5]。嵌合抗原受体包含靶向癌细胞表面抗原的抗体的单链可变片段(scfv),其与铰链、跨膜结构域、共刺激结构域(cd28,41-bb或其他结构域)和cd3激活结构域[1,6]、[7,8]连接。最近,基于两种cd19-car-t细胞疗法(kymriah和yescarta)在临床试验中对急性淋巴细胞白血病和其他血液系统肿瘤的高应答率,fda批准了它们用于治疗相应血液系统肿瘤[3]、[9-11]。还有若干种其他car-t细胞已开展临床试验,例如:用于b细胞淋巴瘤的cd22-car-t细胞[12],用于多发性骨髓瘤的bcma-car-t细胞[13-14]等。
就实体瘤而言,由于脱靶(on-targetoff-tumor)效应、抑制性肿瘤微环境、进入肿瘤的car-t细胞减少、t细胞耗竭和低持久性等原因,car-t细胞疗法在靶向实体瘤方面仍面临许多挑战[15]、[16-18]。靶向实体瘤的car-t细胞面临的主要挑战是,大多数实体瘤的肿瘤抗原均在正常组织中表达。
plap
plap是一种由alpp基因编码的胎盘碱性磷酸酶。plap是一种催化磷酸单酯的水解的金属酶。plap主要在胎盘和子宫内膜组织中表达,在正常组织中不表达。
plap在胎盘中高表达[19],并在除睾丸外的大多数正常组织中不表达[20]。现已发现它在恶性精原细胞瘤、畸胎瘤[20]、[21]、卵巢癌和宫颈癌[22]、[23]、[24],和结肠腺癌[25]中过表达。在肺、胰腺和胃肿瘤中也检测到plap[39]。plap也在非小细胞肺癌患者外泌体中的其他几种膜结合蛋白中被检测到,并有可能成为预后标志物[26]。
人plap是alpp基因编码的535个氨基酸的糖基化蛋白,其1-22为信号肽,然后是胞外结构域(23-506),513-529为跨膜结构域(序列如下所示,跨膜结构域标有下划线)uniprot数据库(www.uniprot.org/uniprot/p05187;nm_001632)。其序列如下所示(seqidno:1)。
存在四种不同但相关的碱性磷酸酶:肠型(alpi)(nm_001631);胎盘;胎盘样型(alppl2)(nm_031313),均由2号染色体上基因编码;肝/骨/肾型(alpl)(组织非特异性)(nm_000478),由1号染色体上的基因编码。
附图说明
图1显示了car的结构。在左侧:第一代(无共刺激结构域)结构,在中间:第二代(一个共刺激结构域cd28或4-bb),在右侧:第三代car(两个或若干个共刺激结构域)[7]。
图2显示了小鼠和人源化的plap-car构建物结构。
图3显示了plap高表达和低表达患者的无复发生存概率与随访月数的关系。
图4a通过facs分析显示了plap在几种plap阴性和plap阳性的结肠癌细胞系中的表达。显示了每种细胞系的mfi(平均荧光强度)/同型比率。图4b通过mrna和蛋白质显示了三种阳性和三种阴性细胞系的plap表达水平。plap阴性:hct116、sw620和ht-29细胞系;plap阳性:lovo、caco-2、ls123细胞系。
图5a显示了与t细胞和模拟-car-t细胞相比,plap-car-t细胞更显著地特异性杀死plap阳性结肠癌细胞。如材料和方法中所述,使用实时细胞毒性测定法(rtca)。car-t细胞与靶细胞的比例(e:t)为10:1。图5b显示了与正常t细胞相比,plap-car-t细胞对lovo和ls-123结肠癌靶细胞具有显著的杀伤活性,但对plap阴性hct116和ht29结肠癌细胞系没有显著的杀伤活性。图5c显示了针对plap阳性细胞的car-t分泌ifn-γ的显著水平。条形图显示了三个独立实验中ifn-γ的平均水平。p<0.05,student’st检验。
图6a显示了用fab抗体通过facs检测到plaph2-和plaph4-car-t阳性细胞。图6b显示了用生物素化的重组plap蛋白通过facs检测到plapcar-t阳性细胞。图6c(1)和图6c(2)显示了如材料和方法中所述的实时细胞毒性的定量。人源化plap-car-t特异性杀死palp-阳性的结肠癌细胞,但没有杀死palp-阴性的结肠癌细胞。p<0.06,student’st-检验,与模拟-car-t细胞相比,plap-car-t细胞的细胞毒性增加。图6d(1)和图6d(2)显示,与模拟-car-t细胞相比,针对plap阳性结肠癌细胞系而不是针对plap阴性结肠癌细胞系,plap-car-t细胞分泌ifn-γ,il-2和il-6的水平显著增加。p<0.05,student’st-检验。
图7a显示了人源化plap-car-t细胞显著地降低lovo异种移植肿瘤生长。使用car-t细胞处理的肿瘤体积明显小于使用经模拟对照细胞处理的肿瘤体积。p<0.05,student’st检验。图7b显示了人源化plap-car-t细胞处理的肿瘤大小明显小于对照小鼠。p<0.05,student’st检验。图7c显示了经hplap-car-t处理的小鼠的肿瘤重量明显低于对照组小鼠。p<0.05,student’st检验。图7d显示了在人源化plap-car-t细胞处理的小鼠的血清中,ast、alt和淀粉酶水平没有受到显著影响。样品按照材料和方法中的描述进行分析。
图8a(1)-8a(3)显示了plaph5-car-t细胞显著杀死plap阳性结肠癌细胞(caco-2细胞和lovo细胞),但不杀死plap阴性结肠癌细胞(hct116)。图8b显示了plaph5-car-t细胞针对plap阳性结肠癌细胞(caco-2细胞和lovo细胞),而非plap阴性结肠癌细胞(hct116)分泌显著更高水平的ifn-γ。
图9a是facs数据的量化,其显示了通过facs分析的plap-car-t处理前后结肠癌细胞系中pdl-1的表达。加入ifn-γ(20/ng/ml)作为pdl-1诱导的阳性对照。相比t细胞和模拟-car-t细胞,plap阳性的lovo细胞响应hplap-car-t细胞而明显诱导pdl-1的表达,而caco-2、hct116、ht29细胞则没有。图9b显示了对不同剂量的car-t细胞,pdl-1表达上调的响应。图9c显示了hplap-car-t细胞诱导的lovo癌细胞中pdl-1的时间依赖性。图9d显示了在与plap阳性靶细胞共孵育后,在car-t细胞中诱导了pd-1表达。利用pd-1抗体的facs分析显示,在与靶细胞共孵育之前和之后car-t细胞的pd-1水平显示显著升高。p<0.05,student’st检验。图9e显示了plap-car-t细胞在与plap阳性细胞共孵育后,lag-3表达上调。相对于不含靶细胞的模拟car-t细胞或car-t细胞,lag-3水平显著上调。p<0.05,student’st检验。图9f显示了plap-car-t细胞与pd-1抗体或lag-3抗体的组合增加了car-t细胞对靶细胞的细胞毒性。rtca分析是plaph2-car-t细胞以3:1的比例单独或者与pd-1抗体或pdl-1抗体联合使用进行的。显示了在与lovo靶细胞过夜共孵育后的rtca的定量。图9g显示了与仅plap-car-t细胞或仅抗体相比,plap-car-t细胞与pd-1抗体或lag-3抗体组合时分泌的ifn-γ显著增加。与plap-car-t细胞加同型抗体相比,*p<0.05,student’s单尾t检验。
具体实施方式
定义
如本文所使用的,“过继细胞疗法”(act)是一种使用癌症患者自身的t淋巴细胞或nk细胞或其他造血细胞(诸如巨噬细胞),诱导具有抗肿瘤活性的多能细胞在体外扩增并重新输入癌症患者体内的治疗方法。
如本文所使用的,“亲和力”是单个分子与其配体结合的强度。亲和力通常通过平衡解离常数(kd或kd)来测量和报告,该常数用于分子间相互作用的强度评估和排序。
如本文所使用的,“嵌合抗原受体(car)”指的是一种融合蛋白,其包括能够与抗原结合的胞外结构域、非该胞外结构域来源的多肽组成的跨膜结构域和至少一个胞内结构域。“嵌合抗原受体(car)”有时被称为“嵌合受体”、“t小体”,或“嵌合免疫受体(cir)”。“能与抗原结合的胞外结构域”指的是能与某一抗原结合的任何寡肽或多肽。“胞内结构域”指的是任何已知作为结构域起作用的寡肽或多肽,该结构域传递信号以激活或抑制细胞内的生物过程。
如本文所使用的,“结构域”指的是多肽中的一个区域,该区域独立于其他区域折叠成特定结构。
如本文所使用的,“单链可变片段(scfv)”指的是来源于保留与抗原结合能力的抗体的单链多肽。scfv的一个示例包括通过重组dna技术形成的抗体多肽,并且其中免疫球蛋白重链(h链)和轻链(l链)片段的fv区域通过间隔序列连接。本领域技术人员已知用于制备scfv的各种方法。
如本文所使用的,“肿瘤抗原”指的是具有抗原性的生物分子,其表达引起癌症。
本发明人已经发现plap是一种独特的肿瘤标志物,并且plap可有利地用于制备可用于car-t细胞疗法或car-nk细胞疗法的plap-car-t细胞或plap-nk细胞,这是因为plap不在正常组织中表达。与其他在正常组织中低水平表达的肿瘤标志物不同,plap靶标的优势在于其不在大多数正常组织中表达而仅在胎盘和睾丸中表达,从而使plap-car-t细胞/plap-nk细胞不与正常组织反应,因此它们是安全的且具有低毒性。
本发明提供了靶向在多种类型的癌症(诸如卵巢癌、精原细胞瘤和结肠癌)中高度过表达的plap肿瘤抗原的car-t细胞和nk细胞。本发明的plap-car-t细胞和plap-nk细胞对结肠癌细胞系和卵巢癌细胞系若干种癌细胞具有高细胞毒性:。
本发明涉及一种嵌合抗原受体融合蛋白,其从n-末端到c-末端包括:(i)含vh和vl的单链可变片段(scfv),该scfv与人plap结合,(ii)跨膜结构域,(iii)共刺激结构域cd28,和(iv)激活结构域。
在一个实施方案中,plap抗体是小鼠抗体,并且vh具有seqidno:5的氨基酸序列,而vl具有seqidno:6的氨基酸序列。
在一个实施方案中,plap抗体是人源化的抗体,并且vh具有seqidno:16、21、26、30或34的氨基酸序列,而vl具有seqidno:22的氨基酸序列。
在一个实施方案中,scfv包括seqidno:8、18、23、27、31或35的氨基酸序列;或者具有至少95%、96%、97%、98%或99%序列一致性的氨基酸序列,条件是该序列变化位于非cdr的框架区域。
在一个实施方案中,car包括seqidno:5、15、20、25、29或33的氨基酸序列;或者具有至少95%、96%、97%、98%或99%序列一致性的氨基酸序列,条件是该序列变化不在cdr区域内。。
序列变化(即氨基酸改变)优选是微小的氨基酸改变,诸如保守氨基酸替换。保守氨基酸替换是本领域技术人员熟知的。
本发明涉及一种用于治疗癌症的过继细胞治疗方法,包括向患有癌症的受试者施用plapcar-t细胞、plapcar-nk细胞或plapcar-巨噬细胞的步骤,其中,所述癌症选自由结肠癌、肺癌、胰腺癌、胃癌、睾丸癌、畸胎瘤、精原细胞瘤、卵巢癌和宫颈癌组成的组,并且所述癌症是plap阳性的。
适用于plapcar的抗体包括抗plap的小鼠plap抗体和抗plap的人源化plap抗体。在一个实施方案中,抗体对plap具有高亲和力。
本发明的car包括特异性结合plap的单链可变片段(scfv)。抗plap抗体的重链(h链)和轻链(l链)片段通过接头序列连接。例如,接头可以是5-20个氨基酸。scfv结构从n-末端到c-末端可以是vl-接头-vh,或vh-接头-vl。
本发明的car包括跨越膜的跨膜结构域。跨膜结构域可以源自天然多肽,或者也可以是人工设计的。源自天然多肽的跨膜结构域能从任何膜结合蛋白或跨膜蛋白中获得。例如,可以使用t细胞受体的α或β链、cd3ζ链、cd28、cd3-ε、cd45、cd4、cd5、cd8、cd9、cd16、cd22、cd33、cd37、cd64、cd80、cd86、cd134、cd137、icos、cd154或gitr的跨膜结构域。人工设计的跨膜结构域是一种主要包括诸如亮氨酸和缬氨酸疏水残基的多肽。优选地,在合成的跨膜结构域的每一端各发现苯丙氨酸、色氨酸和缬氨酸的三联体。在优选的实施方案中,跨膜结构域源自cd28或cd8,其提供了良好的受体稳定性。
在本发明中,共刺激结构域选自下组的蛋白的共刺激信号分子:人cd28、4-1bb(cd137)、icos-1、cd27、ox40(cd137)、dap10和gitr(aitr)。
胞内结构域(激活结构域)是car的信号传递部分。抗原识别后,受体聚集并且信号传递到细胞。最常用的胞内结构域成分是cd3-ζ(cd3z或cd3ζ),它包含3个itam。在抗原结合后向t细胞传递激活信号。cd3-ζ可能不提供完全足够的激活信号,并可能需要额外的共刺激信号。例如,一个或更多个共刺激结构域可以与cd3-ζ一起用于传递增殖/存活信号。
本发明的car可包括在scfvn-末端的信号肽,使得当car在细胞(诸如t细胞、nk细胞或巨噬细胞)内表达时,新生蛋白被导向内质网,并随后被导向细胞表面(新生蛋白在此表达)。信号肽的核心可能包含一长段有形成单个α-螺旋趋势的疏水氨基酸。信号肽可能始于一小段带正电荷的氨基酸,这有助于在转位过程中多肽正确拓扑结构的形成。在信号肽的终端,通常有一段可被信号肽酶识别和切割的氨基酸。信号肽酶可以在转位过程中或转位完成后切割,产生游离信号肽和成熟蛋白,游离的信号肽被特定的蛋白酶消化。例如,信号肽可以来源于人cd8或gm-csf,或者来源于具有1或2个氨基酸突变的变体,条件是该信号肽仍然具有引起细胞表面car表达的功能。
本发明的car可包括一个间隔序列作为铰链区,将scfv与跨膜结构域连接,并将抗原结合结构域与胞内结构域在空间上分开。柔性间隔子使得结合结构域能够以不同方向定向,以使其能够结合肿瘤抗原。间隔序列可以是:igg1fc区域、igg1铰链区、cd8茎(stalk)、或其组合。优选人cd28或cd8茎。
本发明提供了编码上述car的核酸。编码car的核酸可以通过常规方法由特定car的氨基酸序列制备。编码氨基酸序列的碱基序列可以从每个结构域氨基酸序列的前述ncbirefseqid或genbenk登录号获得,并且本发明的核酸可以使用标准分子生物学和/或化学方法制备。例如,基于碱基序列,可以合成核酸,本发明的核酸可以通过使用聚合酶链式反应(pcr)从cdna文库中获得的dna片段来制备。
可以将编码本发明的car的核酸插入到载体中,并且可以将载体导入细胞中。例如,可以使用病毒载体,诸如逆转录病毒载体(包括肿瘤逆转录病毒载体、慢病毒载体和假型载体)、腺病毒载体、腺伴随病毒(aav)载体、猿猴病毒载体、痘苗病毒载体或仙台病毒载体、爱泼斯坦-巴尔病毒(ebv)载体,和hsv载体。作为病毒载体,优选使用缺乏复制能力从而不能在感染细胞中自我复制的病毒载体。
例如,当使用逆转录病毒载体时,本发明的方法可以通过基于载体所具有的ltr序列和包装信号序列选择合适的包装细胞并使用包装细胞制备逆转录病毒颗粒来进行。包装细胞的实例包括pg13(atcccrl-10686)、pa317(atcccrl-9078)、gp+e-86和gp+envam-12,以及psi-crip。逆转录病毒颗粒也可以用具有高转染效率的293细胞或293t细胞制备。基于逆转录病毒和包装细胞生产的多种逆转录病毒载体可用于包装逆转录病毒载体,这些逆转录病毒载体可从许多公司广泛购得。
本发明提供了经修饰以表达上述嵌合抗原受体融合蛋白的t细胞,或nk细胞,或巨噬细胞。本发明的car-t细胞、car-nk细胞或car-巨噬细胞通过car与特定抗原结合,从而将信号传递到细胞中,并且由此激活细胞。表达car的细胞的激活根据宿主细胞的种类和car的胞内结构域而变化,并且可以基于例如细胞因子的释放、细胞增殖速率的提高、细胞表面分子的变化等作为指标来确认。
经修饰以表达car的t细胞或nk细胞或巨噬细胞可用作疾病的治疗剂。所述治疗剂包括表达car的t细胞作为活性成分,并且可以进一步包括合适的赋形剂。赋形剂的实例包括本领域技术人员已知的药学上可接受的赋形剂。
本申请证明了car-t细胞靶向在结肠癌肿瘤中过表达的plap抗原的效力。本申请证明了plap-car-t细胞特异性地降低plap阳性结肠癌细胞的生存能力,但不降低plap阴性癌细胞的生存能力。plap-car-t细胞在与plap阳性结肠癌细胞共孵育后分泌出显著水平的ifn-γ,但在与plap阴性癌细胞共孵育后未分泌出显著水平的ifn-γ。本申请证明plap-car-t细胞显著降低体内lovo(阳性plap-结肠癌细胞)异种移植肿瘤生长。用hplap-car-t细胞处理小鼠之后,小鼠血液中的ast、alt或淀粉酶水平没有增加,小鼠体重也没有下降,这证明hplap-car-t细胞在体内没有毒性作用。此外,hplap-car-t细胞与pd-1或lag-3抗体的组合增加了car-t细胞对抗结肠癌细胞的效力。
发明人发现plap-car-t细胞能显著杀死所有plap阳性的癌细胞,但不能杀死plap阴性的结肠癌。这意味着plap-car-t细胞的高度特异性。此外,lovo和caco-2结肠癌细胞中car-t细胞对pdl-1的上调作用不同。lovo结肠癌细胞响应plap-car-t细胞而诱导产生pdl-1,而caco-2细胞则不会。这两种细胞系都被hplap-car-t细胞有效杀死,与pdl-1表达的诱导无关。人源化plap-car-t细胞杀死lovo细胞比杀死caco-2细胞更快,并且针对lovo结肠癌细胞比针对caco-2细胞分泌更多的ifn-γ。此外,t细胞和模拟car-t细胞在lovo细胞中的活性高于caco-2细胞。这表明hplap-car-t细胞能克服lovo细胞中pdl-1的上调。这表明当用ifn-γ预处理lovo细胞以上调pdl-1时,plap-car-t细胞有效地杀死了lovo细胞。具有kras突变的结肠癌对诸如西妥昔单抗(erbitux)等疗法具有耐药性[40],而hplap-car-t细胞有效地杀死了两种不同的结肠癌细胞系:lovo(密码子13突变:g13d)和ls123(密码子12突变:g12d)。这是hplap-car-t细胞对抗因kras突变导致对其他疗法的耐药性的实体瘤的另一个优势。
plap-car-t细胞在与plap阳性结肠癌细胞系共培养后上调pd-1和lag-3,但在与plap阴性结肠癌细胞系共培养后没有增加。本发明人已经发现,在lovo结肠癌细胞系中,pdl-1响应于plap-car-t细胞呈剂量依赖性上调。pd-1、pdl-1或lag-3抗体与plap-car-t细胞联合可显著增加针对lovo癌细胞的car-t诱导的细胞毒性和ifn-γ分泌。因此,检查点抑制剂可以减少car-t细胞耗竭,并为联合治疗提供依据。
plapscfv-(cd28、ox-40、4-1bb或gitr)-cd3ζ-car-t细胞、car-nk细胞或car-巨噬细胞可与下列不同的化学疗法联合使用:检查点抑制剂;靶向疗法、小分子抑制剂和抗体。
标签(flag标签或其他标签)偶联的plapscfv可用于制备car。
第三代car-t或其他共激活信号结构域可用于car内plap-scfv。
双特异性plap抗原和其他抗原(egfr、her-2、vegfr、ngfr)的car-t细胞、car-nk细胞,或car-巨噬细胞可用于免疫疗法。双特异性car-t细胞的构建体包含靶向plap的第一scfv,和靶向第二肿瘤抗原的第二scfv。具有双特异性抗体的car-t细胞可以更有效和更特异性地靶向过表达两种肿瘤抗原的癌细胞。
plap-car-t细胞、car-nk细胞或car-巨噬细胞与靶向其他肿瘤抗原或肿瘤微环境(如vegfr-1-3)的car-t细胞、car-nk细胞或car-巨噬细胞的组合(即双car-t细胞、car-nk细胞或car-巨噬细胞)可用于增强单一疗法plap-car的活性。
plap-car-t细胞、car-nk细胞或car-巨噬细胞可用于激活吞噬作用和阻断“不吞”信号传导。
plap-car-nk细胞是安全的效应细胞,因为它们可以避免细胞因子风暴、肿瘤溶解综合征,和脱靶效应等潜在的致死性并发症。
抗plap抗体h2、h4和h5vh及vl序列可用作双特异性抗体的一个臂。
plap-car-t细胞和含抗plapvh和vl的双特异性抗体均可与检查点抑制剂(pdl-1抗体、pd-1抗体、lag-3抗体、tim-3抗体、tigit抗体和其他抗体),和化学疗法联合使用,以提高对癌细胞的疗效。
以下实施例进一步说明了本发明。这些实施例仅仅是为了说明本发明,而不是为了限制本发明。
实施例
实施例1、材料与方法
细胞和培养基
在加10%fbs和1%青霉素/链霉素的dulbecco改良eagle培养基(dmem)中,培养来自alstem(richmond,ca)的hek293ft细胞。根据irb批准的方案,使用ficoll-paque溶液(gehealthcare)从来自加利福尼亚州斯坦福市斯坦福医院血液中心的全血中分离出人外周血单核细胞(pbmc)。结肠癌细胞系:plap-阴性:sw620、ht29、hct116和plap-阳性:lovo、caco-2、ls123获自沃尔特·博德莫尔博士(oxford,uk),他的实验室如[28-29]所述使用snp、sequenommassarrayiplex和humanomniexpress-24珠芯片阵列鉴定细胞系,并检测是否存在所述支原体。细胞系在加10%fbs和青霉素/链霉素的dmem中培养。用于plapmrna水平检测的来自w.博德莫尔实验室的117个结肠癌细胞系的列表在补充中显示。
细胞系额外地通过facs使用细胞特异性表面标志物进行鉴定,并在加湿5%co2培养箱中培养。
抗体
单克隆pd-1(eh122h7)、pdl-1(克隆29e2a3)、tigit(克隆a15152g)、lag3(克隆7h2c65)、cd62l(克隆dreg-56)、cd45ro(克隆uchl1)、cd4(克隆rpa-t4)和cd8(克隆rpa-t8)抗体来自biolegend。plap抗体(克隆h17e2)获自thermofisher。其余抗体描述于[30]中。
car构建体
含有cd8α信号肽、plapabscfv[21]、cd8铰链区、cd28共刺激结构域和cd3激活结构域的第二代car从ef1启动子开始往下克隆到经修饰的慢病毒载体pcd510(systemsbioscience)中。用人源化的plapscfv(称为人源化的plap或plaph2、h4(克隆2或4)),和用胞内蛋白的scfv或包含转铁蛋白抗体三个表位的45个氨基酸序列的模拟对照(称为模拟-car)生成相同的构建体。小鼠plap-car是由synbio生成的。人源化的plapscfv序列是由idt合成作为gblock序列,nhei和xhoi限制性位点位于scfv的两侧,并亚克隆到cd8α信号肽和cd8铰链序列之间的慢病毒载体中的这些位点。
plap的人源化
plap的人源化如[31]中所述进行。利用[32、33]所述的计算机生物信息学方法对来自具有最高同源性的人抗体克隆的人编码框来进行人源化配对。将小鼠cdr插入这些克隆中,并使用不同的人源化scfv变体来生成car构建体并进行car-t细胞功能测试。
293ft细胞中的慢病毒的制备
如[34]所述通过使用转染剂(alstem)和慢病毒包装混合物转染293ft细胞,将慢病毒car构建体用于生成慢病毒。根据说明书,使用lenti-xqrt-pcr试剂盒(takara)和7900ht热循环仪(thermofisher)通过rt-pcr检测以pfu/ml为单位的慢病毒滴度。
car慢病毒转导以及car-t细胞的扩增
将pbmc细胞以1×106细胞/ml重悬于包含10%fbs和300u/mlil-2(thermofisher)的aimv-albumax培养基(thermofisher)中。用cd3/cd28dynabeads(invitrogen)激活pbmc,并在24孔板中培养。如[30、31、34]所述,在培养24和48小时时,使用transplus转导增强剂(alstem)将car慢病毒加入pbmc培养物中。car-t细胞培养和扩增14天,其中通过加入新鲜培养基以保持细胞密度为1×106细胞/ml。
荧光激活细胞分选(facs)分析
为了检测car的表达,将5×105细胞悬浮在1×pbs+0.5%bsa缓冲液中,并在冰上与人血清(jacksoninjectorysearch,westgrove,pa)一起孵育10min。然后加入别藻蓝蛋白(apc)标记的抗cd3(ebioscience,sandiego,ca)、7-氨基放线菌素d(7-aad,biolegend,sandiego,ca)、抗f(ab)2或其同型对照,并将细胞在冰上孵育30min。然后用缓冲液冲洗细胞,并在facscalibur(bdbiosciences)上分析,首先进行光散射对比7-aad染色,然后针对cd3染色对比f(ab)2染色或同型对照染色,绘制7-aad阴性活门控细胞图。利用facs检测结肠癌细胞系的plap水平,使用了来自ximbio(london,uk)的小鼠单克隆plap抗体(h17e2),并在facscalibur上进行了分析。
blitzfortebio结合试验
使用如[30]所述的blitzfortebio系统进行plap抗体与来自sinobiological的重组plap胞外结构域蛋白的结合试验。简言之,抗小鼠捕获(amc)生物传感器在动力学缓冲液(pbs,0.1%tween,0.05%bsa)中浸泡10min,并然后与0.1mg/ml的小鼠抗plap抗体在同一缓冲液中浸泡30min。清洗后,使用生物传感器结合不同浓度的plap抗原。用blitz系统软件检测kd。
实时细胞毒性试验(rtca)
将贴壁的结肠癌靶细胞(每孔10000个细胞)接种到96孔e板(aceabiosciences,sandiego,ca)中,并使用基于阻抗的实时细胞分析(rtca)icelligence系统(aceabiosciences)培养过夜。20-24小时后,用包含10%fbs的aimv-albmax培养基中1×105效应细胞(car-t细胞、模拟car-t细胞或未转导t细胞)替换培养基,一式三份。在一些实验中,将10μg/ml的检查点蛋白抗体pd-1、lag-3或同型抗体单独或与car-t细胞联合加入到效应细胞中。在一些系列实验中,靶细胞用20ng/mlifn-γ预处理24h。使用rtca系统监测细胞1-2天,并绘制阻抗(与细胞指数成比例)随时间变化图。细胞毒性计算为(无效应细胞的靶细胞阻抗-有效应细胞的靶细胞阻抗)×100/无效应细胞的靶细胞阻抗。
细胞因子分泌的elisa试验
将靶细胞与效应细胞(car-t细胞或未转导t细胞)一起培养在加有10%fbs的aimv-albumax培养基的u形底部96孔板中,一式三份。16小时后,取出上清液并离心以去除残留细胞。在一些实验中,使用rtca分析后的上清液进行elisa细胞因子试验。将上清液转移到一个新的96孔板中,并根据说明书,使用thermofisher的试剂盒通过elisa分析人细胞因子。
小鼠体内异种移植研究
六周大的雄性nsg小鼠(jacksonlaboratories,barharbor,me)按照机构动物护理和使用委员会(iacuc)方案饲养。每只小鼠皮下注射包含在无菌1×pbs中2×106结肠癌细胞。在第1、7和13天,向小鼠静脉注射car-t细胞(1×107car-t细胞/小鼠)。每周两次使用卡尺测量肿瘤大小,并使用公式w2l/2确定肿瘤体积(单位为mm3),其中w为肿瘤宽度,l为肿瘤长度。结束时,采集0.1ml血液用于毒理学标志物分析。
毒理学标志物。
小鼠血清样本由idexbioanalytics(westsacramento,ca)使用临床化学分析仪(beckman-coulterau680)处理,以检测毒理学标志物的水平:alt(丙氨酸转氨酶)、ast(天冬氨酸转氨酶)、淀粉酶,单位为u/ml。
原发性肿瘤样本
从promab(richmond,ca)的存档载玻片中获取了具有不同类型的正常组织或肿瘤组织的样本。含106个原发性结肠癌腺癌的tma载玻片获自biomax(rockville,md),并与plap抗体一起用于ihc分析。
免疫组织化学(ihc)染色
将原发性肿瘤组织或正常组织切片载玻片或原发性tma载玻片在二甲苯中孵育两次,每次10分钟,然后在乙醇中水合,并在1×pbs中洗涤。使用高压锅在10mmph6.0的柠檬酸盐缓冲液,中进行热诱导抗原修复20分钟。载玻片用pbs洗涤,在3%h2o2溶液中孵育10分钟,然后用1×pbs再次洗涤,并在山羊血清中孵育20分钟。将组织切片载玻片与小鼠单克隆plap(h17e2)一抗在4℃孵育过夜,或在37℃孵育1.5小时。载玻片用pbs洗涤3次,用生物素偶联的二抗孵育10分钟,用pbs洗涤,用链霉亲和素偶联的过氧化物酶孵育10分钟,并用1×pbs缓冲液洗涤3次。载玻片在显微镜下在dab底物溶液中孵育2-5分钟。用水清洗停止反应,用苏木精复染,用水洗涤,并在75%、80%、95%和100%乙醇和二甲苯中脱水。对于阴性对照使用同型抗体,而对于阳性对照使用胎盘样本。使用imagesplus2.0软件在moticdmb5-2231pl显微镜(motic,厦门,中国)上采集图像。使用r2基因组分析和可视化平台(http://r2platform.com/http://r2.amc.nl)进行plap表达与无进展生存期预后的相关性分析。
实施例2、小鼠plap-cd28-cd3ζcar的序列
car构建体是:人cd8信号肽、小鼠scfv或人源化的源自抗体h17e2(vh-接头-3×(ggggs)-vl)、cd8铰链区、cd28跨膜、共激活结构域、cd3ζ激活结构域(图2)。ecor1和xhoi位点内带有car构建体的慢病毒载体序列如下所示。该scfv的两侧带有nhei和xhoi位点,可潜在地重新克隆到其他构建体中。plap-cd28-cd3的核苷酸序列如下所示,
seqidno:2,以xbai位点(斜体)起始的tctagagccgccacc-侧翼载体序列:
seqidno:3(小鼠plapcar,称为pmc262),以atg起始,并以终止密码子taa结束(带下划线),信号肽以粗体显示,具有cdr1、2、3(粗体带下划线)的vh;接头用斜体表示,具有cdr1、2、3(粗体带下划线)的vl;scfv两侧翼是5’nhe和3’xho位点,小字体
seqidno:4:带有ecori位点(斜体)的taggaattc侧翼载体
seqidno:5是seqidno:3(小鼠plap-cd28-cd3ζcar)的氨基酸序列:信号肽-vh-接头(斜体小字体gssss×3)-vl-h-cd28-cd3。粗体序列为小鼠plapscfv;cdr1、2、3加下划线;vh-用斜体显示的接头-vl。
小鼠vh(具有带下划线的cdr1、2、3),seqidno:6
小鼠vl(具有带下划线的cdr1、2、3),seqidno:7
小鼠plapscfv,seqidno:8
car构建体的方案如下所示,其显示了seqidno:3的子结构域序列。
<hucd8信号肽>seqidno:9
atggccttaccagtgaccgccttgctcctgccgctggccttgctgctccacgccgccaggccg
<nhei限制性酶切位点>
gctagc
<小鼠plapscfv(vh-接头-vl)>seqidno:10
<xhoi限制性酶切位点>
ctcgag
<cd8>seqidno:11
<cd28tm/激活>seqidno:12
<cd3ζ>seqidno:13
<ecori限制性酶切位点>
gaattc
实施例3、具有人源化抗体h1的plapcar。
seqidno:14(人h1plapcar),以atg起始,并以终止密码子taa结束(下划线)。序列从信号肽起始,然后是人源化的plapscfvh1。除scfv部分外,核苷酸序列的结构与seqidno:2相同。粗体序列是人源化h1plap-1scfv(cdr1、2、3带下划线)。与小鼠相比,人源化框架区域中的不同核苷酸标有下划线,但未加粗。
seqidno:15为人源化的h1plap-1car氨基酸序列;除scfv部分外,其结构与小鼠plap-car相同;粗体序列是人源化的h1plap-1scfv,cdr1、2、3以斜体和下划线显示;接头是较小字体;cdr区域中的不同氨基酸是常规字体;框架区域中来自小鼠序列的不同氨基酸用下划线标出。
人源化的h1plap-1vh(seqidno:16)
人源化的h1plap-1vl(seqidno:17)
人源化h1plap-1scfv(seqidno:18)
实施例4、具有人源化抗体h2的plapcar。
采用生物信息学方法生成了额外的plapcar人源化版本。对序列进行了密码子优化,以提高car的表达。
序列从信号肽(下划线,经密码子优化)起始,然后是人源化plapscfv(粗体)。除scfv部分外,核苷酸序列的结构与seqidno:3相同。粗体序列为人源化的plap-h2(pmc409)scfv,其余结构与小鼠plap-car(seqidno:5)相同。
人源化的plaph2-car。核苷酸序列(经密码子优化),seqidno:19
人源化的plaph2car氨基酸序列如seqidno:20所示。除scfv部分外,其结构与小鼠plap-car相同;粗体序列是由vl-接头-vl组成的人源化的plapscfv。
人源化的plaph2vh(seqidno:21)
人源化的plaph2vl(seqidno:22),cdr1、2、3标有下划线
人源化的plaph2scfv(seqidno:23)
实施例5、具有人源化抗体h4的plapcar。
密码子优化的人源化的plaph4car(pmc410)的核苷酸序列以信号肽(标有下划线,seqidno:9,经密码子优化)起始,然后是人源化的plapscfv(粗体)。粗体序列是人源的plap-h4(pmc410)scfv,
seqidno:24是人源化的plaph4-car核苷酸序列(经密码子优化)。
seqidno:25是人源化的plaph4car氨基酸序列:scfv序列以粗体显示。
人源化的plaph4vh(seqidno:26),cdr1、2、3标有下划线
人源化的plaph4vl(seqidno:22)
人源化的plaph4scfv(seqidno:27)
实施例6、具有人源化抗体h3的plapcar。
seqidno:28是人源化的plap-h3(pmc407)核苷酸序列:
seqidno:29是plaph3car氨基酸序列(scfv序列加粗)。
人源化的plap-h3vh,seqidno:30
人源化plaph3vl,seqidno:22
人源化的plaph3scfv,seqidno:31
实施例7、具有人源化抗体h5的plapcar。
seqidno:32是人源化的plap-h5scfv核苷酸序列,其插入xho和nhei位点之间:
人源化的plap-h5car氨基酸序列(seqidno:33)
人源化的plap-h5vh,seqidno:34。cdr1、2、3标有下划线
人源化的plaph5vl,seqidno:22
人源化的plaph5scfv,seqidno:35
实施例8、plap在大多数正常组织中呈阴性表达,而在胃肠癌中表达
我们用plap抗体对胎盘、睾丸、结肠癌、卵巢癌和其他来自不同类型癌症的正常或恶性组织进行了ihc染色。胎盘染色最强,睾丸、结肠癌和卵巢癌呈阳性,其他类型癌(乳腺癌、肺癌、前列腺癌)及正常组织:胰腺、扁桃体、直肠、肌肉、食管、脑等组织均呈阴性。此外,我们使用癌细胞系百科全书(ccle)在电脑上评估了1457种不同恶性细胞系(包括63种结肠癌细胞系)的plapmrna表达。plap在胃肠(gi)癌中的表达高:食道癌、上呼吸消化道癌、胃癌、胰腺癌和结肠癌。我们还使用非恶性正常组织中plap表达的基因型-组织表达(gtex)数据库进行分析。plapmrna在许多正常组织中表达极低,许多组织的tmp(每百万kb转录物)mrna水平为0。相反,当我们将epcam作为阳性对照进行分析时,其在许多正常组织中的表达为中-高水平,在结肠为445tmp(每百万kb转录产物)、小肠为391和甲状腺为259,为中等水平表达。因此,与其他肿瘤相关标志物相比,plap在大多数正常组织中呈阴性表达。
实施例9、plap在原发性结肠肿瘤和结肠癌细胞系中表达
我们使用106种原发性结肠癌肿瘤,用小鼠plap抗体进行ihc染色,并在106份样本中的25份中发现plap表达,占所有结肠癌肿瘤的23.8%。我们还通过r2基因组学分析和可视化平台检测了557例原发性结肠癌肿瘤中的plap表达,并与患者结果进行了相关性分析(图3)。plap高表达患者的生存期短于plap低表达患者,这证明plap表达与结肠癌的不良预后相关。这些数据表明,plap在原发性结肠癌肿瘤中过表达。
此外,我们采用微阵列技术检测了117种结肠癌细胞系的plapmrna水平,并检测到21.3%的结肠癌细胞系表达plapmrna。我们进行了facs分析,并在plapmrna高表达的结肠癌细胞系(lovo、caco-2和ls123细胞系)中检测到plap(图4a)。我们在plap阴性结肠癌细胞系(诸如hct116、ht-29和sw620细胞系)中检测到极少量plap表达(图4b)。因此,plapmrna和plap蛋白水平彼此对应(图4b)。为了确认plap抗体h17e2的特异性,我们通过bliblitz分析检测到其识别经纯化重组plap蛋白的kd=3.2nm。plap抗体h17e2还识别在293细胞中表达的plap蛋白。因此,plap在结肠癌中表达,并且plap抗体可以检测plap抗原,表明其可用于car-t治疗。
实施例10、plap-car-t细胞特异性地杀死plap阳性细胞,但不杀死plap阴性细胞。
我们使用小鼠单克隆plap抗体scfv、cd8α铰链、cd28跨膜和共刺激结构域及cd3激活结构域设计了第二代car构建体(图2)。我们制备了具有细胞内蛋白scfv的慢病毒plap-car和模拟car,并转导t细胞以生成car-t细胞。plap-car-t细胞扩增>200倍,与模拟-car-t细胞或t细胞相似。用小鼠fab抗体通过facs检测car-t阳性细胞(图5a)。
利用实时细胞毒性试验(rtca)检测plap-car-t细胞对plap阳性靶结肠癌细胞系:lovo和ls-123;以及对plap阴性结肠癌细胞系:ht29和hct116的杀伤作用。与正常t细胞相比,plap-car-t细胞对lovo和ls-123结肠癌靶细胞具有显著的杀伤活性,但对plap阴性hct116和ht29结肠癌细胞系没有显著的杀伤活性(图5b)。此外,所有car-t细胞系都针对plap阳性靶结肠癌细胞分泌显著水平的ifn-γ,但不针对plap阴性结肠癌细胞分泌显著水平的ifn-γ(图5c)。对于正常293和cho细胞系也不存在ifn-γ的显著分泌(图5c)。这些数据显示了plap-car-t细胞对plap阳性结肠癌细胞系的特异性功能活性。
实施例11、人源化的plap-car-t细胞(h2和h4)特异性地杀死plap阳性细胞
为了改善mplap-car-t细胞,我们对小鼠plapscfv进行人源化,并生成了人源化的plap-car细胞(图2)。采用fab抗体通过facs检测,人源化的plaph2有44.1%的car阳性细胞,而人源化的plaph4car-t细胞有50.6%的car阳性细胞(图6a)。为了确认plap-car-t细胞对plap抗原的特异性,我们使用生物素化的plap重组蛋白进行了facs(图6b)。生物素化plap蛋白识别plap-car以及fab抗体,这证明人源化的plap-scfv与plap抗原特异性结合(图6b)。
与模拟对照car-t细胞相比,plap-car-t细胞(h2和h4)在rtca试验中显著杀死plap阳性细胞,而未显著杀死plap阴性细胞(图6c)。此外,plap-car-t细胞针对plap阳性结肠癌细胞分泌显著水平的ifn-γ、il-2和il-6,但不针对plap阴性结肠癌细胞分泌显著水平的ifn-γ、il-2和il-6(图6d)。这些数据表明,人源化的plap-car-t细胞特异性地并有效地杀死plap阳性结肠癌细胞。
实施例12、人源化的plap-car-t细胞(h2和h4)显著降低结肠癌异种移植肿瘤的生长。
我们在lovo异种移植小鼠模型体内分析了plap-car-t细胞功效(图7)。nsg小鼠皮下注射lovo癌细胞,并然后在第1、7、13天分别注射car-t细胞。人源化plaph2和plaph4-car-t细胞显著降低lovo异种移植肿瘤生长(图7a)。人源化plap-car-t细胞显著减小了肿瘤大小(图7b)和肿瘤重量(图7c)。plap-car-t细胞未降低小鼠体重,这表明car-t细胞毒性为阴性。在第16天,使用抗人cd3抗体在小鼠血液中检测到人t细胞和car-t细胞,证明了人源化的plap-car-t细胞在体内的持久性。
为了测试car-t细胞的毒性,我们对来自小鼠血清的若干种酶(ast、alt、和淀粉酶)进行了分析(图7d)。plap-car-t细胞对这些酶没有毒性作用(图7d),这表明体内plap-car-t细胞没有毒性。因此,plap-car-t细胞在体内具有高效、无毒性的特点。
实施例13、人源化的plap-car-t细胞(h5)特异性地杀死plap阳性细胞
根据实施例1进行实时细胞毒性试验(rcta)和ifn-γ试验。
图8a显示,与仅使用t细胞和靶细胞相比,plaph5-car-t细胞显著地杀死了plap阳性结肠癌细胞(caco-2细胞和lovo细胞)。rtca显示,plaph5-car-t细胞(h5)不会杀死plap阴性结肠癌细胞(hct116)。图8b显示,plaph5-car-t细胞针对plap阳性结肠癌细胞(caco-2细胞和lovo细胞)分泌显著更高水平的ifn-γ,但不针对plap阴性结肠癌细胞(hct116)分泌显著更高水平的ifn-γ。
这些数据表明,人源化的plaph5-car-t细胞特异性且有效地杀死plap阳性结肠癌细胞,并针对plap阳性结肠癌细胞系特异性地分泌ifn-γ。
实施例14、plap-car-t细胞联合检查点抑制剂可增加car-t细胞的活性。
将hplap-car-t细胞和结肠癌靶细胞共培养24小时时后,我们检测了响应于hplap-car-t细胞的结肠癌靶细胞上的pdl-1的表达(图9a)。我们还使用ifn-γ(一种已知在癌细胞中诱导pdl-1的药剂)[35]作为pdl-1诱导表达的阳性对照。plap阴性细胞ht29和hct116细胞响应于hplap-car-t细胞激活了pdl-1,与其对t细胞、模拟car-t细胞和ifn-γ的响应相似(图9a)。与此相反,与对t细胞和模拟car-t细胞的响应相比,plap阳性lovo细胞响应于car-t细胞显著上调pdl-1,且高于对ifn-γ的响应(图9a)。caco-2细胞响应于ifn-γ以及plap-car-t细胞均未激活pdl-1(图9a)。这些数据表明,在plap阳性癌细胞中,car-t细胞引起pdl-1显著上调,并且不同plap阳性癌细胞的pdl-1上调水平不同,并且与模拟car-t细胞和未转导t细胞相比,plap-car-t细胞在plap阴性结肠癌细胞中未引起pdl-1显著上调。
由于lovo细胞对plap-car-t细胞的响应比对ifn-γ的响应更能激活pdl-1(图9a),因此我们更详细地专注于该细胞系中pdl-1的上调。pdl-1的表达在加入car-t细胞后1小时和4小时较低,并在24小时引起pdl-1显著上调(图9b),其水平在49小时没有增加更多(未显示)。我们将不同剂量的hplap-car-t细胞加入到lovo细胞中,共培养24小时,并检测lovo结肠癌靶细胞中pdl-1上调对hplap-car-t细胞的剂量依赖性反应(图9c)。即使在向靶向癌细胞中加入小剂量的plap-car-t细胞时(效应细胞与靶细胞之比,e:t=0.3:1),pdl-1也显著上调(图9c)。
为了评估在与结肠癌细胞共孵育后,car-t细胞中检查点蛋白的上调,我们测试了若干种检查点蛋白:pd-1、tim-3、tigit和lag-3。与共培养前相比,在与阳性结肠癌靶细胞共培养后,只有pd-1在car-t细胞中显著上调(图9d)。在与plap阳性细胞(caco-2和lovo细胞)共培养后pd-1蛋白水平上调,但与plap阴性hct116和ht29细胞共培养后不上调(图9d)。lag-3在与lovo癌细胞系共培养后也显著上调(图9e)。因此,plap阳性靶细胞上调pdl-1,并且plap-car-t细胞上调pd-1或lag-3的表达。
为了测试检查点抑制剂与plap-car-t细胞的组合,我们将plap-h2-car-t细胞与pd-1抗体或lag-3抗体组合,并用lovo靶细胞进行rtca分析(图9f)。与仅使用同型抗体的plap-car-t细胞或与仅使用pd-1抗体或lag3抗体相比,联合使用plap-car-t细胞和pd1抗体或lag3抗体显著上调的细胞毒性(图9f)。在lovo细胞中,与每个单独治疗相比,plap-car-t细胞与pd-1抗体或lag-3抗体联用显著增加了ifn-γ的分泌(图9g)。当plap-car-t细胞和pd-1抗体与治疗前经ifn-g预处理pdl-1上调的靶细胞共培养时,也观察到ifn-γ分泌增加,从而证实了上述数据(未显示)。因此,将hplap-car-t细胞与检查点抑制剂(pd1抗体或lag3抗体)联合使用,是诱导plap-car-t细胞增加ifn-γ分泌对抗结肠癌的有效方法。
参考文献
1.eshhar,z.,etal.cancerj2014,20,123-126.
2.maus,m.v.,etal.clincancerres2016,22,1875-1884.
3.locke,f.l.,etal.lancetoncol2019,20,31-42.
4.locke,f.l.,etal.molther2017,25,285-295.
5.grupp,s.a.bestpractresclinhaematol2014,27,222-228.
6.eshhar,z.,etal.brjcancersuppl1990,10,27-29.
7.golubovskaya,v.,etal.cancers(basel)2016,8.
8.golubovskaya,v.cancers(basel)2017,9.
9.june,c.h.,etal.science2018,359,1361-1365.
10.fraietta,j.a.,etal.nature2018,558,307-312.
11.fraietta,j.a.,etal.natmed2018,24,563-571.
12.fry,t.j.,etal.natmed2018,24,20-28.
13.carpenter,r.o.,etal.clincancerres2013,19,2048-2060.
14.cohen,a.d.,etal.jclininvest2019,130.
15.anurathapan,u.,etal.cytotherapy2014,16,713-733.
16.beatty,g.l.,etal.oncoimmunology2014,3,e970027.
17.chen,n.,etal.oncoimmunology2017,6,e1273302.
18.gilham,d.e.,etal.trendsmolmed2012,18,377-384.
19.durbin,h.,etal.intjcancersuppl1988,2,50-58.
20.tucker,d.f.,etal.brjcancer1985,51,631-639.
21.epenetos,a.a.,etal.brjcancer1985,51,641-644.
22.epenetos,a.a.,etal.lancet1985,2,350-353.
23.orsaria,m.,etal.cancerbiomark2016,17,479-486.
24.vergote,i.b.,etal.tumourbiol1992,13,168-174.
25.harmenberg,u.,etal.tumourbiol1991,12,237-248.
26.sandfeld-paulsen,b.,etal.moloncol2016,10,1595-1602.
27.kalyan,a.,etal.jgastrointestoncol2018,9,160-169.
28.bacac,m.,etal.clincancerres2016,22,3286-3297.
29.hsia,l.t.,etal.procnatlacadsciusa2016,113,e2162-2171.
30.berahovich,r.,etal.cancers(basel)2018,10.
31.golubovskaya,v.,etal.cancers(basel)2017,9.
32.almagro,j.c.,etal.frontbiosci2008,13,1619-1633.
33.gilliland,g.l.,etal.methodsmolbiol2012,841,321-349.
34.berahovich,r.,etal.frontbiosci(landmarked)2017,22,1644-1654.
35.rozman,p.,etal.cytokinegrowthfactorrev2018,41,40-53.
36.waks,a.g.,etal.patholrespract2019,215,251-255.
38.kosmas,c.,etal.oncology1998,55,435-446.
39.muensch,h.a.,etal.cancer1986,58,1689-1694.
40.dunn,e.f.,etal.oncogene2011,30,561-574.
- 还没有人留言评论。精彩留言会获得点赞!