具有方酸连接的标记前体的制作方法
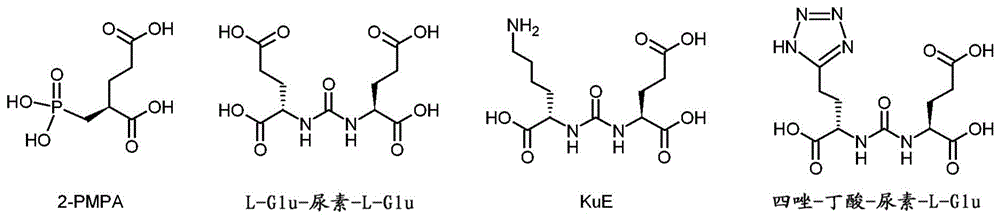
本发明涉及标记前体,其包含螯合剂或氟化基团和一个或两个靶向载体,所述靶向载体各自经由连接基和任选的间隔基连接至螯合剂或氟化基团。根据本发明的标记前体旨在用于癌症的核医学成像型放射诊断和治疗(治疗诊断学),其中肿瘤细胞表达前列腺特异性膜抗原(psma)、成纤维细胞活化蛋白(fap)或法尼基焦磷酸合酶(fpps)。在临床治疗中,大约15年以来,一直使用成像型放射诊断方法,如正电子发射断层扫描法(pet)和单光子发射计算机断层扫描法(singlephotonemissioncomputedtomography,spect)。近来,治疗诊断学方法也变得越来越重要。在放射诊断学和治疗诊断学中,将肿瘤细胞用放射性同位素例如68ga或177lu进行标记或照射。在此,使用标记前体,其共价(18f)或配位(68ga、99mtc、177lu)结合各自的放射性同位素。在金属放射性同位素的情况下,标记前体包含作为必需化学组分的用于有效和稳定地络合放射性同位素的螯合剂以及作为功能性组分的结合在肿瘤组织中的靶结构上的生物靶向载体。通常,生物靶向载体对肿瘤细胞的细胞膜上的受体、蛋白质或酶具有高亲和力。在静脉注射至血液循环中之后,被放射性同位素标记的标记前体积聚在肿瘤细胞之上或之中。为了使健康组织中的在诊断检查时的辐射剂量最小化,使用了少量具有几小时至几天的短半衰期的放射性同位素。通过螯合剂或氟化基团修饰靶向载体的构型和化学性质,并且通常强烈影响其对肿瘤细胞的亲和力。因此,螯合剂或氟化基团与至少一种靶向载体之间的连接在复杂的反复试验或所谓的生化筛选中进行定制。在此合成大量包含螯合剂或氟化基团以及至少一个靶向载体的标记前体,并且尤其定量对肿瘤细胞的亲和力。螯合剂或氟化基团以及与靶向载体的化学连接对于各个标记前体的生物学和放射学效力具有决定性作用。除了高亲和力之外,标记前体还必须满足另外的要求,如-各个放射性同位素快速和有效的络合或共价键合;-相对于健康组织而对肿瘤细胞的高选择性;-体内稳定性,即生理条件下血清中的生化抗性。前列腺癌对于发达国家的男性而言,前列腺癌是最常见的癌症类型和第三常见致命癌症。在该疾病的情况下,肿瘤生长进展缓慢,并且在早期诊断的情况下,5年的生存率接近100%。如果在肿瘤已转移才发现该疾病,则生存率会急剧下降。针对肿瘤的过早和过于积进的方法反而可能不必要地损害患者的生活质量。例如手术切除前列腺可能导致尿失禁和阳痿。疾病的可靠的诊断和分期信息对于以患者的高生活质量的成功治疗而言至关重要。除了由医师触诊前列腺之外,一种广泛的诊断工具是测定患者血液中的肿瘤标记物。前列腺癌的最突出的标志是血液中的前列腺特异性抗原(psa)的浓度。然而,psa浓度的效力尚无定论,因为具有稍微升高的值的患者经常没有前列腺癌,而15%的前列腺癌患者并不显示出血液中升高的psa浓度。用于诊断前列腺肿瘤的另一靶结构是前列腺特异性膜抗原(psma)。与psa相反,血液中不能检测到psma。它是具有酶活性的膜结合糖蛋白。其任务在于从n-乙酰天冬氨酰谷氨酸(naag)和叶酸(聚)-γ-谷氨酸中裂解c端谷氨酸。psma几乎不出现在正常组织中,但在前列腺癌细胞中强烈过度表达,其中该表达与肿瘤疾病的分期密切相关。同样,40%的前列腺癌的淋巴结转移和骨转移显示出psma的表达。psma的分子靶向的一种策略在于,用抗体结合psma的蛋白质结构。另一种方式在于利用熟知的psma的酶促活性。在psma的酶结合袋中存在两个结合谷氨酸的zn2+离子。在具有两个zn2+离子的中心之前存在一个芳族结合袋。该蛋白质能够扩展并且适应结合配偶体(诱导契合),使得除了naag之外,它还可以结合叶酸,其中使蝶酸基团停靠在芳香族结合袋中。利用psma的酶促亲和力可以使得底物被吸收到细胞中(内吞作用),而与底物的酶促裂解无关。因此,psma抑制剂尤其良好地适合作为成像型诊断学和诊断治疗学放射性药物或者放射性示踪剂的靶向载体。经放射性标记的抑制剂结合在酶的活性中心,但不在那里被转化。因此,不解开抑制剂与放射性标记之间的键。借助内吞作用,使具有放射性标记的抑制剂被吸收到细胞中并且在肿瘤细胞内积累。具有对psma的高亲和力的抑制剂通常含有谷氨酸基序和不可酶促裂解的结构。高效的psma抑制剂是2-膦酰基甲基-戊二酸或者2-膦酰基甲基-戊烷二酸(2-pmpa),其中的谷氨酸基序与不能被psma裂解的膦酸酯基团结合。用于临床相关的放射性药物psma-11和psma-617中的另一组psma抑制剂是基于脲的抑制剂。经证实有利的是,除了解决谷氨酸基序的结合袋以外,还解决psma的芳族结合袋也是有利的。例如,在高效放射性药物psma-11中,l-赖氨酸-脲-l-谷氨酸盐(kue)结合基序通过己基(己基连接基)结合到芳香族hbed螯合剂(n,n'-双(2-羟基-5-(亚乙基-β-羧基)苄基)乙二胺n,n'-二乙酸酯)。反之,如果l-赖氨酸-脲-l-谷氨酸盐(kue)结合至非芳香族螯合剂dota(1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸盐),则发现肿瘤组织中降低的亲和力和累积。为了依然可以将dota螯合剂用于具有治疗性放射性同位素如177lu或225ac的psma仿射放射性药物,必须改造连接基。借助通过各种芳香结构有目的地取代己基,发现作为当前的黄金标准的高效放射性药物psma-617。(图1:psma抑制剂;图2:标记前体psma-11;图3:标记前体psma-617)。肿瘤间质许多肿瘤包括恶性上皮细胞,并被多个非癌性细胞群包围,包括活化的成纤维细胞、内皮细胞、周细胞、免疫调节细胞和细胞外基质中的细胞因子。围绕肿瘤的这些所谓基质细胞在癌的发展、生长和转移中起重要作用。基质细胞的很大一部分是活化的成纤维细胞,其被称为癌症相关的成纤维细胞(caf)。随着肿瘤的进展,caf改变其形态和生物学功能。这些改变是由癌细胞与caf之间的细胞间通讯诱导的。在此,caf形成有利于癌细胞生长的微环境。已显示,单独地靶向癌细胞的疗法是不充分的。有效的疗法必须将肿瘤的微环境,即caf列入。在所有人类癌症的大于90%的情况下,由caf过度表达成纤维细胞活化蛋白(fap)。因此,fap代表放射诊断和治疗诊断学的有前途的攻击点。适合作为fap标记前体的仿射靶向载体的尤其是类似于psma的fap抑制剂(fapi或fapi)。fap具有双峰的、经由二肽基肽酶(dpp)和脯氨酰寡肽酶(prep)相同的活性中心催化的活性。因此考虑抑制fap的dpp活性和/或prep活性的两种类型的抑制剂。已知的fap的prep活性的抑制剂具有对fap的低选择性。在过度表达fap以及prep的癌症类型中,尽管prep抑制剂的fap选择性低,但它们也适合作为靶向载体。图4显示了dota缀合的fap标记前体,其中螯合剂经由喹啉上的4-氨基丁氧基官能团连接到药效团((s)-n-(2-(2-氰基-4,4-二氟吡咯烷-1-基)-2-氧代乙基)-6-(4-氨基-丁氧基)-喹啉-4-甲酰胺。(图4:dota缀合的fap标记前体)骨转移骨转移表达法尼基焦磷酸合酶(fpps),其为hmg-coa还原酶-(甲羟戊酸)-途径中的一种酶。通过抑制fpps抑制了法尼基的产生,这是信号蛋白与细胞膜对接的重要分子。结果,诱导了致癌骨细胞的凋亡。fpps被双膦酸盐,如阿仑膦酸盐、帕米膦酸盐和唑来膦酸盐抑制。例如在治疗骨转移时通常使用示踪剂bpamd与靶向载体帕米膦酸盐。作为用于治疗骨转移的治疗诊断学的特别有效的示踪剂,经证实有效的是唑来膦酸盐(zol),其为具有杂芳族n单元的羟基双膦酸盐。与螯合剂nodaga和dota缀合的唑来膦酸盐(图5)目前是最有效的骨转移放射治疗诊断学(图5:示踪剂dota-唑来膦酸盐(左)和nodaga-唑来膦酸盐(右))。现有技术中已知大量采用放射性同位素的用于癌症疾病的诊断和治疗诊断学的标记前体。wo2015055318a1公开了用于前列腺或上皮癌的诊断和治疗诊断学的放射性示踪剂,如尤其是图3中所示的化合物psma-617。本发明的任务在于提供对于前列腺癌和基质癌的诊断和治疗诊断有效的放射性示踪剂的标记前体,其具有高的肿瘤选择性和剂量。该任务通过结构(a)、(b)、(c)、(d)、(e)、(f)、(g)、(h)、(i)、(j)、(k)或(l)的标记前体得以解决,其中(a)=ch-l1-qs-tv1,(b)=ch-l1-qs-s1-tv1,(c)=ch-l1-qs-s1-qs-tv1,(d)=ch-l1-qs-s2-qs-s1-tv1,(e)=tv2-qs-l2-ch-l1-qs-tv1,(f)=tv2-s3-qs-l2-ch-l1-qs-s1-tv1,(g)=tv2-qs-s4-qs-l2-ch-l1-qs-s2-qs-tv1,(h)=tv2-s3-qs-s4-qs-l2-ch-l1-qs-s2-qs-s1-tv1,(i)=fg-l1-qs-tv1,(j)=fg-l1-qs-s1-tv1,(k)=fg-l1-qs-s2-qs-tv1,(l)=fg-l1-qs-s2-qs-s1-tv1;其包含-螯合剂ch,选自:edta(乙二胺四乙酸盐)、edtmp(二亚乙基三胺五(亚甲基膦酸))、dtpa(二亚乙基三胺五乙酸盐)及其衍生物、dota(十二碳-1,4,7,10-四胺-四乙酸盐)、dotaga(2-(1,4,7,10-四氮杂环十二烷-4,7,10)-戊烷二酸)和其它dota衍生物、trita(十三碳-1,4,7,10-四胺-四乙酸盐)、teta(十四碳-1,4,8,11-四胺-四乙酸盐)及其衍生物、nota(九碳-1,4,7-三胺-三乙酸盐)及其衍生物如notaga(1,4,7-三氮杂环壬烷,1-戊二酸,4,7-乙酸盐)、nopo(1,4,7-三氮杂环壬烷-1,4-双[亚甲基(羟基甲基)次膦酸]-7-[亚甲基(2-羧基乙基)次膦酸])、pepa(十五碳-1,4,7,10,13-五胺-五乙酸盐)、heha(十六碳-1,4,7,10,13,16-六胺-六乙酸盐)及其衍生物、hbed(羟基苄基-亚乙基-二胺)及其衍生物、dedpa及其衍生物如h2dedpa(1,2-[[6-(羧酸盐)吡啶-2-基]甲基胺]乙烷)、dfo(去铁胺)及其衍生物、三羟基吡啶酮(thp)及其衍生物如ym103、trap(三氮杂环壬烷次膦酸)、teap(四氮杂环癸烷次膦酸)及其衍生物、aazta(6-氨基-6-甲基全氢-1,4-二氮杂-n,n,n',n'-四乙酸盐)和衍生物如data((6-戊酸)-6-(氨基)甲基-1,4-二氮杂三乙酸盐);sarar(1-n-(4-氨基苄基)-3,6,10,13,16,19-六氮杂双环[6.6.6]-二十碳-1,8-二胺)及其盐、氨基硫醇和以下类型的它们的衍生物或-氟化基团fg,其选自-一个或两个连接基l1和l2,其彼此独立地选自酰胺残基、羧酰胺残基、次膦酸酯/盐残基、烷基残基、三唑残基、硫脲残基、亚乙基残基、马来酰亚胺残基、-(ch2)m-、-(ch2ch2o)m-和-(ch2)mnh-,其中m=1、2、3、4、5、6、7、8、9或10;-一个或更多个方酸残基qs-任选地一个、两个、三个或四个间隔基sj,其中1≤j≤4,其彼此独立地选自酰胺残基、羧酰胺残基、次膦酸酯/盐残基、烷基残基、三唑残基、硫脲残基、亚乙基残基、马来酰亚胺残基、-(ch2)n-、-(ch2)-ch(cooh)-nh-、-(ch2ch2o)n-和-(ch2)n-nh-,其中n=1、2、3、4、5、6、7、8、9或10;和-一个或两个靶向载体tv1和tv2,其彼此独立地选自结构[1]至[41]的化合物的残基,其中其中,y为保护基,和x'=cl、br或i,和靶向载体[1]-[41]的虚线键表示与离去基团的键合位点。根据本发明的标记前体的有利的实施方案的特征在于-标记前体含有正好一个靶向载体tv1;-标记前体含有两个靶向载体tv1和tv2,其中tv1≠tv2,它们彼此不同;-标记前体含有两个相同的靶向载体tv1和tv2,其中tv1=tv2;-保护基y选自叔丁氧羰基(叔丁基)、三烷基甲硅烷基基团、三甲基甲硅烷基(-si(ch3)3)、三乙基甲硅烷基(-si(ch2ch3)3)、异丙基二甲基甲硅烷基(-si(ch3)2c(ch3)2)、叔丁基二甲基甲硅烷基(-si(ch3)2c(ch3)3)和叔丁氧基二甲基甲硅烷基(-si(ch3)2oc(ch3)3);-连接基l1和l2相同(l1=l2);-连接基l1和l2彼此不同(l1≠l2);-间隔基s1和s3相同(s1=s3);-间隔基s1和s3彼此不同(s1≠s3);-间隔基s2和s4相同(s2=s4);和/或-间隔基s2和s4彼此不同(s2≠s4)。具有螯合剂ch或氟化基团fg的根据本发明的标记前体旨在用选自下组的放射性同位素进行标记:44sc、47sc、55co、62cu、64cu、67cu、66ga、67ga、68ga、89zr、86y、90y、90nb、99mtc、111in、135sm、140pr、159gd、149tb、160tb、161tb、165er、166dy、166ho、175yb、177lu、186re、188re、213bi和225ac,或者18f、131i或211at。因此,本发明另外涉及包含上述标记前体之一的放射性示踪剂化合物,所述标记前体具有:-螯合剂ch和选自下组的络合的放射性同位素:44sc、47sc、55co、62cu、64cu、67cu、66ga、67ga、68ga、89zr、86y、90y、90nb、99mtc、111in、135sm、140pr、159gd、149tb、160tb、161tb、165er、166dy、166ho、175yb、177lu、186re、188re、213bi和225ac;或-氟化基团fg和共价键合的放射性同位素18f、131i或211at或者含有18f、131i或211at的共价键合的基团,尤其是-cf218f(三氟甲基)。此外,本发明涉及上述标记前体用于制备放射性药物的用途。在有利的实施方案中,将上述标记前体用于制备用44sc、47sc、55co、62cu、64cu、67cu、66ga、67ga、68ga、89zr、86y、90y、90nb、99mtc、111in、135sm、140pr、159gd、149tb、160tb、161tb、165er、166dy、166ho、175yb、177lu、186re、188re、213bi、225ac、18f、131i或211at标记的放射性药物。在有利的实施方案中,将上述标记前体用于制备用于正电子发射断层(pet)成像诊断的放射性药物。在有利的实施方案中,将上述标记前体用于制备用于单光子发射计算机断层扫描(spect)成像诊断的放射性药物。在有利的实施方案中,将上述标记前体用于制备用于治疗癌性肿瘤的放射性药物。此外,本发明的任务在于,提供用于合成标记前体的简单和有效的方法,所述标记前体用于诊断和治疗诊断用psma和/或fap表达的癌性肿瘤。该任务通过包括下述步骤的方法得以解决:-使螯合剂ch或氟化基团fg与连接基l1缀合成前体p1=ch-l1或者p1=fg-l1,或使螯合剂ch或氟化基团fg与连接基l1和方酸qs缀合成前体p2=ch-l1-qs或者p2=fg-l1-qs,或使螯合剂ch与连接基l1和l2缀合成前体p3=l2-ch-l1,或使螯合剂ch与连接基l1、l2和方酸qs缀合成前体p4=qs-l2-ch-l1-qs;-任选地,使靶向载体tv1与方酸qs缀合成前体p5=tv1-qs,或使靶向载体tv1与方酸qs和间隔基s2缀合成前体p6=tv1-qs-s2,或使靶向载体tv1与间隔基s1缀合成前体p7=tv1-s1,或使靶向载体tv1与间隔基s1和方酸qs缀合成前体p8=tv1-s1-qs,或使靶向载体tv1与间隔基s1、方酸qs和间隔基s2缀合成前体p9=tv1-s1-qs-s2;-任选地,使靶向载体tv2与方酸qs缀合成前体p10=tv2-qs,或使靶向载体tv2与方酸qs和间隔基s4缀合成前体p11=tv2-qs-s4,或使靶向载体tv2与间隔基s3缀合成前体p12=tv2-s3,或使靶向载体tv2与间隔基s3和方酸qs缀合成前体p13=tv2-s3-qs,或使靶向载体tv2与间隔基s3、方酸qs和间隔基s4缀合成前体p14=tv2-s3-qs-s4;-使靶向载体tv1与所述前体p2缀合成或使前体p1和p5缀合成结构ch-l1-qs-tv1或fg-l1-qs-tv1的标记前体,或使前体p1和p8或p2和p7缀合成结构ch-l1-qs-s1-tv1或fg-l1-qs-s1-tv1的标记前体,或使前体p2和p9缀合成结构ch-l1-qs-s2-qs-s1-tv1或fg-l1-qs-s2-qs-s1-tv1的标记前体;或-使所述前体p3、p5和p10缀合成结构tv2-qs-l2-ch-l1-qs-tv1的标记前体,或使前体p3、p8和p13或p4、p7和p12缀合成结构tv2-s3-qs-l2-ch-l1-qs-s1-tv1的标记前体,或使前体p4、p6和p11缀合成结构tv2-qs-s4-qs-l2-ch-l1-qs-s2-qs-tv1的标记前体,或使前体p4、p9和p14缀合成结构tv2-s3-qs-s4-qs-l2-ch-l1-qs-s2-qs-s1-tv1的标记前体;其中,-螯合剂ch选自edta(乙二胺四乙酸盐)、edtmp(二亚乙基三胺五(亚甲基膦酸))、dtpa(二亚乙基三胺五乙酸盐)及其衍生物、dota(十二碳-1,4,7,10-四胺-四乙酸盐)、dotaga(2-(1,4,7,10-四氮杂环十二烷-4,7,10)-戊烷二酸)和其它dota衍生物、trita(十三碳-1,4,7,10-四胺-四乙酸盐)、teta(十四碳-1,4,8,11-四胺-四乙酸盐)及其衍生物、nota(九碳-1,4,7-三胺-三乙酸盐)及其衍生物如notaga(1,4,7-三氮杂环壬烷,1-戊二酸,4,7-乙酸盐)、nopo(1,4,7-三氮杂环壬烷-1,4-双[亚甲基(羟基甲基)次膦酸]-7-[亚甲基(2-羧基乙基)次膦酸])、pepa(十五碳-1,4,7,10,13-五胺-五乙酸盐)、heha(十六碳-1,4,7,10,13,16-六胺-六乙酸盐)及其衍生物、hbed(羟基苄基-亚乙基-二胺)及其衍生物、dedpa及其衍生物如h2dedpa(1,2-[[6-(羧酸盐)吡啶-2-基]甲基胺]乙烷)、dfo(去铁胺)及其衍生物、三羟基吡啶酮(thp)及其衍生物如ym103、trap(三氮杂环壬烷次膦酸)、teap(四氮杂环癸烷次膦酸)及其衍生物、aazta(6-氨基-6-甲基全氢-1,4-二氮杂-n,n,n',n'-四乙酸盐)和衍生物如data((6-戊酸)-6-(氨基)甲基-1,4-二氮杂三乙酸盐);sarar(1-n-(4-氨基苄基)-3,6,10,13,16,19-六氮杂双环[6.6.6]-二十碳-1,8-二胺)及其盐、氨基硫醇和以下类型的它们的衍生物所述氟化基团fg选自-所述连接基l1和l2彼此独立地选自酰胺残基、羧酰胺残基、次膦酸酯/盐残基、烷基残基、三唑残基、硫脲残基、亚乙基残基、马来酰亚胺残基、-(ch2)m-、-(ch2ch2o)m-和-(ch2)mnh-,其中m=1、2、3、4、5、6、7、8、9或10;-所述间隔基sj彼此独立地选自酰胺残基、羧酰胺残基、次膦酸酯/盐残基、烷基残基、三唑残基、硫脲残基、亚乙基残基、马来酰亚胺残基、-(ch2)n-、-(ch2)-ch(cooh)-nh-、-(ch2ch2o)n-和-(ch2)nnh-,其中n=1、2、3、4、5、6、7、8、9或10,其中1≤j≤4;和-所述靶向载体tv1和tv2彼此独立地选自结构[1]至[41]的化合物其中,y为保护基,和x'=cl、br或i,和靶向载体[1]-[41]的虚线键表示与离去基团的键合位点。根据本发明的方法的有利的实施方案的特征在于,-靶向载体tv1和tv2彼此不同(tv1≠tv2);-靶向载体tv1和tv2相同(tv1=tv2);-保护基y选自叔丁氧羰基(叔丁基)、三烷基甲硅烷基基团、三甲基甲硅烷基(-si(ch3)3)、三乙基甲硅烷基(-si(ch2ch3)3)、异丙基二甲基甲硅烷基(-si(ch3)2c(ch3)2)、叔丁基二甲基甲硅烷基(-si(ch3)2c(ch3)3)和叔丁氧基二甲基甲硅烷基(-si(ch3)2oc(ch3)3);-连接基l1和l2相同(l1=l2);-连接基l1和l2彼此不同(l1≠l2);-间隔基s1和s3相同(s1=s3);-间隔基s1和s3彼此不同(s1≠s3);-间隔基s2和s4相同(s2=s4);和/或-间隔基s2和s4彼此不同(s2≠s4)。氟化基团fg包括标记有18f、131i或211at之一的离去基团x。离去基团x为下述的残基:溴(br)、氯(cl)、碘(i)、对甲苯磺酰基(-so2-c6h4-ch3;缩写为"ts")、硝基苯磺酸酯或硝基苯磺酸盐(-oso2-c6h4-no2;缩写为"nos")、2-(n-吗啉代)乙磺酸(-so3-(ch2)2-n(ch2)4o;缩写为"mes")、三氟甲磺酸盐或者三氟甲烷磺酰基(-so2cf3;缩写为"tf")或九氟丁磺酸盐(-oso2-c4f9;缩写为"non")。在本发明的范围内,使用以下简称或缩写:psma.........前列腺特异性膜抗原;fap.............成纤维细胞活化蛋白;fpps...........法尼基焦磷酸合酶;2-pmpa......2-膦酰基甲基戊二酸;kue............l-赖氨酸-脲-l-谷氨酸盐;dota.qs.psma标记前体,尤其是具有根据图8的结构式,包括dota(十二碳-1,4,7,10-四胺-四乙酸盐)作为螯合剂,所述螯合剂通过一个或两个连接基、方酸基团和间隔基与一个或两个具有结构式[1]、[2]、[3]和/或[4]的psma靶向载体连接;nota.qs.psma标记前体,尤其是具有根据图9的结构式,包括nota(九碳-1,4,7-三胺-三乙酸盐)作为螯合剂,所述螯合剂通过一个或两个连接基、方酸基团和间隔基与一个或两个具有结构式[1]、[2]、[3]和/或[4]的psma靶向载体连接;aazta.qs.psma标记前体,尤其是具有根据图9的结构式,包括aazta作为螯合剂,所述螯合剂通过一个或两个连接基、方酸基团和间隔基与一个或两个具有结构式[1]、[2]、[3]和/或[4]的psma靶向载体连接;data.qs.psma标记前体,尤其是具有根据图9的结构式,包括data作为螯合剂,所述螯合剂通过一个或两个连接基、方酸基团和间隔基与一个或两个具有结构式[1]、[2]、[3]和/或[4]的psma靶向载体连接;dotaga.qs.psma标记前体,包括dotaga(2-(1,4,7,10-四氮杂环十二烷-4,7,10)戊烷二酸)作为螯合剂,所述螯合剂通过一个或两个连接基、方酸基团和间隔基与一个或两个具有结构式[1]、[2]、[3]和/或[4]的psma靶向载体连接;notaga.qs.psma标记前体,包括notaga(1,4,7-三氮杂环壬烷,1-戊二酸,4,7-乙酸盐)作为螯合剂,所述螯合剂通过一个或两个连接基、方酸基团和间隔基与一个或两个具有结构式[1]、[2]、[3]和/或[4]的psma靶向载体连接;trap.qs.psma标记前体,包括trap(三氮杂环壬烷次膦酸)作为螯合剂,所述螯合剂通过一个或两个连接基、方酸基团和间隔基与一个或两个具有结构式[1]、[2]、[3]和/或[4]的psma靶向载体连接;nota.qs.pam标记前体,包括nota作为螯合剂,所述螯合剂通过一个或两个连接基、方酸基团和间隔基与一个或两个具有结构式[40]的帕米膦酸盐靶向载体连接。另外的在本发明范围内使用的缩写对应于上述缩写,其中另一个螯合剂、另一个氟化基团和/或另一个靶向载体-尤其是根据结构式[5]至[41]的fap的靶向载体-以类似的方式通过其相应的缩写或首字母缩写来表示。例如,在骨转移中用于靶向法尼基焦磷酸合酶(fpps)的类似衍生物根据双膦酸盐的类型,将帕米膦酸盐缩写为"pam"和将唑来膦酸盐缩写为"zol"。根据本发明的标记前体任选地包含一个或更多个间隔基sj,其中1≤j≤4,即,一个间隔基s1、两个间隔基s1和s2、三个间隔基s1、s2和s3或四个间隔基s1、s2、s3和s4。在靶向载体的结构式[1]-[41]中,以虚线示出了根据本发明的标记前体的为了与方酸基团或间隔基s1或s2缀合的靶向载体。通过虚线键缀合的基团是离去基团,其在将靶向载体与方酸基团或间隔基s1或s2连接时解离。在下文借助附图和实施例更详细地阐述本发明。图6显示了一些根据本发明使用的螯合剂ch的结构(图6:根据本发明使用的螯合剂)。实施例1:psma标记前体的合成策略在合成根据本发明的标记前体时,优选使用方酸二酯。由此可借助简单的反应方法表现大量、有时非常复杂的标记前体。方酸二酯的特征在于它们与胺的选择性反应性,从而在连接螯合剂、连接基、间隔基和靶向载体时不需要保护基。此外,可通过ph值控制连接。首先,合成psma的靶向载体(参见图7),并且在含水介质中在ph=7纯化之后,用方酸二酯转化成义肢(prostethisch)基团或者与螯合剂连接的前体(参见图8)。(图7:qs-kue前体的合成方案)在psma的靶向载体的情况下,例如借助已知方法合成psma抑制剂l-赖氨酸-脲-l-谷氨酸盐(kue)。在此,使键合至固相,尤其是聚合物树脂并且用叔丁氧羰基(叔丁基)保护的赖氨酸与双重叔丁基保护的谷氨酸反应。在通过三光气活化经保护的谷氨酸和连接至固相结合的赖氨酸之后,将l-赖氨酸-脲-l-谷氨酸盐(kue)借助tfa解离并且同时完全脱保护。随后可以将产物借助半制备hplc与游离赖氨酸分离。前述反应基于赖氨酸计的收率大于50%(图8:qs-kue前体连接至dota)。使qs-kue前体在磷酸盐缓冲液中在9的ph值下与螯合剂dota缀合成标记前体dota.qs.psma。为了psma标记前体的放射性标记,将68ga用来自ithembage/ga的发生器的0.6mhcl稀释,并且借助含水乙醇洗脱液在阳离子交换柱上进行处理。放射性标记取决于螯合剂,在介于3.5与5.5之间的ph值和介于25℃与95℃之间的温度进行。借助hplc和iptc记录反应进程,以计算反应的动力学参数。实施例2:标记前体nota.qs.psma.aaztaqs.psma和data.qs.psma借助根据实施例1中描述的策略的合成,使用螯合剂nota、aazta和data代替dota而制备图9中所示的标记前体nota.qs.psma、aazta.qs.psma和data.qs.psma(图9:标记前体nota.qs.psma、aazta.qs.psma和data.qs.psma)。实施例3:放射性卤化物的psma标记前体借助根据图7的合成方案稍微修改的反应,制备图10中示出的用于用卤化物同位素如18f和131i放射性标记的psma标记前体。在图11中示出了对应的用18f标记的放射性示踪剂(图10:用于放射性卤化物的方酸缀合的psma标记前体;图11:在解离叔丁基保护基之前的用于psma的方酸缀合的18f放射性示踪剂)。实施例4:用于99mtc的psma标记前体借助根据实施例1中描述的策略的合成,制备图12中示出的用于用同位素99mtc进行放射性标记的psma标记前体(图12:99mtc的方酸缀合的psma标记前体与“n3s”类型的基于氨基硫醇的螯合剂)。实施例5:fap标记前体的合成策略(图13:fap标记前体的合成策略)图14显示了两个fap标记前体,其根据在图13中所示的合成策略制备。(图14:具有方酸连接的fap标记前体)实施例6:qs作为络合助剂对于临床应用而言非常重要的是,在低的温度有效地进行络合。方酸络合游离金属,并且因此可以保护螯合剂中心免受非特异性配位影响。可以在trap.qs的放射性标记中在不同温度观察到这种效果。trap在室温下定量络合。相比之下,在相同条件下在trap.qs的情况下测量的rcy值仅为50%。如果提高温度,则trap.qs的标记收率升高至定量值。这证明了方酸对络合的影响。图15中所示的该效果使得借助于螯合剂aazta.qs稳定地络合具有高配位数的金属例如锆成为可能(图15:借助aazta.qs配位)。实施例7:二聚标记前体,其各自具有两个kue和fapi靶向载体(图16:二聚标记前体)(i)具有两个胺侧基的do2a单元的合成:(图17:具有两个胺基团的doa2的合成)(ii)kue-qs基序的合成:(图18:kue-qs的合成)(iii)fapi-qs的合成,连接4,4-二氟脯氨酸-喹啉-4-羧酸基序与qs:(图19:fapi-qs的合成)(iv)连接do2a单元与kue-qs和分别的fapi-qs:(图20:二聚体的合成)实施例8:68ga-dota.qs.psma临床前研究借助pet,在右后腿上患有lncap肿瘤的nmrinu/nu小鼠上进行采用68ga-dota.qs.psma、68ga-psma-11和68ga-psma-617型放射性示踪剂的临床前对比试验。图21示出了在注射根据本发明的示踪剂68ga-dota.qs.psma之后60min的pet照片,其中部分照片(a)和(b)示出了未受阻的和分别借助共同注射2-pmpa阻断的肿瘤小鼠的pet图像。表1:psma示踪剂的标准化的摄入值(suv)从图22中示出的pet照片可看出,示踪剂在肿瘤中强烈富集。从pet数据计算,68ga-dota.qs.psma在肿瘤中的标准化的摄取值(standardizeduptakevalue,suv)为0.73。通过提取器官和测量体重和活性确定的生物分布数据显示,对于68ga-dota.qs.psma,与68ga-psma-11和68ga-psma-617相比,其肿瘤活性稍低或相等。相比之下,肾脏的脱靶活性明显低于68ga-psma-11的情形。与其他已知放射性示踪剂相比,68ga-dota.qs.psma在肾脏和肝脏中的脱靶富集明显降低。68ga-dota.qs.psma具有对肿瘤组织的高的亲和力,并且改进影像学pet诊断pca原发性肿瘤并且尤其是骨盆区域中受pca影响的淋巴结的对比度和信噪比。还减少了肾脏和邻近器官的辐射暴露,这为诊断治疗学治疗提供了显著优势。采用64cutrap.qs.psma和68ga-notaga.qs.psma的类似试验提供了相当的结果。此外,将dota.qs.psma用177lu和225ac标记。该示踪剂的放射学和生理学稳定性的初步结果表明其适用于诊断治疗学。由于psma的芳族结合袋对psma抑制剂的亲和力的影响,psma示踪剂的亲脂性具有一定的重要性。研究表明,提高的亲脂性还促进示踪剂在肿瘤细胞中的摄取或者内吞作用。因此,根据本发明的示踪剂trap.qs.psma和dota.qs.psma的亲脂性借助donovan和pescatore的hplc方法(s.f.donovan,m.c.pescatore,j.chromatogr.a2002,952,47-61)来测定。为此,在odp-hplc柱中以甲醇/水梯度在7的ph值下测量trap.qs.psma、dota.qs.psma和某些具有已知亲脂性的校准标准品的保留时间。表2连同psma-11和psma-617的文献值一起显示了trap.qs.psma和dota.qs.psma通过保留时间的线性回归计算的logd值。因为dota.qs.psma在odp-hplc柱上没有保留,所以对于logd仅给出最大值。trap.qs.psma、psma-11和psma-617具有相当的亲脂性。出人意料地,与psma-11和psma-617相比,肾脏中的trap.qs.psma的摄取明显降低。通过各个logd值的细微差异并不能解释此观察结果。显然,不仅亲脂性影响亲和力和内吞作用,而且其他相互作用如酶结合袋中的π-π积聚同样起作用。在此,方酸由于相比于苯基小的尺寸而显得有利。相比之下,dota.qs.psma相较于与psma-617相当的在肾脏中的摄取显示出高得多的亲脂性。表2:psma示踪剂的亲脂性示踪剂logd[nm]trap.qs.psma-1.5±0.5dota.qs.psma≤-3.5psma-11-1.7±0.6psma-617-2.0±0.3此外,借助pet,采用[68ga]ga-dota.qs.psma、[68ga]ga-psma-11和[68ga]ga-psma-617型放射性示踪剂在患有lncap肿瘤的nmrinu/nu裸小鼠上进行临床前离体试验。图23显示了相应化合物的器官分布。获得的结果强调了在体内试验中获得的结果。通过提取器官并且测量体重和活性确定的生物分布数据显示,[68ga]ga-dota.qs.psma的肿瘤活性与[68ga]ga-psma-11和[68ga]ga-psma-617相比略低或相同。相比之下,肾脏中的脱靶活性明显低于[68ga]ga-psma-11的情况。实施例9:fpps示踪剂双膦酸盐,如阿仑膦酸盐、帕米膦酸盐和唑来膦酸盐(分别为结构式[39]、[40]和[41])抑制法尼基焦磷酸合酶(fpps)并且诱导骨转移中的细胞凋亡。为了标记骨转移,根据实施例1中描述的策略合成具有螯合剂nota和靶向载体帕米膦酸盐(结构式[40])的方酸连接的示踪剂nota.qs.pam。图24示出了借助螯合剂nodaga的应用的合成方案(图24:nodaga.qs.pam的合成)。将根据本发明的示踪剂nota.qs.pam和在临床应用中建立的参考示踪剂dotazol用68ga标记,将其注射至年轻健康的wistar大鼠,并且在注射之后5min、60min和120min的时间间隔记录pet扫描。图25显示了在注射之后120min的示踪剂68ga-nota.qs.pam和68ga-dotazol的相应pet照片。两种示踪剂均显示具有提高的重塑速率的在骨骼区域的特异性摄取,尤其是在幼鼠仍在生长的骨骺中。除骨骼之外,膀胱也具有提高的活动性,并且指示优先的肾脏排泄。相比之下,在软组织中的保留极其低。与68ga-dotazol相比,68ga-nota.qs.pam的肾脏排泄量略有减少。该观察结果与psma示踪剂的肾脏排泄一致。这是靶组织中提高的累积与游离的、非特异性结合的示踪剂的加速的肾脏排泄相关的结果。在药理动力学方面,根据本发明的方酸连接的示踪剂具有相对于已知示踪剂的有利之处。在图26中,以条形图的方式示出了在注射之后5min和60min的间隔内不同器官中68ga-nota.qs.pam和68ga-dotazol的吸收值(suv)。活动性的器官分布显示,这两种示踪剂均显示出在骨骼中的高的摄取和保留,其中68ga-nota.qs.pam具有略高的股骨suv(suv股骨:68ga-dotazol=3.7±0.4;68ga-nota.qs.pam=4.5±0.2)。两种示踪剂的特征都在于肾脏排泄和在剩余组织中的低保留率。实施例10:qs.psma为了阐明qs的活性,如在dota.qs.psma情况下采用nodaga.qs.psma进行了相当的试验。图27和28显示了分别用放射性dc(图27)和放射性hplc(图28)测量的具有68ga的化合物的放射性标记。收率超过95%。在人血清中和在pbs缓冲液中进行了相应的稳定性测试。该化合物在pbs中和在hs中在2h之后显示出大于95%的稳定性。图29显示了借助hplc测量的稳定性。另外,在体内和离体试验了三个化合物dotaga.qs.psma、nodaga.qs.psma和trap.qs.psma。图30显示了分别用68ga标记的三个化合物的离体结果。从图30可以看出,所有三种化合物均在肿瘤组织中富集,其中nodaga.qs.psma显示出在肾脏中的低富集。实施例11:trap.qs.psma图31a-b、32-36显示了放射性示踪剂[68ga]ga-trap.qs.psma的合成以及放射性标记、稳定性和体内试验的测量结果。该结果与实施例8和10的结果相当。用68ga和64cu进行体内研究。从照片中可以看出,与实施例8和10相比,两种放射性示踪剂在肾脏中的富集均提高。这归因于不同的、由于长链连接基引起的实施例11的放射性示踪剂的亲脂性。离体对比显示,与[68ga]ga-psma-11相比,肿瘤组织中的富集提高并且在肾脏中明显降低。实施例12:[68ga]ga-dataqs.psma另外的根据本发明的化合物是data.qs.psma型的那些,其结构对应于其它所列出的化合物,其中螯合剂data使得更简单和更温和的标记成为可能。在图37中示出的合成的情况下实现了约70%的收率。在采用68ga的放射性标记的情况下,达到大于95%的收率(图38)。如从图39可以看出,化合物在人血清(hs)以及磷酸盐缓冲的盐水(pbs)中均具有高稳定性。在采用lncap细胞的化合物的体外试验中,获得51.5nm的ic50值,这与psma-11和psma-617相当(表3)。此外,将data.qs.psma型化合物与psma-11在同一动物模型中在体内进行比较。mip图像(图40)清楚地显示了对于[68ga]ga-data.qs.psma而言,也观察到在肿瘤中非常好的富集。此外,由图40中描绘的时间/活性曲线可以看出,[68ga]ga-data.qs.psma通过肾脏排泄在第一个小时中明显比[68ga]-psma-11的情况下更快地进行。在肿瘤组织中的富集是相当的。表3:未标记的化合物的ic50值化合物ic50[nm]psma-1126.1±1.2psma-61715.1data.qs.psma51.1±5.5离体试验的结果(图41-43)与体内观察结果一致。如从表4可以看出,两种采用68ga标记的化合物在肿瘤中的%id/g值是相当的,而在[68ga]ga-data.qs.psma的情况下,在肾脏和唾液腺中的富集是显著降低。唾液腺中的低富集是非常有利的,因为唾液腺在用于放射性药物治疗前列腺癌的已知方法中暴露于升高的剂量并且其功能显著受到损害。此外,采用2-pmpa的阻断研究表明,根据本发明的化合物对于psma而言具有增加的特异性。表4:离体活性实施例13:[44sc]sc-aazta.qs.psma与data类似,螯合剂aazta也能够在温和条件下用放射性核素,例如44sc和68ga标记。在该实施例中将44sc用于标记,并且研究了放射性示踪剂[44sc]sc-aazta.qs.psma的性质。在图44示出的合成可以简单地并且以高收率进行。如在图45中所示,在放射性标记的情况下也获得高收率。[44sc]sc-aazta.qs.psma的稳定性在24h的时间段内为大于95%(图46)。此外,在三只各自携带lncap肿瘤的小鼠中以体内方式研究放射性示踪剂[44sc]sc-aazta.qs.psma。此外,对小鼠之一进行了阻断试验。表5和图47中所示的离体结果表明,用44sc标记的aazta衍生物也高度富集在肿瘤组织中。此外,可以采用2-pmpa阻断肿瘤中的大部分活性。这也适用于肾脏。这些结果与相应的体内研究一致。图48和49显示了在注射后1h没有阻断的肿瘤活性和注射后40(静态记录20min)通过共注射2-pmpa而阻断的肿瘤活性。表5:离体活性实施例14:用于18f标记的化合物对于采用18f的pet诊断,合成了各种标记前体,并且在lncap细胞中进行了体外试验。在此,对于几种试验的化合物,观察到低的、对应于psma-11和psma-617的ic50值。图50中显示了三种这种类型的化合物及其ic50值(图50:用于18f放射性示踪剂的化合物)。实施例15:dota.fapi和data.fapi在图51中示出的dota.qs.fapi的合成类似于实施例6进行并且包括以下步骤:(a)多聚甲醛、meoh、amberlysta21;(b)pd/c、ch3cooh、绝对etoh、k2co3;(c)溴乙酸叔丁酯、mecn、k2co3;(d)福尔马林(37重量%)、ch3cooh、nabh4、mecn;(e)1mlioh、1,4-二氧六环/h2o(2:1);(f)n-boc-乙二胺、hatu、hobt、dipea、mecn;(g)(i)80%tfa于dcm中,(ii)3,4-二乙氧基环丁-3-烯-1,2-二酮、磷酸盐缓冲液ph7、1mnaoh;(h)3,磷酸盐缓冲液ph9、1mnaoh(图51:合成data.qs.fapi)。采用68ga的标记快速并且以高收率进行(图52和53)。在人血清(hs)和nacl溶液中的稳定性在2h的时间段内为大于98%(表6)。表6:在hs、etoh和nacl中的稳定性fapic50值的测量采用z-gly-pro-7-氨基-4-甲基香豆素(amc)进行。prepic50值的测定采用n-琥珀酰基-gly-pro-amc进行。选择性指数与文献值相当(jansenetal.jmedchem,2014,7,3053)。测量值示于表7中。表7:ic50值和选择性指数在患有结肠癌(ht29)的小鼠中采用[68ga]ga-dota.qs.fapi的体内以及离体研究中显示在肿瘤组织中的高富集(图54和55)。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1