利用无细胞DNA样本中的小变异的多层分析的癌症组织来源预测的制作方法
本公开总体上涉及预测受试者中的癌症组织来源,并且更具体地涉及对获自受试者的测试样本进行一种或多种物理和/或计算测定以便预测癌症组织来源。
背景技术:
使用下一代测序(ngs)对循环的诸如无细胞dna(cfdna)的无细胞核苷酸的分析被认为是癌症的检测和诊断的有价值的工具。与传统的肿瘤活检方法相比,分析cfdna可能更有利;但是,在肿瘤来源的cfdna中进行标识面临着严峻的挑战,特别是对于癌症的早期检测和癌症组织来源的早期预测等目的,其中癌症指示性信号尚未被表明。通过cfdna的使用以足够的灵敏度和特异性来准确预测受试者癌症的特征和来源面临着各种挑战。
技术实现要素:
所描述的实施例提供了一种方法,该方法除了生成癌症存在或不存在的预测之外,还基于从受试者获得的测试样本中的cfdna来生成对一个或多个受试者的癌症组织的预测。这样,本公开还可以除了生成一个或多个受试者中癌症存在的检测的预测之外,还用于解决癌症的来源组织问题。
具体地,使用一种或多种测序测定法,在本文中也称为物理测定法,对来自受试者的cfdna进行测序以生成序列读数,其示例包括小变异测序测定法。通过对应的计算分析来处理物理测定的序列读数,其中使用计算测定和/或物理测定来提取包括小变异特征和/或拷贝数特征的特征。因此,物理和计算分析输出了序列读数的特征的值,该值对于生成癌症组织来源的预测具有指导意义。作为示例,小变异特征(例如由小变异测序测定法生成的序列读数得到的特征)可以包括体细胞变异的总数,并且拷贝数特征可以包括焦点拷贝数。可以进一步生成和分析不是从基于测序的方法得到的其他特征,诸如可以参考临床症状和患者信息的基线特征。
在一些实施例中,一个或多个特征或特征类型的类型(例如小变异特征、拷贝数特征等)可被提供给预测模型,该预测模型生成对癌症组织来源的预测和/或是否存在癌症的预测。在一些实施例中,可以将不同特征和/或特征类型的值分别提供给不同的预测模型。每个单独的预测模型可以输出一个得分,然后作为对输出癌症预测的整体模型的输入。
本文公开的实施例描述了一种用于确定受试者的癌症来源组织的方法,该方法包括:在处理来自受试者的无细胞脱氧核糖核酸(cfdna)样本后,访问数据集,数据集包括对cfdna样本应用物理测定所生成的序列读数;在数据集上执行计算测定以生成一组特征的值;用预测模型处理该一组特征,以从候选组织来源集合中生成该受试者的癌症来源组织的预测,该预测模型通过函数将该一组特征的值转换为预测;并返回与受试者中癌症的存在有关的组织来源的预测。在一些实施例中,该方法确定输出的预测中的置信度,并基于该置信度将预测提供给相关实体。
在一些实施例中,预测模型是多层模型,其将受试者分类为第一子模型中的癌变组或非癌变组,并且在应用第二子模型时生成组织来源的预测。在一些实施例中,第一子模型是二项分类模型。在一些实施例中,第二子模型是多项回归模型(例如惩罚多项回归模型)。然而,在替代实施例中,第一子模型和/或第二子模型可以包括其他模型架构。
在一些实施例中,该方法从候选组织来源中预测与癌症的存在有关的组织来源,所述候选组织来源包括以下一项或多项:子宫组织来源、甲状腺组织来源、肾脏组织来源、前列腺组织来源、胰腺组织来源、卵巢组织来源、多发性骨髓瘤组织来源、淋巴瘤组织来源、肺组织来源、白血病组织来源、肝胆组织来源、头部组织来源、颈部组织来源、胃组织来源、食道组织来源、直肠组织来源、宫颈组织来源、乳腺组织来源和膀胱组织来源、另一组织来源以及组织来源的任意组合或分组(例如女性生殖系统组织来源、头颈部组织来源、胃肠道组织来源等)。
在一些实施例中,受试者是无症状的。在一些实施例中,无细胞核酸包含无细胞dna(cfdna)。在一些实施例中,序列读数是从下一代测序(ngs)过程生成的。在一些实施例中,使用合成测序从大规模平行测序程序生成序列读数。
在一些实施例中,测试样本是血液、血浆、血清、尿液、脑脊液、粪便、唾液、胸膜液、心包液、宫颈拭子、唾液或腹膜液样本。
附图说明
图1a描绘了根据一个或多个实施例的基于从受试者获得的cfdna样本所得到的特征来生成与癌症的存在有关的组织来源的预测的总体流程处理。
图1b描绘了根据一个或多个实施例的使用至少从受试者获得的cfdna样本来确定与癌症的存在有关的组织来源的预测的总体流程图。
图1c描绘了根据一个或多个实施例的图1b的变体,其使用至少从受试者获得的cfdna样本,利用子模型来确定与癌症的存在有关的组织来源的预测。
图1d描绘了根据一个或多个实施例的用于基于各种输入特征和子模型来确定组织来源的预测和/或其他预测的总体流程图。
图1e描绘了根据一个或多个实施例的基于由多个预测模型分别处理的多种类型的输入特征来确定组织来源的预测的总体流程图。
图2a描绘了根据一个或多个实施例的用于执行测序测定以生成序列读数的方法的流程处理。
图2b描绘了根据一个或多个实施例的用于执行测序测定以生成序列读数的图2a的变体。
图3a是根据一个或多个实施例的用于执行数据工作流程以分析由小变异测序测定所生成的序列读数的示例流程处理。
图3b描绘了根据一个或多个实施例的通过质量标准的应用来生成特征向量作为预测模型的输入的流程处理。
图4a描绘了根据一个或多个实施例的用于处理特征向量以预测组织来源的模型架构的示例。
图4b描绘了根据一个或多个实施例的与组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的实施例。
图4c描绘了根据一个或多个实施例的将预测模型的实施例应用于从来自受试者的样本中得到的特征向量以返回组织来源预测的流程处理。
图5a描绘了根据一个或多个实施例的与表1-表22中所示的组织来源的预测有关的预测模型的精度度量输出的示例。
图5b描绘了根据一个或多个实施例的与表1-表22中所示的组织来源的预测有关的预测模型的召回度量输出的示例。
图6a描绘了根据一个或多个实施例的与乳腺组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6b描绘了根据一个或多个实施例的与直肠组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6c描绘了根据一个或多个实施例的与肺组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6d描绘了根据一个或多个实施例的与非癌症分组的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6e描绘了根据一个或多个实施例的与胰腺组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6f描绘了根据一个或多个实施例的与膀胱组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6g描绘了根据一个或多个实施例的与对未知原发组织来源的癌症的预测有关的与不同基因相关联的特征的模型系数输出的示例。
图6h描绘了根据一个或多个实施例的与宫颈组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6i描绘了根据一个或多个实施例的与食道组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6j描绘了根据一个或多个实施例的与对胃组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6k描绘了根据一个或多个实施例的与头部/颈部组织来源的预测有关、的与不同的基因相关联的特征的模型系数输出的示例。
图6l描绘了根据一个或多个实施例的与肝胆组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6m描绘了根据一个或多个实施例的与淋巴瘤组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6n描绘了根据一个或多个实施例的与黑色素瘤组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6o描绘了根据一个或多个实施例的与多发性骨髓瘤组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6p描绘了根据一个或多个实施例的与对另一组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6q描绘了根据一个或多个实施例的与卵巢组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6r描绘了根据一个或多个实施例的与预测前列腺组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6s描绘了根据一个或多个实施例的与肾脏组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6t描绘了根据一个或多个实施例的与甲状腺组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图6u描绘了根据一个或多个实施例的与子宫组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。
图7描绘了用于实现本公开的各种方法的示例计算机系统。
具体实施方式
附图和以下描述仅通过说明的方式涉及优选实施例。应当注意,从下面的讨论中,在不背离所要求保护的原理的情况下,本文公开的结构和方法的替代实施例将容易地被认为是可以采用的可行替代方案。
现在将详细参考几个实施例,其示例在附图中示出。要注意的是,在可行的情况下,附图中可以使用相似或类似的附图标记,并且可以指示相似或类似的功能。例如在诸如“预测模型160a”的附图标记之后的字母表示该文本专门指代具有该特定附图标记的元素。文本中没有后续字母的附图标记,诸如“预测模型160”,是指图中带有该附图标记的任意或所有元素(例如文本中的“预测模型160”是指图中的附图标记“模型160a”和/或“预测模型160b”)。
术语“个体”是指人类个体。术语“健康个体”是指假定没有癌症或疾病的个体。术语“受试者”是指已知患有或潜在患有癌症或疾病的个体。
术语“序列读数”是指从获自个体的样本中读数的核苷酸序列。序列读数可通过本领域已知的各种方法获得。
术语“读段”或“读数”是指任意核苷酸序列,包括从个体获得的序列读数和/或从个体获得的样本中读数的初始序列得到的核苷酸序列。例如,读段可以指对齐的序列读数、折叠的序列读数或缝合的读数。此外,读段可以指单个核苷酸碱基,诸如单核苷酸变异。
术语“单核苷酸变异”或“snv”是指在例如从个体读数的序列的核苷酸序列的位置(例如位点)上,一个核苷酸到不同的核苷酸的取代。从第一核碱基x到第二核碱基y的取代可以表示为“x>y”。例如胞嘧啶对胸腺嘧啶snv可以表示为“c>t”。
术语“插入缺失(indel)”是指在读数的序列中具有长度和位置(也可以称为锚定位置)的一个或多个碱基的任意插入或删除。插入对应于正长度,而删除对应于负长度。
术语“突变(mutation)”是指一个或多个snv或插入缺失。
术语“候选变异”、“称为变异”或“假定变异”是指核苷酸序列的一种或多种检测到的核苷酸变异,例如在基因组中被确定为突变的位置(即,候选snv)或一个或多个碱基的插入或删除(即候选插入缺失)。通常,基于序列读数或折叠读数中替代性等位基因的存在,将核苷酸碱基视为所谓的变异,其中一个或多个位置上的核苷酸碱基与参考基因组中的核苷酸碱基不同。另外,候选变异可以称为真阳性或假阳性。
术语“真阳性”是指指示真实生物学的突变,例如个体中潜在的癌症、疾病或种系突变的存在。真阳性不是由健康个体中自然发生的突变(例如反复突变)或诸如在核酸样本的测定制备过程中的处理错误的其他人工来源引起的。
术语“假阳性”是指被错误地确定为真阳性的突变。通常,当处理序列读数与平均噪声率更高或噪声率不确定性更大相关联时,更容易出现假阳性。
“cfnas”的术语“无细胞核酸”是指可以在细胞外的诸如血液、汗液、尿液或唾液的体液中发现的核酸分子。无细胞核酸可互换用作循环核酸。
术语“无细胞脱氧核糖核酸”、“无细胞dna”或“cfdna”是指在诸如血液、汗液、尿液或唾液的体液中循环并且源自一种或多种健康细胞和/或源自一个或多个癌细胞的脱氧核糖核酸片段。
术语“循环肿瘤dna”或“ctdna”是指源自肿瘤细胞或其他类型癌细胞的脱氧核糖核酸片段,其可以由于生物过程,诸如死亡细胞的凋亡或坏死,被释放到个体的体液中,诸如血液、汗液、尿液或唾液中,或由活的肿瘤细胞主动释放。
术语“循环肿瘤rna”或“ctrna”是指源自肿瘤细胞或其他类型癌细胞的核糖核酸片段,其其可以由于生物过程,诸如死亡细胞的凋亡或坏死,被释放到个体的体液中,诸如血液、汗液、尿液或唾液中,或由活的肿瘤细胞主动释放。
术语“基因组核酸”、“基因组dna”或“gdna”是指包括源自一个或多个健康细胞的染色体dna的核酸。
术语“替代等位基因”或“alt”是指相对于参考等位基因例如对应于已知基因具有一个或多个突变的等位基因。
术语“测序深度”或“深度”是指从给定位置、区域或基因座处的个体获得的样本中读数的片段的总数。在一些实施例中,深度是指整个基因组或目标测序组的平均测序深度。
术语“交替深度”或“ad”是指样本中支持alt的多个读数,例如包括alt的突变。
术语“参考深度”是指样本中的读数的数量,其在候选变异位置处包括参考等位基因。
术语“替代频率”或“af”是指给定alt的频率。af可以通过将样本的对应ad除以给定alt的样本深度来确定。
术语“变异”或“真正的变异”是指基因组中某个位置的突变的核苷酸碱基。这样的变异可以导致个体中癌症的发展和/或进展。
术语“边缘变异”是指位于序列读数的边缘附近的突变,例如在距读数的序列的边缘的核苷酸碱基的阈值距离内。
术语“非边缘变异”是指未被确定为例如通过使用本文所述的边缘变异滤波方法而由人工过程导致的候选变异。在一些情况下,非边缘变异可能不是真正的变异(例如基因组中的突变),因为与一种或多种人工过程相反,由于不同的原因,非边缘变异可能会出现。
术语“拷贝数畸变(aberrations)”或“cna”是指体肿瘤细胞中拷贝数的改变。例如cna可以指实体瘤中的拷贝数改变。
术语“拷贝数改变”或“cnv”是指得到自种系细胞的拷贝数改变或得到自非肿瘤细胞的体细胞拷贝数改变。例如cnv可指由于克隆性造血作用(clonalhematopoiesis)而引起的白细胞中的拷贝数改变。
术语“拷贝数事件”是指拷贝数畸变和拷贝数变异中的一者或两者。
1.生成癌症预测
1.1整体流程
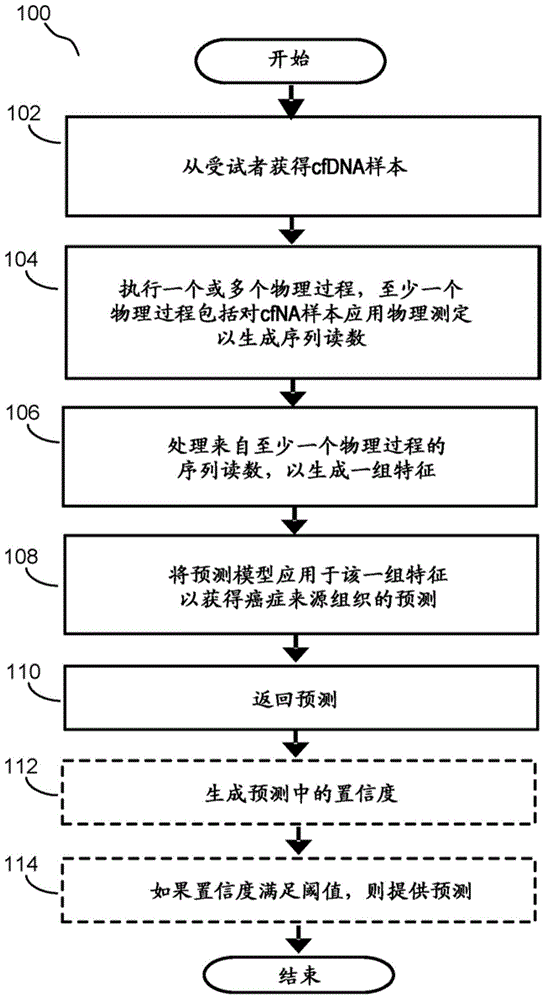
图1a描绘了根据一个实施例的基于源自从个体获得的cfdna样本的特征来生成癌症组织来源的预测的总体流程100。将进一步参考图1b-图1e,每个图均描绘了根据实施例的用于使用至少从个体获得的cfdna样本来确定癌症预测的总体流程图。
在步骤102,从个体(例如从采样设备、从自动采样设备)获得测试样本。通常,样本可以来自健康受试者、已知患有或怀疑患有癌症的受试者、或以前没有已知信息的受试者(例如无症状受试者)。测试样本可以是以下一种或多种的样本:血液、血浆、血清、尿液、粪便和唾液样本。备选地,测试样本可以包括以下一种或多种的样本:全血、血液得分、组织活检、胸膜液、心包液、脑脊髓液和腹膜液。
如图1b-1e的每一个所示,测试样本可以包括cfdna115。在各种实施例中,测试样本可以另外或替代地包括基因组dna(gdna)。如图1b-1e所示,gdna来源的一个示例是白细胞(wbc)dna120。
在步骤104中,执行一个或多个物理过程分析(例如通过包括测序系统的实验室设备),其中至少一个物理过程分析包括对cfdna115进行基于测序的分析以生成序列读数。参考图1b-图1c,物理过程分析的示例可以包括小变异测序测定134。参考图1d-图1e,另外的物理过程分析可以包括以下一项或多项:基线分析130、全基因组测序测定132、拷贝数分析136和甲基化测序分析138。
小变异测序测定法是指通常通过可用于确定小变异的靶向基因测序板来生成序列读数的物理测定法,其示例包括单核苷酸变异(snv)和/或插入或删除。备选地,也可以使用完整的基因组测序方案或完整的外显子组测序方案对小变异进行评估。如下所述,并且相对于图1c、图1d和图1e,具有计算分析140c的性能的小变异测序测定134的输出可用于生成小变异特征和/或拷贝数特征156,具有或不具有关于图1d和1e所述的拷贝数测定。在示例中,计算分析可以涉及本文描述的实施例的任意数量的训练过的模型(“贝叶斯层次模型”、“联合模型”等)或过滤器。
个体110的基线分析130可以包括个体110的临床分析,并且可以由医师或医学专家来执行。在一些实施例中,基线分析130可以包括对个体110的cfdna115中可检测到的种系改变的分析。在一些实施例中,基线分析130可以利用附加信息来进行种系改变的分析,诸如对上调或上调基因的标识。可以通过诸如图1d-1e中所描绘的计算分析140a的计算分析来提供这样的附加信息。基线分析130在下面进一步详细描述。
全基因组测序测定法是指生成整个基因组或整个基因组的实质部分的序列读数的物理测定法。这样的物理测定可以采用全基因组测序技术或全外显子组测序技术。
拷贝数测定法是指一种物理测定法,其从序列读数生成描述较大规模变异(或较长序列的变异)的输出,诸如拷贝数变异或拷贝数畸变。这样的物理测定法可以采用整个基因组或整个外显子组测序技术,或可操作用于获取样本的拷贝数变异特性的其他测序技术。
甲基化测序测定法是指生成序列读数的物理测定法,其可用于确定基因组中多个cpg位点的甲基化状态或甲基化模式。这种甲基化测序测定的一个示例可以包括cfdna的亚硫酸氢盐处理,以将未甲基化的胞嘧啶(例如cpg位点)转化为尿嘧啶(例如使用ezdna甲基化-金或ezdna甲基化-闪电试剂盒(可从zymoresearch公司获得))。备选地,可以使用酶促转化步骤(例如使用胞嘧啶脱氨酶(诸如apobec-seq(可从nebiolabs获得)))将未甲基化的胞嘧啶转化为尿嘧啶。转化后,可以通过整个基因组测序过程或靶向基因测序小组对转化的cfdna分子进行测序,并使用序列读数来评估在多个cpg位点的甲基化状态。基于甲基化的测序方案是本领域已知的(例如参见us2014/0080715,其通过引用并入本文)。在另一个实施例中,dna甲基化可以在其他情况下在胞嘧啶中发生,例如chg和chh,其中h为腺嘌呤、胞嘧啶或胸腺嘧啶。还可以使用本文公开的方法和程序来评估5-羟甲基胞嘧啶形式的胞嘧啶甲基化(参见,例如wo2010/037001和wo2011/127136,其通过引用并入本文)。在一些实施例中,甲基化测序测定不需要执行碱基转化步骤来确定整个基因组中cpg位点的甲基化状态。例如这种甲基化测序测定法可以包括pacbio测序或oxfordnanopore测序。
小变异测序测定134和/或其他测定是由cfdna115上的各个系统组件进行的,以生成和处理cfdna115的序列读数。在各个实施例中,小变异测序测定134和/或全基因组测序测定132、拷贝数测定136和甲基化测序测定138中的一个或多个可以进一步由wbcdna120上的各个系统组件进行以生成wbcdna120的序列读数。在每个测定中进行的处理步骤是关于图2更详细地描述。
在步骤106,处理由于执行基于测序的测定而生成的序列读数,以确定特征值。特征通常是可从物理测定和/或计算分析中获得的信息类型,其可用于预测受试者的癌症和/或癌症的存在的组织来源。通常,用于标识个体中组织来源和/或癌症存在的预测是基于将输入特征作为一种或多种模型架构的组成部分转换成预测输出。
通过应用一个或多个计算分析来处理序列读数,结合图1b-图1e更详细地描述。通常,每个计算分析140表示可由计算机的处理器可执行的算法,此后称为处理系统。因此,每个计算分析都会分析序列读数并基于序列读数来输出值特征。每种计算分析对于给定的基于测序的测定都是特定的,因此,每种计算分析都会输出特定类型的特征,该特定类型的特征对于基于测序的测定是特定的。
如图1b-图1e所示,从应用小变异测序测定法生成的序列读数使用计算分析140c被处理,否则被称为小变异计算分析。计算分析140c输出小变异特征154。另外或可替代地,从全基因组测序测定132的应用生成的序列读数使用计算分析140b被处理,否则被称为全基因组计算分析。计算分析140b输出整个基因组特征152。另外或可替代地,从拷贝数测定136的应用生成的序列读数使用计算分析140d被处理,否则被称为拷贝数计算分析。计算分析140d输出拷贝数特征156(其也可以由计算分析140c输出)。另外地或替代地,从甲基化测序测定法的应用生成的序列读数使用计算分析140e被处理,否则被称为甲基化计算分析。计算分析140e输出甲基化特征158。另外或可替代地,计算分析140a分析来自基线分析130的信息并输出基线特征150。
在步骤108,将预测模型应用于特征以生成与个体110的与癌症的存在有关的组织来源的预测。组织来源的预测的示例包括对以下一项或多项的预测:子宫组织来源、甲状腺组织来源、肾脏组织来源、前列腺组织来源、胰腺组织来源、卵巢组织来源、多发性骨髓瘤组织来源、淋巴瘤组织来源、肺组织来源、白血病组织来源、肝胆组织来源、头部组织来源、颈部组织来源、胃组织来源、食道组织来源、直肠组织来源、宫颈组织来源、乳腺组织来源和膀胱组织来源。癌症组织来源的预测的示例可以附加地或替代地包括对受试者中癌症来源的一组组织来源的预测,包括以下一项或多项:胃肠道组织来源的分组(例如包括胃组织、包括食道组织等)、女性生殖系统组织来源(例如包括卵巢组织,包括乳腺组织、包括宫颈组织等)、男性生殖系统组织来源(例如包括前列腺组织等)、头和颈部组织来源(例如包括头部组织、包括颈部组织等)、循环系统组织来源、神经系统组织(例如脑组织、脊髓组织等)以及其他分组。另外地或可替代地,预测模型可以在生成预测的不同阶段,输出指示癌症的存在或不存在、严重性、阶段、癌症的等级、癌症亚型、治疗决策以及对治疗的反应的可能性,如下文更详细描述。
在各个实施例中,预测模型的预测输出是具有置信度值的得分,诸如可能性或概率,其指示受试者中癌症的来源组织。预测输出可以附加地或可替代地包括具有置信度值的得分,用于以下一项或多项的预测:癌症的存在与否、严重性、阶段、癌症的等级、癌症亚型、治疗决策和对治疗有反应的可能性。在表征来自特定组织来源的癌症的存在/不存在、表征来自一组组织来源的癌症的存在/不存在、或通常表征癌症的存在/不存在时、得分可能是单数的。替代地,这样的得分可以是多个,使得预测模型的输出可以包括针对类别集合中的每一个的得分(例如组织来源、组织来源的分组、癌症的存在、癌症的不存在、等),其针对每个类别具有置信度值。为了描述的清楚起见,预测模型的输出通常被称为得分集合,该集合包括取决于哪个预测模型被配置为确定的一个或多个得分。
在步骤110,系统返回预测模型的输出,具有与每个预测输出关联的相关联置信度值112。在步骤114,如果各个输出的置信度满足阈值条件,则系统随后提供预测模型的输出。在一些实施例中,该方法可以还包括生成用于预测模型的输出的置信度参数的值,并且在通过该值确定阈值条件的满足之后,将预测提供给实体(例如医疗保健提供者等),用于为用户供应与癌症组织来源和/或癌症存在的预测有关的护理。
可以根据输入到预测模型中的特定特征、和/或根据在生成预测的不同阶段提供的预测模型的输出,来配置预测模型的结构,如关于以下的图1b-1d更详细地描述。在下文中,关于生成预测模型所接收的一种或多种类型的特征的值的处理工作流程来描述每个特别结构化的预测模型。如下文所使用的,工作流程过程是指物理过程分析、计算分析和预测癌症模型的应用的执行。
在一个实施例中,如图1b所示,预测模型160可以接收第一类型的输入特征,诸如小变异特征154,并输出组织来源预测190。另外,预测模型160可以接收第二类型的输入特征,诸如拷贝数特征156,并且在处理小变异特征154和拷贝数特征156中的至少一个时,输出组织来源预测190。
如图1c所示,在图1b所示实施例的变型中,可以用多个子模型构造预测模型。在图1c所示的实施例中,预测模型包括第一子模型161a,该第一子模型161a接收一个或多个小变异特征154和拷贝数特征156作为输入,并输出与属于癌变组190a或非癌变组190b的受试者相关联的预测得分。第一子模型161a还可以输出与不确定的预测相关联的预测得分。预测模型还包括第二子模型162a,第二子模型162a基于小变异特征154、拷贝数特征156和/或第一子模型161a的输出,输出指示受试者的癌症组织来源190c的一个或多个预测。
如此,如图1c所示,在应用预测模型的第一子模型161a时,并且在确定受试者被分入癌变组时,预测模型可以将受试者分为癌变组190a和非癌变组中的一个,并应用该预测模型的第二子模型162b来生成该受试者的癌症组织来源190c的预测。然而,在图1c所示的实施例的变型中,预测模型可以在不依赖于第一子模型161的输出的情况下应用第二子模型162和/或以任意其他合适的顺序来应用子模型。此外,在一些示例中,用作第一子模型161a的输入的相同特征也用作第二子模型162a的输入。可以使用计算分析作为第二子模型162a的输入,从cfdna样本中得到附加和/或替代特征。在某些情况下,附加和/或替代特征是在确定受试者被分为癌变组190a之后和/或根据确定受试者被分为癌变组190a而得到的。
在图1d所示的实施例中,可以构建预测模型以接收其他类型的输入特征,诸如基线特征150、全基因组特征152、小变异特征154、甲基化特征156和/或其他以上简要描述的特征148。类似于图1c所示的实施例,图1d所示的实施例中的预测模型包括第一子模型161b,该第一子模型161b接收基线特征150、全基因组特征152、小变异特征154、拷贝数特征156、甲基化特征158和其他特征148中的一个或多个作为输入,并输出与属于癌变组190a或非癌变组190b的受试者相关联的预测得分。第一子模型161b还可以输出与不确定的预测相关联的预测得分。预测模型还包括第二子模型162b,其基于基线特征150、全基因组特征152、小变异特征154、拷贝数特征156、甲基化特征158和其他特征148、和/或第一子模型的输出161b,输出一个或多个预测,该一个或多个预测指示受试者的癌症组织来源190c。这样,如图1d所示,在应用预测模型的第一子模型161b时,并且在确定受试者是癌症受试者时,预测模型可以将受试者分为癌变组190a和非癌变组190b之一,并且应用预测模型的第二子模型162b来为受试者生成癌症来源组织190c的预测。然而,在图1d所示的实施例的变型中,预测模型可以应用第二子模型162b而不依赖于第一子模型161b的输出和/或以任意其他合适的顺序来应用子模型。此外,在一些示例中,用作第一子模型161b的输入的相同特征也用作第二子模型162b的输入。可以使用计算分析作为第二子模型162b的输入,从cfdna样本中获取附加和/或替代特征。在某些情况下,在确定受试者被分组为癌变组190a之后,得到附加和/或替代特征。
此外,如图1d所示,系统可以基于第一子模型161b的输出,生成与受试者的健康状况相关的另一个预测190d和/或对来自受试者的样本进行额外的分析。例如基于第一子模型161b的输出,系统可以对来自受试者的预留样本执行反射测定。然后,基于反射分析,系统可以生成受试者的健康状况的另一种预测和/或以增加的置信度来输出将受试者分组为癌变组和非癌变组之一的预测(例如基于另一种基于测序的分析的实现)。仅作为示例,对个体(例如对个体的血液样本)的基线分析130可以提供各种临床症状和/或患者信息,其可用于证实来自预测模型160的癌症预测和/或用于提供特征以输入到预测模型160以生成癌症预测或其他预测190d。例如可以将个体的血液样本用于全血细胞计数(“cbc”),以测量个体血液中的几种成分和特征(例如非基于序列的特征)。一些特征可以包括wbc计数,当wbc计数高时,wbc计数可以用于从预测模型160增强对白血病的预测,和/或当血小板计数低时,可用于增加肝癌或肝功能衰竭的预测,或其他肝病预测190d。。
如图1d所示,在执行具有上述小变异测序分析134的输出的计算分析140c时,可以提取拷贝数特征156。可以相对于其他物理和/或计算分析,在对来自受试者的一个或多个样本上执行的拷贝数分析136的输出上执行计算分析140d时,可以附加地或替代地提取拷贝数特征156。
在一些实施例中,如图1e所示,该系统可以包括用于应用单独的预测癌症模型的架构,每个预测癌症模型被构造为处理一种类型的输入特征。在该实施例中,在第一阶段,将从每个计算分析(即,计算分析140a-140e)输出的特征的值分别输入到与每种特征类型相关联的各个子模型(160a-160e)中。然后,每个单独的子模型的输出用于生成受试者的组织来源预测190c。更详细地,如图1e所示,提供以下一项或多项:基线特征150作为输入被提供给预测模型160a,全基因组特征152作为输入被提供给预测模型160b,小变异特征154作为输入被提供给预测模型160c,拷贝数特征156作为输入被提供给预测模型160d,以及甲基化特征158作为输入被提供给预测模型160e。然后可以对预测模型160a-160e中的每一个的输出进行共处理以生成受试者的组织来源预测190c。
尽管图1e描绘了五个单独的预测模型160a-160e的输出用于生成受试者的组织来源预测190c,但是在各种实施例中,在生成组织来源来源预测190c中可以涉及附加或更少的预测模型。例如在一些实施例中,预测模型160a-160e中的任意一个、两个、三个、四个或五个,具有被配置为处理其他输入特征的任意其他合适的预测模型,可以用于输出用于生成组织来源预测190c的信息。
此外,在各个实施例中,由每个预测模型160a-160e输出的得分的数量可以不同。例如图1e中所示的预测模型160c可以输出一个得分集合(以下称为“变异基因得分”和“顺序得分”),和/或预测模型160a、160b、160d和/或图1e中所示的预测模型160a、160b、160d和160e中的任何一个或多个可以输出相应的得分集合。
在关于图1b-图1e所描述和示出的预测模型的每个不同实施例中,每个预测模型可以用包括以下一项或多项的子模型架构来构造:二项式模型和多项模型,在下面更详细地描述。附加地或替代地,子模型架构可以包括以下一项或多项:决策树、整体(例如装袋(bagging)、提升、随机森林)、梯度提升机、线性回归、朴素贝叶斯、神经网络或逻辑回归。每个预测模型都包括用于与不同组织来源相关的回归函数的学习系数。备选地,关于不同的模型架构,预测模型或子模型可以包括与训练相关联的学习权重。术语权重在此通常用于表示与模型的任意给定特征相关的学习量,而不论哪种特定的机器学习技术被使用。
在训练期间,训练数据被处理以生成用于特征的值,所述用于特征的值用于训练预测模型函数的系数和/或权重。例如训练数据可以包括从训练样本获得的cfdna和/或wbcdna以及输出标签。例如标签可以指示与从中获取训练样本的受试者中癌症的存在有关的实际组织来源,可以指示训练样本的受试者是否已知癌变或无癌变(例如健康),和/或可以指示与训练样本相关联的癌症严重程度。取决于图1b-1e所示的特定实施例,预测模型接收从与要训练的模型有关的一种或多种物理测定和计算分析中获得的一种或多种特征的值。根据训练中模型输出的得分与训练数据的输出标签之间的差异,预测模型功能的系数或权重被优化,使预测模型能够做出更准确的预测。
所训练的预测癌症模型可以被存储,并且随后在需要时例如在图1a的步骤108中的部署期间被检索。
1.2物理测定
图2a是根据更详细地描绘图1a的步骤104的一个实施例的用于执行物理测定以制备用于测序的核酸样本并生成序列读数的方法的流程图。方法104a包括但不限于以下步骤。例如方法104a的任意步骤可以包括用于质量控制或本领域技术人员已知的其他实验室测定方法的定量子步骤。
在步骤210a中,从受试者获得包含多个核酸分子(dna或rna)的测试样本,并从测试样本中提取和/或纯化核酸。在本公开中,除非另有说明,否则dna和rna可以互换使用。即,在变异调用和质量控制中使用错误源信息的以下实施例可以适用于dna类型的核算序列和rna类型的核算序列两者。但是,为了清楚和解释起见,本文描述的实施例可以集中在dna上。提取的样本中的核酸可以包括整个体类基因组或人类基因组的任意子集,包括整个外显子组。备选地,样本可以是人类转录组的任意子集,包括整个转录组。可以从已知患有或怀疑患有癌症的受试者获得测试样本。在一些实施例中,测试样本可以包括血液,血浆,血清,尿液,粪便,唾液,其他类型的体液或其任意组合。备选地,测试样本可以包括选自由全血,血液成分,组织活检,胸膜液,心包液,脑脊髓液和腹膜液构成的组的样本。在一些实施例中,用于抽取血液样本的方法(例如注射器或手指刺)的侵入性可以比用于获得组织活检的过程的侵入性小,所述组织活检可能需要手术。提取的样本可以包含cfdna和/或ctdna。对于健康的个体,人体可以自然清除cfdna和其他细胞碎片。通常,可以使用本领域中任意已知的方法从测试样本中提取和纯化无细胞核酸。例如可以使用一种或多种已知的市售方案或试剂盒,诸如qiaamp循环核酸试剂盒
在步骤220a中,制备测序文库。在文库制备过程中,例如通过接头连接(使用t4或t7dna连接酶)或本领域其他已知方式,将包括独特分子标识符(umi)的测序接头连接添加至核酸分子(例如dna分子)。umi是短核酸序列(例如4-10个碱基对),其被添加到dna片段的末端并用作独特的标签,其可用于鉴定源自特定dna片段的核酸(或序列读数)。加入接头后,例如使用聚合酶链反应(pcr)来扩增衔接子-核酸构建体。在pcr扩增过程中,umi与连接的dna片段一起被复制,这提供了一种用于在下游分析中鉴定来自相同原始片段的序列读数的方法。可选地,如本领域中众所周知的,测序接头还可以包括通用引物、样本特异性条形码(用于多重化)和/或一种或多种测序寡核苷酸,以用于随后的簇生成和/或测序(例如用于合成测序(sbs)的已知p5和p7序列(加利福尼亚圣地亚哥的
在步骤230a中,从文库中富集靶向的dna序列。根据一些实施例,在靶向富集期间,杂交探针(在本文中也称为“探针”)用于靶向和下拉已知是或可以是关于癌症(或疾病)的存在或不存在、癌症状态或癌症分类(例如,癌症类型或来源组织)的信息的核酸片段。对于给定的工作流程,可以将探针设计为与dna或rna的目标(互补)链退火(或杂交)。靶链可以是“阳性”链(例如转录成mrna、然后被转换成蛋白质的链)或互补的“阴性”链。探针的长度范围可以是10s、100s或1000s的碱基对。在一些实施例中,基于基因组设计探针,以分析怀疑对应于某些癌症或其他类型疾病的基因组(例如人或另一生物的)的特定突变或靶区域。而且,探针可以覆盖靶区域的重叠部分。如本领域技术人员将容易理解的,本领域中任意已知的手段都可以用于靶向富集。例如探针可以被生物素化并且链霉亲和素包被的磁珠用于富集探针捕获的靶核酸。参见例如duncavage等人,jmoldiagn.13(3):325-333(2011);和newman等人,natmed.20(5):548-554(2014)。通过使用目标基因组而不是对整个基因组进行测序(“全基因组测序”),基因组的所有表达基因(“全外显子组测序”或“全转录组测序”),方法100可用于增加目标区域的测序深度,其中深度是指样本内给定目标序列已被测序的次数的计数。增加测序深度允许样本中稀有序列变异的检测和/或增加测序过程的通量。杂交步骤之后,杂交的核酸片段被捕捉,并且也可以使用pcr被扩增。
在步骤240a中,从富集的核酸分子(例如dna分子)生成序列读数。可以通过本领域已知的方法从富集的核酸分子中获取测序数据或序列读数。例如方法100可以包括下一代测序(ngs)技术,包括合成技术
在各种实施例中,富集的核酸样本215a被提供给测序器245a以进行测序。如图2a所示,测序器245a可以包括图形用户界面250a,该图形用户界面使用户能够与特定任务(例如,启动测序或终止测序)以及一个或多个装载站155交互,用于提供测序盒,测序盒包括富集片段样本和/或进行测序测定所需的缓冲液。因此,一旦用户已经向测序器245a的装载站255a提供了必要的试剂和富集的片段样本,用户就可以通过与测序器245a的图形用户界面250a交互来启动测序。在步骤240a中,测序器245a执行测序并从核酸样本215输出富集的片段的序列读数。
在一些实施例中,测序器245a与一个或多个计算设备260a通信地耦合。每个计算设备260a可以处理诸如变异调用或质量控制的各种应用的序列读数。测序器245a可以将bam文件格式的序列读数提供给计算设备260a。每个计算设备260a可以是个体计算机(pc)、台式计算机、膝上型计算机、笔记本、平板pc或移动设备之一。计算设备260a可以通过无线、有线或无线和有线通信技术的组合通信地耦合到测序器245a。通常,计算设备260a被配置有处理器和存储计算机指令的存储器,该计算机指令在由处理器执行时使处理器处理序列读数或执行本文公开的任意方法或过程中的一个或多个步骤。
在一些实施例中,可以使用本领域已知的方法将序列读数与参考基因组对齐,以确定对齐位置信息。例如在一些实施例中,序列读数与人类参考基因组hg19对齐。人类参考基因组的序列hg19可以从genomereferenceconsortium获得,参考号为grch37/hg19,也可以从santacruzgenomicsinstitute提供的genomebrowser中获得。对齐位置信息可以指示参考基因组中与给定序列读数的起始核苷酸碱基和终止核苷酸碱基相对应的区域的起始位置和终止位置。对齐位置信息还可以包括序列读数长度,其可以从起始位置和终止位置确定。参考基因组中的区域可以与基因或基因的片段相关。
在各种实施例中,例如当使用配对末端测序过程时,序列读数包括标记为r1和r2的读数对。例如可以从双链dna(dsdna)分子的第一端测序第一读数r1,而可以从双链dna(dsdna)的第二端测序第二读数r2。因此,第一读数r1和第二读数r2的核苷酸碱基对可以与参考基因组的核苷酸碱基一致地对齐(例如以相反的方向)。从读数对r1和r2中得到的对齐位置信息可以包括参考基因组中对应于第一读数(例如r1)的末端的起始位置和参考基因组中对应于第二读数(例如r2)的末端的终止位置。换句话说,参考基因组中的起始位置和终止位置代表核酸片段所对应的参考基因组中的可能位置。具有sam(序列对齐图)格式或bam(二进制)格式的输出文件可以被生成,并被输出以进行进一步的分析,诸如变异调用。
图2b是根据更详细地描绘图1a的步骤104的另一实施例的用于执行物理测定(例如测序测定)以生成序列读数的方法的流程图。方法104b包括但不限于以下步骤。例如方法104b的任意步骤可以包括用于质量控制或本领域技术人员已知的其他实验室测定方法的定量子步骤。
通常,步骤(例如步骤205b-235b)的各种子组合用于小变异测序测定法和/或以下一项或多项:全基因组测序测定法和甲基化测序测定法。例如可以执行步骤205b和215b-235b进行小变异测序测定。另外,在一些实施例中,可以对全基因组测序测定法执行步骤205b、215b、230b和235b。另外,在一些实施例中,执行步骤205b-235b中的每一个用于甲基化测序测定。例如采用靶向基因组亚硫酸氢盐测序的甲基化测序测定法采用步骤205b-235b中的每一个。备选地,在一些实施例中,进行步骤205b-215b和230b-235b用于甲基化测序测定。例如采用全基因组亚硫酸氢盐测序的甲基化测序测定不需要执行步骤220b和225b。
在步骤205b,例如通过纯化过程从测试样本中提取核酸(例如cfdna)。通常,可以使用本领域中任意已知的方法来纯化dna。例如可以通过在管中沉淀和/或沉淀核酸来分离核酸。提取的核酸可以包括cfdna,或者其也可以包括gdna,诸如wbcdna。
在步骤210b中,处理cfdna片段以将未甲基化的胞嘧啶转化为尿嘧啶。在一些实施例中,该方法对dna使用亚硫酸氢盐处理,其将未甲基化的胞嘧啶转化为尿嘧啶而不转化甲基化的胞嘧啶。例如亚硫酸氢盐的转化使用商业试剂盒,诸如ezdnamethylation-gold、ezdnamethylation-direct或ezdnamethylation-lightning试剂盒(可从zymoresearchcorp,irvine,ca获得)。在另一个实施例中,使用酶促反应完成未甲基化的胞嘧啶向尿嘧啶的转化。例如该转化可以使用可商购的试剂盒,用于将未甲基化的胞嘧啶转化为尿嘧啶,诸如apobec-seq(nebiolabs、ipswich、ma)。
在步骤215b,制备测序文库。在文库制备期间,例如接头包括一种或多种测序寡核苷酸,用于后续的簇生成和/或测序(例如已知用于合成测序(sbs)的p5和p7序列(illumina,圣地亚哥,加利福尼亚))通过接头连接将其与核酸片段的末端连接。在一些实施例中,在接头连接期间将独特的分子标识符(umi)添加至提取的核酸。umi是短的核酸序列(例如4-10个碱基对),其在接头连接期间被添加到核酸的末端。在一些实施例中,umi是简并碱基对,其充当可用于鉴定从核酸获得的序列读数的独特标签。如后面所述,在扩增过程中,umi可以与连接的核酸一起进一步复制,这提供了一种在下游分析中鉴定源自相同原始核酸片段的序列读数的方法。
在步骤220b中,使用杂交探针来丰富所选核酸组的测序文库。可以设计杂交探针以靶向并与靶向核酸序列杂交,以下拉和富集靶向核酸片段,这些片段可以为癌症(或疾病)的存在与否,癌症状态或癌症分类(例如癌症类型或来源组织)提供信息。根据该步骤,可以将多个杂交下拉探针用于给定的靶序列或基因。探针的长度范围可为约40至约160个碱基对(bp),约60至约120bp,或约70bp至约100bp。在一些实施例中,探针覆盖靶区域或基因的重叠部分。在一些实施例中,杂交探针被设计为富集已处理(例如使用亚硫酸氢盐)以将未甲基化的胞嘧啶转化为尿嘧啶的dna分子(即,探针被设计为富集后转化的dna分子)。在其他实施例中,杂交探针被设计为富集未经处理(例如使用亚硫酸氢盐)以将未甲基化的胞嘧啶转化为尿嘧啶的dna分子(即,探针被设计为富集预转化的dna分子)。对于靶向基因组测序,杂交探针被设计为靶向和拉下核酸片段,核酸片段源自靶基因组中包括的特定基因序列。对于整个外显子组测序,杂交探针设计为靶向和拉下源自参考基因组中外显子序列的核酸片段。
在杂交步骤220b之后,使经杂交的核酸片段富集225b。例如可以使用pcr捕获并扩增杂交的核酸片段。可以富集靶序列以获得可以随后测序的富集序列。这提高了序列读数的测序深度。
在步骤230b中,对核酸测序以生成序列读数。序列读数可以通过本领域已知的手段获得。例如许多技术和平台直接从平行的数百万个单独的核酸(例如诸如cfdna或gdna的dna)分子中获得序列读数。这样的技术可以适合于执行靶向基因组测序、全外显子组测序、全基因组测序、靶向基因组亚硫酸氢盐测序和全基因组亚硫酸氢盐测序中的任意一种。
作为第一个示例,通过合成测序技术依赖于荧光核苷酸的检测,因为它们被掺入到与待测序模板互补的新生dna链中。在一些方法中,将长度为30-50个碱基的寡核苷酸在5'端共价锚定至玻璃盖玻片。这些锚定的股线执行两个功能。首先,如果模板被配置有与表面结合的寡核苷酸互补的捕获尾,它们将充当目标模板链的捕获位点。它们还充当模板导向引物延伸的引物,引物延伸形成了序列读数的基础。捕获引物用作固定位置位点,可使用多个合成、检测和染料连接物化学裂解的周期来确定序列,以去除染料。每个循环包括添加聚合酶/标记的核苷酸混合物、漂洗、成像和染料裂解。
在替代方法中,聚合酶用荧光供体分子修饰并被固定在载玻片上,而每个核苷酸均用与伽马-磷酸酯相连的受体荧光部分进行颜色编码。当核苷酸掺入从头链时,该系统检测荧光标记的聚合酶和荧光修饰的核苷酸之间的相互作用。
任意合适的合成测序平台都可用于鉴定突变。合成测序平台包括roche/454lifesciences的genomesequencer、illumina/solexa的genomeanalyzer、appliedbiosystems的solid系统以及来自helicosbiosciences的heliscope系统。pacificbiosciences和visigenbiotechnologies也已经描述了合成测序平台。在一些实施例中,被测序的多个核酸分子结合至支持物(例如固体支持物)。为了将核酸固定在支持物上,可以在模板的3'和/或5'端添加捕获序列/通用引物位点。通过使捕获序列与共价附于支持物的互补序列杂交,可以使核酸与支持物结合。捕获序列(也称为通用捕获序列)是与连接到支持物上的序列互补的核酸序列,所述支持物可以双重用作通用引物。
作为捕获序列的替代,可以将偶联对的成员(例如抗体/抗原,受体/配体或抗生物素蛋白-生物素对)可以连接到每个片段以在涂有该偶联对的相应第二成员的表面上捕获。捕获之后,可以例如通过单分子检测/测序来分析序列,包括依赖于模板的合成测序。在合成测序中,将表面结合的分子在聚合酶存在下暴露于多种标记的三磷酸核苷酸。模板的序列由掺入生长链3'端的标记核苷酸的顺序决定。这可以实时完成,也可以分步重复进行。为了进行实时分析,可以对每个核苷酸掺入不同的光学标记,并且可以利用多个激光来刺激掺入的核苷酸。
大规模并行测序或下一代测序(ngs)技术包括合成技术、焦磷酸测序、离子半导体技术、单分子实时测序、通过连接测序、纳米孔测序或双末端测序。大规模并行测序平台的示例有illuminahiseq或miseq、ionpersonalgenomemachine、pacbiorsii测序器或sequelsystem、qiagen的genereader和oxfordminion。可以使用其他类似的当前大规模并行测序技术,以及这些技术的后代。
在步骤235b,可以使用本领域中已知的方法将序列读数与参考基因组对齐,以确定对齐位置信息。对齐位置信息可以指示参考基因组中与给定序列读数的起始核苷酸碱基和终止核苷酸碱基相对应的区域的起始位置和终止位置。对齐位置信息还可以包括序列读数长度,其可以从起始位置和终止位置确定。参考基因组中的区域可以与基因或基因的片段相关。
在各种实施例中,序列读数包括表示为r1和r2的读数对。例如可以从核酸片段的第一末端测序第一读数r1,而可以从核酸片段的第二末端测序第二读数r2。因此,第一读数r1和第二读数r2的核苷酸碱基对可以与参考基因组的核苷酸碱基一致地对齐(例如以相反的方向)。从读数对r1和r2中获得的对齐位置信息可以包括参考基因组中对应于第一读数(例如r1)的末端的起始位置和参考基因组中对应于第二读数(例如r2)的末端的终止位置。换句话说,参考基因组中的起始位置和终止位置代表核酸片段所对应的参考基因组中的可能位置。具有sam(序列对齐图)格式或bam(二进制对齐图)格式的输出文件可以被生成并被输出以进行进一步分析。
在步骤235b之后,使用诸如上文所述并在图1d中示出的计算分析140b、140c或140d的计算分析来处理对齐的序列读数。小变异计算分析140c、全基因组计算测定140b、甲基化计算分析140d和基线计算分析中的每一个在下面进一步详细描述。
2.小变异计算分析
2.1小变异特征
上面关于图1b-1e所述的小变异计算分析140c接收由小变异测序测定134生成的序列读数,并基于序列读数确定小变异特征154的值,其中小变异特征的值154可以被组装成向量。
小变异特征154的实例包括以下任一项:受试者的cfdna中的体细胞变异总数、非同义变异总数、同义变异总数、样本中每个基因代表的变异数量、在基因组中每个基因是否存在体细胞变异、与癌症相关的特定基因的体细胞变异是否存在、在基因组中每个基因的变异的等位基因频率(af)、公开数据库指定的每个类别的体细胞变异、诸如oncokb、另一种致癌相关特征、与基因相关联的非同义变异的最大变异等位基因频率、按照体细胞变异的af的顺序排列、基于体细胞变异的af的其他与顺序统计相关的特征(例如,相对顺序统计特征,其表示第一变异的等位基因频率与至少一种其他变异的等位基因频率的比较)和/或与热点突变或突变类型相关的特征,诸如无义或错义类型突变。
小变异特征的其他示例可以包括描述以下一项或多项的特征:基于等位基因频率的已知与癌症相关的体细胞变异的分类、描述一个或多个基因的第一突变和第二突变的联合存在的突变相互作用(例如,被表示为对应于第一突变和第二突变的特征值的乘积的平方根)。关于通过利用预测模型处理小变异特征而生成的预测,在检测到上述特征之一或特征组合(或从上述特征得到)时,预测模型可以优先返回一个候选组织来源而不是其他候选组织来源。
通常,小变异特征154的特征值是基于对体变异的准确标识而预测的,该体变异可以指示与受试者中癌症的存在有关的组织来源。小变异计算分析140c标识候选变异,并从候选变异中区分可能存在于个体基因组中的体细胞变异和不太可能预测与受试者中癌症存在相关的组织来源的假阳性变异。更具体地,小变异计算分析140c鉴于干扰信号(例如噪声)和/或可归因于基因组来源(例如,来自gdna或wbcdna)的变体,标识存在于cfdna中的候选变体可能来自体细胞来源。另外,可以过滤候选变异以去除可能由于伪像而出现的假阳性变异,并且因此不能指示个体中的癌症。例如假阳性变异可以是在序列读数的边缘处或附近检测到的变异,其由于自发的胞嘧啶脱氨和末端修复错误而生成。因此,在滤除假阳性变异之后仍保留的体变异及其特征可用于确定小变异特征。
对于体细胞变异总数的特征,小体变异计算分析140c可以将整个基因组或基因组中鉴定出的体细胞变异总计。因此,对于从个体获得的cfdna样本,体细胞变异总数的特征可以表示为样本的cfdna中鉴定的体细胞变异总数的单个数值。
对于非同义变异总数的特征,小变异计算分析140c可以进一步过滤所标识的体变异以标识为非同义变异的体变异。如本领域众所周知的,核酸序列的非同义变异导致与该核酸序列相关的蛋白质的氨基酸序列的改变。例如非同义变异可以改变个体的一种或多种表型或引起(或使个体更易患)个体发展为癌症、癌细胞或其他类型的疾病。因此,小变异计算分析140c通过确定对三核苷酸的一个或多个核苷酸碱基的修饰将导致基于修饰的三核苷酸生成不同的氨基酸,来确定候选变异将导致非同义的变异。通过将整个基因组中标识出的非同义变异相加来确定非同义变异总数的特征值。因此,对于从个体获得的cfdna样本,非同义变异总数的特征可以表示为单个数值。
对于同义变异总数的特征,同义变异表示未被归类为非同义变异的其他体细胞变异。换句话说,小变异计算分析140c可以执行所标识的体变异的过滤,如关于非同义变异所描述的,并在整个基因组或基因组中标识同义变异。因此,对于从个体获得的cfdna样本,同义变异总数的特征被表示为单个数值。
对于每个基因是否存在体细胞变异的特征可以涉及cfdna样本的多个特征值。例如靶向基因组可以在组中包括500个基因,因此,小变异计算分析140c可以生成500个特征值,每个特征值代表该组中基因的体细胞变异的存在或不存在。例如如果基因中存在体细胞变异,则特征的值为1。相反,如果基因中不存在体细胞变异,则特征的值为0。通常,可以使用任意大小基因组。例如基因组可以包含遍及基因组的100、200、500、1000、2000、10000或更多个基因靶标。在一些实施例中,基因组可包含约50至约10,000个基因靶标、约100至约2000个基因靶标或约200至约1000个基因靶标。
对于已知/与癌症相关联的特定基因的体变异的存在/不存在的特征,可以从诸如oncokb的公共数据库访问已知与癌症相关的特定基因。已知与癌症相关的基因的示例包括tp53、lrp1b和kras。已知与癌症相关的每个基因都可以与特征值相关,诸如1(表示基因中存在体细胞变异)或0(表示基因中不存在体细胞变异)。
可以通过访问诸如oncokbchakravarty等人,jcopo2017的公共数据库来确定表示每个类别的体细胞变异的af的特征。例如oncokb将基因的临床信息分类为四个不同类别之一,诸如fda批准、标准护理、新兴临床证据和生物学证据。每个此类类别可以是其自己的特征,具有自己的对应值。可以访问以确定特征的其他公共可用数据库包括由美国国家癌症研究所的基因组数据共享中心(gdc)支持的癌症体细胞突变目录(cosmic)和癌症基因组图谱(tcga)。福布斯等cosmic:somaticcancergeneticsathigh-resolution,nucleicacidsresearch,第45卷,第d1期,2017年1月4日,第d777-d783页。在一些实施例中,将每个类别特征的体细胞变异的af的值确定为跨该类别中的基因的体细胞变异的最大af。在另一个实施例中,每个类别特征的体细胞变异的af值被确定为跨该类别中的基因的体细胞变异的平均af。也可以使用除每个类别的最大af和每个类别的平均af以外的措施。
代表每个基因的体细胞变异的af的特征(例如在靶基因组中)是指与特定基因相关的序列读数中体细胞变异的频率的量度。通常,该特征由基因组的每个基因或整个基因组中的每个基因的一个特征值表示。该特征的值可以是该基因的体细胞变异的af的统计值。用于给特征指定值的精确测量值可以根据实施例而改变。在一些实施例中,将该特征的值确定为基因中每个位置(例如基因组中)中所有体细胞变异的最大af。在一些实施例中,将该特征的值被确定为该基因每个位置的所有体细胞变异的平均af。因此,对于具有500个基因的示例靶向基因组,存在代表每个基因的体细胞变异的af的500个特征值。除了最大自动af或平均自动af以外,还可以使用其他措施。
每个类别的体细胞变异的af可以根据由诸如oncokb的公开数据库指定的类别来确定。例如oncokb将基因分类为四个不同类别之一。在一些实施例中,每个类别的体细胞变异的af是跨该类别中的基因的体细胞变异的最大af。在一些实施例中,每个类别的体细胞变异的af是跨该类别中的基因的体细胞变异的平均af。
根据体变异的af,体变异的排名顺序是指体变异的前n个等位基因频率。通常,变异等位基因频率的值可以从0到1,其中变异等位基因频率0表示在该位置没有任意序列具有替代等位基因,而变异等位基因频率1表示所有序列在该位置具有备用等位基因。在其他实施例中,可以使用变异等位基因频率的其他范围和/或值。在各种实施例中,排序次序特征独立于体细胞变异本身,而是仅由前n个变异等位基因频率的值表示。前5个等位基因频率的排名顺序特征的示例可以表示为:[0.1,0.08,0.05,0.03,0.02],其表示与体细胞变异无关的5个最高等位基因频率范围为0.02到0.1。
2.2小变异计算分析过程概述
处理系统,诸如计算机的处理器,执行用于执行小变异计算分析140c的代码。
图3a是根据一些实施例的用于从序列读数确定体细胞变异的方法300的流程图。在步骤305a,处理系统折叠对齐的序列读数。在一些实例中,折叠序列读数包括使用umi,以及任选地来自输出文件的测序数据的对齐位置信息,以将多个序列读数折叠成共有序列,以确定核酸片段或其一部分的最可能序列。独特的序列标签的长度可以是约4至20个核酸。由于umi通过富集和pcr与连接的核酸片段一起复制,因此序列处理器205可以确定某些序列读数源自核酸样本中的相同分子。在一些实施例中,具有相同或相似的对准位置信息(例如阈值偏移内的开始和结束位置)并且包括共同的umi的序列读数被折叠,并且处理系统生成折叠的读数(在本文中也称为“共同读数”)。以代表核酸片段。如果对应的折叠读数对具有共同的umi,则处理系统会将共有读数指定为“双链体”,这表示原始核酸分子的正链和负链均被捕获;否则,折叠后的读数将被指定为“非双链体”。在一些实施例中,作为折叠序列读数的替代或补充,处理系统可以对序列读数执行其他类型的纠错。
在步骤305b,处理系统基于对应的对准位置信息来缝合折叠的读数。在一些实施例中,处理系统比较第一序列读数和第二序列读数之间的对齐位置信息,以确定第一序列读数和第二序列读数的核苷酸碱基对在参考基因组中是否重叠。在一个用例中,响应于确定第一序列读数和第二序列读数之间的重叠(例如给定数量的核苷酸碱基)大于阈值长度(例如核苷酸碱基的阈值数量),处理系统指定第一序列读数和第二序列读数为“已缝合”;否则,折叠后的读数将被指定为“未缝合”。在一些实施例中,如果重叠大于阈值长度并且如果该重叠不是滑动重叠,则缝合第一序列读数和第二序列读数。例如滑动重叠可包括均聚物游动(例如单个重复核苷酸碱基)、二核苷酸游动(例如两个核苷酸碱基序列)或三核苷酸游动(例如三个核苷酸碱基序列),其中均聚物运行、二核苷酸运行或三核苷酸运行具有至少阈值长度的碱基对。
在步骤305c,处理系统将读数组装到路径中。在一些实施例中,处理系统组装读数以生成针对靶区域(例如基因)的有向图,例如debruijn图。有向图的单向边缘代表目标区域中k个核苷酸碱基的序列(在本文中也称为“k-mers”),并且这些边缘通过顶点(或节点)相连。处理系统将折叠的读数与有向图对齐,以便可以通过边缘和对应的顶点的子集按顺序表示任意折叠的读数。
在一些实施例中,处理系统确定描述有向图的参数集合并处理有向图。另外,该参数集合可以包括从折叠的读数到由有向图中的节点或边表示的k-mer的成功对齐的k-mer的计数。处理系统存储有向图和相应的参数集合,其可以被取回以更新图或生成新图。例如处理系统可以基于参数集合来生成有向图的压缩版本(例如或修改现有图)。在一些示例使用情况中,为了滤除具有较低重要性级别的有向图的数据,处理系统去除(例如“剪枝”或“修剪”)具有小于阈值的计数的节点或边缘,并且保持计数大于或等于阈值的节点或边。
在步骤305d,处理系统从组装的读数中标识候选的小变异特征。在一些实施例中,处理系统通过将有向图(其可能已经在步骤305b中被修剪边缘或节点压缩)与基因组靶区域的参考序列进行比较来标识候选的小变异特征。处理系统可使有向图的边缘与参考序列对齐,并记录错配边缘的基因组位置和与边缘相邻的错配核苷酸碱基的基因组位置作为候选小变异的位置。在一些实施例中,边缘不匹配的边缘和边缘左边和右边的不匹配核苷酸碱基的基因组位置被记录为被称为变异的位置。另外,处理系统可以基于目标区域的测序深度来生成候选的小变异。特别地,例如由于大量的序列读数有助于解决(例如使用冗余)序列之间的错配或其他碱基对变异,因此处理系统在确定具有更大测序深度的靶区域中的变异上可以更有信心。
在一些实施例中,处理系统使用模型来标识候选小变异特征,以确定用于从受试者的序列读数的预期噪声率。该模型可以是贝叶斯分层模型,尽管在一些实施例中,处理系统使用一种或多种不同类型的模型。此外,贝叶斯分层模型可以是可用于生成候选变异并且彼此相关的许多可能的模型架构之一,因为它们都对位置特定的噪声信息进行建模,以便于提高变异调用的灵敏度/特异性。更具体地说,处理系统使用来自健康个体的样本来训练模型,以对序列读数的每个位置的预期噪声速率进行建模。
此外,可以将多个不同的模型存储在数据库中或将其检索以用于应用程序后训练。例如训练第一模型以对snv噪声率建模,并且训练第二模型以对插入缺失噪声率进行建模。此外,处理系统可以使用模型的参数来确定序列读数中一个或多个真阳性的可能性。处理系统可以基于可能性来确定质量得分(例如以对数标度)。例如,质量得分是phred质量得分q=-10·log10p,其中p是不正确的候选变异调用(例如假阳性)的可能性。诸如联合模型的其他模型可以使用一个或多个贝叶斯层次模型的输出来确定不同样本(例如每个位置)的序列读数中核苷酸突变的预期噪声。
在步骤305e,处理系统用质量截止标准来分析小变异特征,并且在步骤305f,传递满足质量截止标准的小变异特征,其中关于图3b描述了质量截止标准操作的实施例。在步骤305g中,处理系统应用预测模型(例如相对于以上图1a-图1e描述的预测模型的实施例)以生成指示癌症存在或不存在的预测,并且在步骤305h中,处理系统应用预测模型(例如相对于以上图1a-图1e描述的预测模型的实施例)以生成与受试者中癌症存在相关的组织来源的预测。图3b描绘了根据一个实施例的图3a所示的步骤305e的流程图,该步骤305e用于将质量截止标准应用于候选的小变异特征。在步骤310,处理系统按基因聚集小变异。然后,对于每个变异,处理系统在步骤320中应用质量截止标准,其中如果满足质量标准,则将小变体特征的值设置为非零值(如上文关于小变体特征值所述)。在一些实施例中,如果满足质量标准,则将小变异特征的值设置为最大等位基因频率(max(af))。相反,如果不满足质量标准,则处理系统会将小变异特征的值设置为零。然后,在步骤330a中,处理系统生成具有对应于各个基因的变异值的变异特征向量。在一些变型中,取决于质量标准的满足程度,可以将权重应用于小变异特征的值,其中例如在很大程度上满足质量标准的小变异特征具有更大的权重的值。此外,在一些实施例中,质量截止标准仅被应用于序列的编码区域;例如然而,质量截止标准可以附加地或替代地被应用于序列的非编码区域。
在各种实施例中,可以根据实施例实现在联合模型中生成候选变体和/或执行计算分析以处理测序测定的输出,这些实施例在2018年11月27日提交的题为“modelsfortargetedsequencing”的美国申请号16/201,912中描述,该申请现在作为美国申请公开号2019/0164627公开,其全文并入本文。
此外,如上所述,处理系统可以使用用于处理小变异测序测定的输出的计算分析的输出来得到相关的拷贝数特征。在实施例中,拷贝数特征集合可以包括突变的焦点拷贝数,该焦点拷贝数描述了以低于cfdna样本的序列的阈值比例表示的遗传变异的重复。拷贝数特征集合可以附加地或替代地包括与融合或结构变异相关联的拷贝数特征。
3.其他功能的计算分析
其他特征的计算分析可以根据以下各项中描述的实施例来被执行:2018年4月13日提交的题为“multi-assaypredictionmodelforcancerdetection”的美国申请号62/657,635,其现在通过优先权要求被包括在2019年4月15日提交的题为“multi-assaypredictionmodelforcancerdetection”的美国申请公开号2019/0316209中;以及于2019年5月20日提交的标题为“inferringselectioninwhitebloodcellmatchedcell-freednavariantsand/orinrnavariants”的美国申请号16/417,336,所有这些文献的内容均全文并入本文。
4.预测模型架构
4.1第一子模型
关于用于生成癌症预测的预测模型的不同子模型(以上相对于图3a,步骤305g进行了描述),第一子模型可以被构造为二元分类模型(例如弹性网分类程序包的一部分),无论有无相关置信度,它都会输出将样本标识为癌变或非癌变的预测。二元分类可以允许非负系数输出,其中系数的大小对应于分类为癌变疾病的可能性增加。在某些情况下,二元分类仅限于非负系数输出。仍然,在一些示例中,二元分类还可以允许负系数输出,该负系数输出对应于针对癌症状况分类的降低的可能性。然而,在替代变型中,二元分类可以以任意其他合适的方式输出具有对应于癌变或非癌变状况的系数方向和/或大小的系数。
此外,二元分类模型可以包括阿尔法参数,阿尔法参数被配置为在岭回归模式和lasso回归模式之间调整第一子模型的性能,其中该该方法可以实现用于评估每个一组小变异特征对预测的贡献并基于这些贡献调整阿尔法参数的架构。关于阿尔法参数,针对岭回归模式的阿尔法调整可以通过减少模型系数的幅度来惩罚二项式分类模型系数的高值,从而最大程度地减少对训练的模型的影响。关于阿尔法参数,针对lasso回归模式的阿尔法调整可以相对于模型行为,通过将不相关系数的高值设置为零来惩罚二项式分类模型的系数的高值。这样,二元分类模型可以是惩罚二项式分类模型,其可通过阿尔法参数调整,用于包括将样本强分类为癌变或非癌变的特征。
关于第一子模型的二元分类架构的预测得分输出,可以基于将一组特征(例如小变异特征)作为输入特征进行处理来生成预测得分,其中一组特征与癌症的存在与否有关。然后可以将预测得分与阈值条件进行比较,其中阈值条件的满足指示癌症的存在,阈值条件的不满足指示癌症的不存在。
二元分类模型还可以包括表征癌症信号强度的特异性条件,其中该特异性条件为来自具有高度特异性癌症信号的个体的样本提供了初始过滤器。特异性条件可以是阈值特异性(例如99.9%的特异性、99%的特异性、98%的特异性、95%的特异性等),其中如果特定条件由二元分类的输出满足在模型中,样本是用预测模型的第二子模型(例如如下所述的多项模型)处理的。在一些示例中,基于非癌症群体来选择二项式阈值特异性(例如从针对非癌症样本的二元分类模型预测的预测得分的分布中选择),并且使用多项分类模型进一步检查得分高于对应于阈值特异性的得分的任何样本。
然而,可以用其他过滤器或条件(例如灵敏度条件、非特异性条件、非灵敏度条件)构建二元分类模型,以在不同阶段生成预测模型的得到输出。此外,第一子模型可以具有另一种架构(例如随机森林模型架构、梯度提升机架构等)。
4.2.第二子模型
关于预测模型的不同子模型,第二子模型可以被构造为多项分类模型(例如,作为弹性网络分类包的一部分),无论有无相关置信度,它都会输出标识癌症的组织来源属于候选组织来源集合中的一个或多个的预测。多项分类模型可以是输出一组值的多项回归模型,每个值指示与样本相关的癌症来源于与该值相关联的候选组织来源集合中的一个的概率。
图4a描绘了用于处理特征向量(例如具有小变异特征的特征向量)以预测组织来源的模型架构的示例。在图4a所示的示例中,以惩罚的多项回归模型来处理被布置为向量的一组特征。在图4a所示的示例中,惩罚多项回归模型被布置为回归集合,其中回归系数矩阵(β1,1到βn,k),应用于包含建议解释特征值(例如,对应于不同感兴趣基因的小变体特征)的变体特征向量(f1到fk)生成一个得分(score([f],too1)到得分([f],toon)的向量,用于将特征分配给组织来源组。在图4a所示的示例中,有n个可能的组织来源分组和k个感兴趣的特征。通常,该模型可以被构建为score=β*f,其中,基于通过样本处理观察到的特征,该得分可以指示样本属于特定组织来源组的概率。
在通过惩罚多项回归模型的训练来确定系数时,处理系统可以针对n个可能的组(对应于组织的来源)运行n-1个二元回归模型,其中对于每个二元回归模型,一个组织来源组用作“枢轴”,其余的n-1组织来源组分别抵制“枢轴”。更详细地,对于多项回归的一个二元回归的特定示例,乳腺组织来源可以用作其他组织来源(例如直肠,头颈,卵巢等)被回归所针对的“枢轴”。然后,基于所有概率必须加一的条件,确定与每个回归相关联的得分(或概率)。在求解概率中,估计β的系数(例如使用最大后验(map)估计,使用最大似然方法,使用另一种方法)。跨训练数据集执行与每个组织来源分组的小变异(或其他)特征相对应的得分和估计系数的确定,其中已知与训练样本相关联的组织来源。
惩罚式多项回归模型因此定义了具有由数据集训练的系数集合的函数集合,其中训练数据集可以从一组受试者的cfdna样本中得到。这些功能可以是物流功能或其他功能。对于候选组织来源集合中的每一个,可以用至少八个cfdna样本训练多项回归模型;然而,可以用任意其他合适数量的训练样本来训练多项回归模型。在一些示例中,已知具有多种癌症(例如一种以上癌症类型)的样本被去除以将训练数据集限制到可以合理地训练来源组织的样本。此外,在一些示例中,训练数据集还可以包括来自组织样本(即,gdna)的训练数据。
类似于二元分类模型架构的描述,多项回归模型可以包括阿尔法参数,阿尔法参数被配置为在岭回归模式和lasso回归模式之间调整第二子模型的性能,其中该方法可以实现用于评估一组小变异特征中的每一个对预测的贡献并基于该贡献来调整阿尔法参数的架构。关于阿尔法参数,针对岭回归模式的阿尔法调整可以通过减少这些系数的幅度来惩罚多项回归模型系数的高值,从而最大程度地减少对训练的模型的影响。关于阿尔法参数,针对lasso回归模式的阿尔法调整可以相对于模型行为,通过将不相关系数的高值设置为零来惩罚多项回归模型的系数的高值。这样,多项回归模型可以是可通过阿尔法参数来调整的惩罚多项回归模型,以包括针对将样本分类为不同组织来源组的特征。
多项回归模型还可包括表征该多项回归模型的性能的特异性条件。特异性条件可以是阈值特异性(例如99.9%特异性、99%特异性、98%特异性、95%特异性等)。多项回归模型还可以包括表征该多项回归模型的性能的灵敏度条件。灵敏度条件可以是阈值灵敏度(例如灵敏度为40%、灵敏度为50%、灵敏度为60%、灵敏度为70%等)。此外,基于预测模型的应用,可以通过不同的特异性条件和/或灵敏度条件来评估预测模型的性能。例如当使用模型进行筛选时,特异性条件和/或灵敏度条件可能会有所不同,这与使用模型评估较高风险和/或较高频率的受试者人群相反。在一些实例中,当将预测模型用于筛选目的时,预测模型的性能以99%特异性的至少50%灵敏度为特征。在其他示例中,当将预测模型应用于较高风险和较高频率的人群时,预测模型的性能以至少95%的灵敏度和60%的灵敏度为特征。在一些示例中,多类和/或二元分类器的特异性和/或灵敏度可以由用户设置或由用户以其他方式调整。
然而,可以用其他滤波器或条件(例如灵敏度条件,非特异性条件,非灵敏度条件)构造多项模型,以评估模型性能。此外,第二子模型可以具有另一种架构。例如第二子模型可以包括支持向量机,该支持向量机具有用于针对候选组织来源集合中的其他候选组织来源来评估候选组织来源集合中的每一个的架构。可替代地,第二子模型可以包括随机森林分类器,该随机森林分类器具有从受试者群的样本中得到的学习权重。可替代地,第二子模型可以包括梯度提升机。
图4b描绘了与组织来源的预测有关的与不同基因相关联的特征的模型系数输出的实施例。在图4b中,沿着y轴描绘了与基因集合(gene1至genem)相对应的特征,并且在x轴上描绘了回归模型系数。如图4b所示,对于组织来源组集合中的每一个,训练的预测模型可以针对与相关基因集合(例如gene1至genem)相对应的一组特征中的每一个包括系数集合,该系数集合对应于组织来源(即枢轴)的一组特征相对于其他组织来源的回归。如图4b所示,对于组织来源组1(too组1),模型包括与gene1至genem相关联的每个特征的系数值(在图中表示为正方形)。类似地,对于组织来源组2(too组2),模型包括与gene1至genem相关联的每个特征的系数值(在图中以三角形表示)。类似地,对于组织来源组3(too组3),模型包括与gene1至genem相关联的每个特征的系数值(在图中以圆圈表示)。类似地,对于组织来源组n(too组n),模型包括与gene1到genem相关联的每个特征的系数值(在图中以星形表示)。对于每个系数、幅度和方向(例如正或负方向)指示系数相关的可能性。更详细地,并且如图4b所示,预测模型可以允许:负系数输出对应于对来源组织来源集合的第一组织来源的分类的降低的可能性(例如,对于图4b中的too组1和gene1的特征),零系数输出对应于不确定分类(例如对于图2b中的too组2和gene6的特征),以及正系数输出对应于对候选组织源集合的第一个组织源的分类的增加的可能性(例如对于图4b中的too组3和gene2的特征)。关于系数的大小和方向,在预测模型的系数值的确定过程中,可以根据惩罚过程,取决于与预测的生成相关的特征,将系数的幅度减小或设置为零,如上关于阿尔法参数所述。
4.3.预测模型的应用
图4c描绘了根据一些实施例的用于将预测模型的实施例应用于从来自受试者的样本得到的特征向量以返回组织来源预测的流程处理。对于非训练样本,图4c描绘了过程400,该过程400用于处理样本以提取感兴趣的特征,然后将诸如上述预测模型的实施例的预测模型应用于从样本中提取的特征,以便生成与癌症存在相关联的组织来源预测(以上关于图3a步骤305g和/或305h所述)。更详细地,如图4c所示,在步骤402中,处理系统(诸如以上相对于图3a所述的处理系统)处理从受试者的cfdna样本中的序列读数以生成特征向量(例如如上面关于图3a的步骤305a-305g所述的小变异特征、副本编号特征等)。可以如上所述执行处理cfdna样本。
然后,在步骤404中,处理系统应用预测模型(例如用于生成癌变与非癌变预测的第一子模型和用于生成组织来源预测的第二子模型)。更详细地,在步骤406中,处理系统在使用训练的预测模型的第一子模型从cfdna样本中处理一组特征时提取得分。然后,处理系统在步骤408中,将为样本确定的得分与对应于癌变组与非癌变组的阈值条件进行比较。如果cfdna样本的得分满足与癌变组相关的阈值条件,则预测模型输出将样本与癌变组相关联的预测。相反,如果cfdna样本的得分不满足癌变组的阈值条件,则预测模型会输出将样本与非癌变组相关联的预测。
在步骤410中,处理系统在处理cfdna样本中的一组特征(其中该一组特征可以是相同的特征,也可以是与上述通过第一子模型处理的特征不同的特征)后提取系数集合,并将系数集合与预测模型的训练的第二子模型的系数进行比较。然后,处理系统在步骤408中确定为样本确定的系数与和组织来源组集合中的每一个相对应的系数集合之间的距离。样本相对应的系数集合和与组织来源集合中的每一个相对应的系数集合可以被布置为向量,其中向量之间的距离可以根据欧几里得距离计算或另一种合适的方法来被确定。如果cfdna样本的系数与特定组织来源的系数之间的距离小于cfdna样本的系数与其他组织来源分组的系数之间的距离,则预测模型会输出预测,预测将样本与和得分中的最小距离相对应的特定的组织来源相关联。
关于系数幅度和方向,预测模型可以基于单个特征的值或多个特征的值来生成预测。例如预测模型可以包括与一组特征的特征(例如特定基因的小变异特征)相对应的正系数(例如具有与其他组织来源的正系数不同的高幅度的正系数),并处理该一组特征以从cfdna样本生成组织来源预测的来源,可以包括:从cfdna样本中标识与正系数相关联的特征相对应的信号,并根据与cfdna样本相关联的特征的存在,从预测模型输出候选组织来源集合中的候选组织来源作为预测。
在另一示例中,预测模型可以包括与一组特征中的特征(例如,特定基因的小变异特征)相对应的负系数(例如,具有与其他组织来源的不同的高幅度的负系数),并处理该一组特征以从该cfdna样本生成组织来源预测可以包括:从该cfdna样本中标识与负系数相关联的特征相对应的信号,并基于与cfdna样本相关联的特征的存在,从该预测中排除候选组织来源集合中的候选组织来源。
5.不同组织来源的示例预测模型系数
下面的表3-表23中所示的示例模型系数是通过使用从训练样本中获得的训练数据集对多项回归模型进行训练而确定的。如表1所示,训练样本(n=1453)是从被诊断患有癌症的个体(n=879)和没有癌症诊断的健康个体(n=574)收集的血液样本。无细胞dna从样本中被提取,被测序,并被针对特征进行分析(例如基因内的非同义信息变异),以生成用于训练数据集的训练数据。表2提供了按癌症类型分类的癌症样本(n=879)的分类。基于质量控制阈值或问题,过滤最终训练数据集以移除一些样本,诸如数据集中包括的不可靠流细胞的发现。
5.1膀胱组织来源系数示例
表3提供了与膀胱组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例,其中模型系数是从样本数据集确定的,并且训练数据集是从至少8个cfdna样本确定的。如表3所示,多项回归模型可以具有与不同基因的小变异特征相对应的系数,这是在小变异特征和膀胱组织相对于其他组织组之间的回归中。表3中示出了与基因集合的小变异特征(例如基于绝对值排名前14位的特征)相对应的代表性系数值,其中正系数值表示膀胱组织来源相对于组织来源的证据,并且负系数值表示与组织来源有关的另一种类型癌症的证据。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表3中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成膀胱组织作为组织来源的预测。在一些示例中,基因组(例如,用于生成膀胱组织来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表3中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.2示例乳腺组织来源系数
关于与乳腺组织来源的预测有关的与不同基因相关的特征和对应于基因集合的小变异特征(例如,基于绝对值排名前14的特征)的代表性系数值的模型系数输出的示例在表4中被示出。例如,如表4所示,与pik3ca变异相关的特征为乳腺癌类型提供了阳性证据,而与lrp1b变异相关的特征提供了阴性证据(即,组织来源可能不是乳腺癌,而是另一种癌症),此外,与kras变异相关的特征的存在提供组织来源很可能不是乳腺的强有力的阴性证据(例如,极端负系数)。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表4中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成乳腺组织作为组织来源的预测。在一些示例中,基因组(例如,用于生成乳腺组织来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表4中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.3示例宫颈组织来源系数
关于与宫颈组织来源的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前14的特征)的代表性系数值的模型系数输出的示例在表5中被示出。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表5中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成宫颈组织作为组织来源的预测。在一些示例中,基因组(例如,用于生成宫颈组织来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表5中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.4示例直肠组织来源系数
关于与直肠组织来源的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前14的特征)的代表性系数值的模型系数输出的示例在表6中被示出。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表6中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成直肠组织作为组织来源的预测。在一些示例中,基因组(例如,用于生成直肠组织来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表6中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.5示例食道组织来源系数
关于与食道组织来源的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前14的特征)的代表性系数值的模型系数输出的示例在表7中被示出。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表7中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成食道组织作为组织来源的预测。在一些示例中,基因组(例如,用于生成食道组织来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表7中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.6示例胃组织原始来源系数
关于与胃组织来源的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前14的特征)的代表性系数值的模型系数输出的示例在表8中被示出。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表8中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成胃组织作为组织来源的预测。在一些示例中,基因组(例如,用于生成胃组织来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表8中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.7示例头/颈部组织来源系数
关于与头/颈部组织来源的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前14的特征)的代表性系数值的模型系数输出的示例在表9中被示出。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表9中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成头/颈部组织作为组织来源的预测。在一些示例中,基因组(例如,用于生成头/颈部组织来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表9中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.8示例肝胆组织来源系数
关于与肝胆组织来源的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前14的特征)的代表性系数值的模型系数输出的示例在表10中被示出。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表10中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成肝胆组织作为组织来源的预测。在一些示例中,基因组(例如,用于生成肝胆组织来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表10中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.9示例白血病来源系数
关于与白血病来源的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前14的特征)的代表性系数值的模型系数输出的示例在表11中被示出。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表11中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成白血病作为组织来源的预测。在一些示例中,基因组(例如,用于生成白血病来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表11中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.10示例肺组织来源系数
关于与肺组织来源的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前14的特征)的代表性系数值的模型系数输出的示例在表12中被示出。例如,如下表12所示,lrp1b变体的存在为肺癌类型提供了阳性证据,这与例如上表4一致,其中lrp1b变异的系数相对于乳腺癌类型为强负值.
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表12中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成肺组织作为组织来源的预测。在一些示例中,基因组(例如,用于生成肺组织来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表12中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.11示例淋巴瘤的来源系数
关于与淋巴瘤来源的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前14的特征)的代表性系数值的模型系数输出的示例在表13中被示出。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表13中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成淋巴瘤作为组织来源的预测。在一些示例中,基因组(例如,用于生成淋巴瘤来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表13中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.12.示例黑色素瘤来源系数
关于与黑色素瘤来源的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前11的特征)的代表性系数值的模型系数输出的示例在表14中被示出。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表14中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成黑色素瘤组织作为组织来源的预测。在一些示例中,基因组(例如,用于生成黑色素瘤来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表14中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.13示例多发性骨髓瘤来源系数
关于与多发性骨髓瘤来源的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前14的特征)的代表性系数值的模型系数输出的示例在表15中被示出。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表15中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成多发性骨髓瘤作为组织来源的预测。在一些示例中,基因组(例如,用于生成多发性骨髓瘤来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表15中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.14示例非癌症分组系数
关于与非癌症分组的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前14的特征)的代表性系数值的模型系数输出的示例在表16中被示出。例如,如下表16所示,tp53变异的存在为癌症提供了阳性证据,正如其与非癌症相关的强负系数所证明的那样,而kras变异的存在提供了样本可能不是无害的并且应该与癌症分组被分组的阳性证据。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表16中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成癌症/非癌症的预测。在一些示例中,基因组(例如,用于生成癌症/非癌症预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表16中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.15示例卵巢组织来源系数
关于与卵巢组织来源的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前14的特征)的代表性系数值的模型系数输出的示例在表17中被示出。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表17中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成卵巢组织作为组织来源的预测。在一些示例中,基因组(例如,用于生成卵巢组织来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表17中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.16示例胰腺组织来源系数
关于与胰腺组织来源的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前14的特征)的代表性系数值的模型系数输出的示例在表18中被示出。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表18中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成胰腺组织作为组织来源的预测。在一些示例中,基因组(例如,用于生成胰腺组织来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表18中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.17示例前列腺组织来源系数
关于与前列腺组织来源的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前14的特征)的代表性系数值的模型系数输出的示例在表19中被示出。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表19中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成前列腺组织作为组织来源的预测。在一些示例中,基因组(例如,用于生成前列腺组织来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表19中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.18示例肾脏组织来源系数
关于与肾脏组织来源的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前14的特征)的代表性系数值的模型系数输出的示例在表20中被示出。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表20中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成肾脏组织作为组织来源的预测。在一些示例中,基因组(例如,用于生成肾脏组织来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表20中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.19示例甲状腺组织来源系数
关于与甲状腺组织来源的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前10的特征)的代表性系数值的模型系数输出的示例在表21中被示出。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表21中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成甲状腺组织作为组织来源的预测。在一些示例中,基因组(例如,用于生成甲状腺组织来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表21中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.20示例子宫组织来源系数
关于与宫颈组织来源的预测有关的与不同基因相关联的特征和对应于基因集合的小变异特征(例如排名前14的特征)的代表性系数值的模型系数输出的示例在表22中被示出。
这样,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估与表22中列出的一组小变异特征中的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个相对应的一组特征的值时,可以生成子宫组织作为组织来源的预测。在一些示例中,基因组(例如,用于生成子宫组织来源预测的靶向测序组)可以包括基因和/或基因特征,该基因和/或基因特征对应于表22中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。
5.21组织来源预测的示例精度和召回度量
图5a描绘了与表1-22中所示的一部分组织来源的预测有关的预测模型的精度量输出的示例,其中度量输出是由样本数据集和每个组织来源至少8个cfdna样本的训练数据集确定的。更详细地,图5a包括精度图,一部分样本用实际上属于该组织来源的给定的组织来源被分类,从而表征了针对每个组织来源确定的真阳性与总阳性的比例。例如,图5a示出了被预测模型分类为淋巴瘤的样本中大约70%实际上是淋巴瘤样本,而被预测模型分类为多发性骨髓瘤的样本中大约50%实际上是多发性骨髓瘤样本。
在使用上述预测模型的实施例处理一组特征之后生成和/或返回预测时,处理子系统可以输出与该一组特征相对应并满足预测模型的训练期间的精度条件的组织来源,精度条件是对一组受试者的cfdna样本进行评估的。精度条件可以在与预测模型的开发相关联的训练受试者种群中具有第一条件值,并且在与预测模型的使用相关联的使用中受试者种群中具有第二条件值,从而与预测模型的使用相比,在预测模型的训练中提供不同的精度条件。
图5b描绘了与表1-表22中所示的一部分组织来源的预测有关的预测模型的召回度量输出的示例。更详细地,图5b包括召回图,一部分样本属于实际上与该组织来源分类的组织来源,从而将真阳性的一部分表征为针对每个组织来源确定的真阳性和假阴性的总和。例如图5b示出了大约1/3的实际白血病样本被预测模型正确分类为白血病。结合图5a,可以推断出,当预测模型将样本分类为白血病时,该分类是正确的(例如,参见图5a显示“白血病”为100%),然而,大约2/3的剩余实际白血病样本被归类为其他癌症类型。
在使用上述预测模型的实施例处理一组特征之后生成和/或返回预测时,处理子系统可以输出对应于一一组特征并在预测模型训练期间满足召回条件的候选组织来源,该召回条件在一组受试者的cfdna样本中被评估。召回条件可以在与预测模型的开发相关联的训练受试者种群中具有第一条件值,并且在与预测模型的使用相关联的使用受试者种群中具有第二条件值,从而与使用预测模型相比,在预测模型的训练中提供不同的召回条件。此外,关于根据上述方法步骤的实施例输出预测,处理系统可以在评估表2-表22中的任意一个的一个或多个中列出的一组特征的值后,生成对组织来源的预测。例如,基因组(例如,靶向测序组)可以包括表2-表22任一项以及来自这些表的任何组合中列出的一个或多个基因和/或基因特征。仅作为示例,基因组可以包括从表2-表22的一个或多个、两个或多个、三个或多个、四个或更多、五个或更多、八个或更多、或十个或更多的每个表中列出的一个或多个、两个以上、三个或更多、四个或更多、五个或更多、八个或更多、或十个或更多基因。
6.针对不同组织来源的附加示例预测模型系数
图6a-图6u描绘了关于与多个组织来源的预测相关的与不同基因相关联的特征(例如小变异特征)的模型系数输出的另一示例。下面的示例模型系数是通过使用从训练样本中获得的训练数据集对多项回归模型进行训练来确定的。如表23所示,训练样本(n=1435)是从被诊断为癌症的个体(n=859)和没有癌症诊断的健康个体(n=576)收集的血液样本。无细胞dna从样本中被提取、被测序、并被分析其特征(例如基因内的非同义信息变异),以生成用于训练数据集的训练数据。表24中提供了按癌症类型分类的癌症样本(n=859)的细分。
注意,尽管在该示例中使用的训练样本和先前示例在表1-22处包括的训练样本中存在一些重叠,但是在如下所述的某些情况下,训练数据集也存在一些差异,生成了与组织来源的预测相关联的不同模型系数和/或基因特征。图6a-6u的当前分析与表1-22的先前分析之间的其他差异包括生成特征的差异,诸如对构成基因内“非同义”信息变异的不同分析以及不同的交叉验证折叠集合。例如在表1-22的分析中生成的系数和基因特征使用了一个交叉验证折叠集合,而在下面的图6a-6u的分析中生成的系数和基因特征使用了不同的交叉验证折叠集合,从而对两组不同折叠的比较显示n=132个样本相等,n=1280个样本不相等,n=64不适用于仅存在于两个折叠之一中的样本。
图6a描绘了与乳腺组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的另一示例。如图6a所示,在小变异特征和乳腺组织与其他组织组之间的回归中,多项回归模型可以具有对应于不同基因的小变异特征的系数。在图6a中描绘了代表性的系数值,其中正系数值指示与组织来源有关的乳腺组织来源的证据,负系数值指示与组织来源有关的另一种癌症的证据。例如如图6a所示,pik3ca变异的存在(正系数)表明组织来源是乳腺癌,而apc变异的存在(负系数)表明组织来源不是乳腺癌。通常,包括fgf4、gata3、pik3ca、notch2、flt1、fancd2、c11orf30、notch3、stat4、tp53和epha5的基因中的变异的检测为乳腺组织来源提供了阳性证据,而包括smarca4、fancl、pbrm1、apc、jak2、pdgfrb、braf、foxo1、keap1、slit2、tnfrsf14、ptprt、smad4、lrp1b、erbb1和fat1的基因中的变异的检测为乳腺组织来源提供了阴性证据。
图6b描绘了与直肠组织来源的预测有关的、与不同基因相关联的特征的模型系数输出(例如代表性系数值)的示例。例如如图6b所示,apc变异的存在(正系数)增加了来源组织为直肠的估计概率。通常,包括apc、pten、kras、pik3ca、ncor1、ctnnb1、runx1t1、lrp1b、esr1、braf、epha7、pdgfra、jak2和dnmt3a的基因中的变异的检测可为直肠组织来源提供阳性证据,而包括idh1、btg1、arid1a和cd74的基因的变异的检测为直肠组织来源提供了阴性证据。
图6c描绘了与肺组织来源的预测有关的、与不同基因相关联的特征的模型系数输出示例。例如如图6c所示,keap1、lrp1b和/或egfr变异的存在可以表明来源组织是肺,而apc和/或pik3ca变异的存在表明来源组织不是肺。通常,包括keap1、lrp1b、egfr、ikzf1、arid2、fat1、grm3、erbb4、il7r、bcorl1、atm、smad4、kmt2c、pak7、tet2、kdm6a、pole、irf4、atr、kras、tafpms1、chek2、syk、nras、alk和pold1的基因中的变异的检测提供了肺组织来源的阳性证据,而包括apc和pik3ca的基因变异的检测提供了肺组织来源的阴性证据。
图6d描绘了与非癌症分组的预测有关的、与不同基因相关联的特征的模型系数输出的示例。例如如图6d所示,tp53变异的存在(负系数)强烈表明癌症而不是非癌症。应当注意,图6d中的正系数基因变异(例如fancl、hist1h3i、rps6kb2、phox2b)可能是由于在可能真正患有癌症的非癌变组中存在污染样本所致,并且改善的临床状况将有所改善训练集合。如图6d所示,根据其负系数,指示癌症的其他基因变异包括pbrm1、atr、alk、stag2、ctnnb1、mga、kat6a、kdr、smad4、erbb4、ptprt、arid1a、egfr、braf、notch1、dnmt3a、crebbp、apc、kmt2d、pik3ca、kras和lrp1b。
图6e描绘了与胰腺组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。例如如图6e所示,kras变异指示来源组织是胰腺。通常,包括kras、u2af1、kmt2d、smad4、tgfbr1、fance和tp53的基因中的变异的检测为胰腺组织来源提供了阳性证据,而包括flt4和dnmt1的基因中的变异的检测为胰腺组织来源提供了阴性证据。
图6f描绘了与膀胱组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。如图6f所示,jak2、kdm6a和alox12b基因变异具有正系数,并为膀胱组织来源提供了阳性证据。
图6g描绘了与未知的原发组织来源的癌症的预测有关的、与不同基因相关联的特征的模型系数输出的示例。如图6g所示,stk11、smarca4、kras、tp53、spta1、lrp1b、epha7、idh1和inpp4b基因变异具有正系数,并为未知的原发组织来源的癌症提供了阳性证据。
图6h描绘了与宫颈组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。如图6h所示,ccnd3和rfwd2基因变异具有正系数,并为宫颈组织来源提供了阳性证据。
图6i描绘了与食道组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。如图6i所示,lrp1b、erbb4、spta1、igf1r、egfr、spen、fgfr1、dot1l、fyn、igf1、runx1、foxo1、ptch1、ar、ptprt和ercc3基因变异具有正系数,并为食道组织来源提供了阳性证据。
图6j描绘了与预测胃组织来源的来源相关的、与不同基因相关联的特征的模型系数输出的示例。如图6j所示,kras、dnmt1和prex2基因变异具有正系数,并为胃组织来源提供了阳性证据。
图6k描绘了关于与头颈部组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。如图6k所示,klhl6、notch1、pbrm1、pik3cb、kmt2d、zrsr2、hist1h1c、spta1、npm1、smarca4、b2m和ctnna1基因变异具有正系数,并为头颈部组织来源提供了阳性证据。
图6l描绘了与肝胆组织来源的预测有关的、与不同基因相关的特征的模型系数输出的示例。如图6l所示,ccne1、pik3c2g、ctnnb1、slit2、tshr、tcf7l2、tgfbr2和rptor基因变异具有正系数,并为肝胆组织来源提供了阳性证据。
图6m描绘了与淋巴瘤组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。如图6m所示,crebbp、socs1、bcl2、kmt2d、pdgfrb、tnfrsf14、bcr、rel和amer1基因变异具有正系数,并为淋巴瘤组织来源提供了阳性证据。
图6n描绘了与黑色素瘤组织来源的预测有关的、与不同基因相关的特征的模型系数输出的示例。如图6n所示,dnmt3b和epha3基因变异具有正系数,并为黑素瘤组织来源提供了阳性证据。
图6o描绘了与多发性骨髓瘤组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。如图6o所示,braf、fubp1、idh2和irf4基因变异具有正系数,并为多发性骨髓瘤组织来源提供了阳性证据。
图6p描绘了与被认为是“其他”的组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例,诸如图6a-6u中未示出的其他癌症类型。如图6p所示,pax3、cxcr4和kmt2c基因变异具有正系数,并为其他组织来源类别提供了阳性证据。
图6q描绘了与卵巢组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。如图6q所示,atr、tp53、tnfrs14、fancc、klf4、msh2、fat1和brca2基因变异具有正系数,并为卵巢组织来源提供了阳性证据。
图6r描绘了关于与前列腺组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。如图6r所示,tbx3、grin2a、mga和spen基因变异具有正系数,并为前列腺组织来源提供了阳性证据,而ptprd、spta1、notch、kmt2d、pik3ca、kmt2c、apc、lrp1b和kras基因变异具有负系数,并为前列腺组织来源提供阴性证据。
图6s描绘了与肾脏组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。如图6s所示,vhl、mst1r、idh2、tsc1、notch1、ep300和sncaip基因变异具有正系数,并为肾脏组织来源提供了阳性证据。
图6t描绘了与甲状腺组织来源的预测有关的、与不同基因相关联的特征的模型系数输出的示例。如图6t所示,braf基因变异具有正系数并提供甲状腺组织来源的阳性证据,而tp53基因变异具有负系数并提供甲状腺组织来源的阴性证据。
图6u描绘了与子宫组织来源的预测有关的与不同基因相关联的特征的模型系数输出的示例。如图6u所示,cdc73、sf3b1、pten、tet1和ephb1基因变异具有正系数,为子宫组织来源提供了阳性证据,而tp53基因变异具有负系数,为子宫组织来源提供了阴性证据。
关于根据本文描述的方法步骤的实施例的输出预测,处理系统可以在评估与该特征相关的一组特征中的一个或多个特征的值时,生成组织类型的预测作为组织来源。例如对于特定的组织或癌症类型,处理系统可以评估图6a-6u中针对该癌症类型列出的任何小变异特征的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个。在一些实例中,基因组(例如用于生成组织类型的预测作为组织来源的预测的靶向测序组)可以包括基因和/或基因特征,基因和/或基因特征对应于图6a-6u中在其对应组织或癌症类型中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征。更进一步,来源组织评估和/或基因组(例如靶向的基因组)可以通过针对每个感兴趣的组织来源评估其对应图6a-6u中列出的一个或多个、两个或多个、三个或多个、四个或多个、五个或多个、八个或多个、或十个或多个基因特征的任意组合,生成对上面列出的组织来源的任意组合的预测。
7.示例计算机系统
图7示出了根据一个实施例的用于实现本文描述的过程的各种方法的示例计算机系统的示意图。特别地,图7是示出了示例计算机器组件的框图,该示例计算机器组件能够从计算机可读介质读数指令并使用处理器(或控制器)执行它们。本文描述的计算机可以包括如图7所示的单个计算机、虚拟机、包括图7所示的计算机的多个节点的分布式计算系统、或者计算设备的任意其他合适的布置。
作为示例,图7示出了计算机系统700的示例形式的计算机器的示意图,其中行指令724(例如,软件、程序代码或机器代码)可以被存储在计算机可读介质中以,用于使机器执行本文讨论的任何一个或多个处理。在一些实施例中,计算机器作为独立设备运行,或者可以连接(例如联网)到其他计算机。在联网部署中,该机器可以在服务器-客户端网络环境中以服务器机器或客户端机器的身份运行,或者在对等(或分布式)网络环境中作为对等机器运行。
图7中描述的计算机器的结构可以对应于任意软件、硬件或组合的组件(例如图5a和5b所示的组件或本文描述的处理单元),包括但不限于任意引擎、模块计算服务器、用于执行本文所述的一个或多个过程的机器。尽管图7示出了各种硬件和软件元件,但是本文描述的每个组件可以包括更多或更少的元件。
举例来说,计算机器可以是个体计算机(pc)、平板计算机、机顶盒(stb)、个体数字助理(pda)、蜂窝电话、智能手机、网络设备、网络路由器、物联网(iot)设备、交换机或网桥、或任意能够执行指令724的机器,这些指令指定了该机器要执行的操作。此外,虽然仅示出了单个机器,但是术语“机器”和“计算机”也可以被认为包括单独地或共同地执行指令724以执行本文所讨论的任意一种或多种方法的机器的任意集合。
示例计算机系统700包括一个或多个处理器702,诸如cpu(中央处理单元)、gpu(图形处理单元)、tpu(张量处理单元)、dsp(数字信号处理器)、系统在芯片(soc)、控制器、状态设备、专用集成电路(asic)、现场可编程门阵列(fpga)或它们的任意组合上。计算系统700的各部分还可以包括存储器704,该存储器704存储包括指令724的计算机代码,当指令由处理器702直接或间接执行时,该指令可以使处理器702执行某些动作。指令可以是可以以不同形式被存储的任何指令、命令或命令,诸如设备可读指令、包括源代码的编程指令以及其他通信信号和命令。指令可以在一般意义上被使用,并且不限于机器可读代码。
本文所述的一种或多种方法提高了处理器702的运行速度并减小了存储器704所需的空间。例如本文描述的机器学习方法通过应用一种或多种新技术来简化训练步骤、达到收敛和生成处理器702的结果,降低处理器702的计算的复杂性。本文描述的算法还可以减小模型和数据集的大小,以减少存储器704的存储空间要求。
某些操作的性能可以分布在一个以上的处理器之间,不仅驻留在单个计算机内,而且可以跨多个计算机部署。在一些示例实施例中,一个或多个处理器或处理器实现的模块可以位于单个地理位置中(例如在家庭环境、办公室环境或服务器场中)。在其他示例实施例中,一个或多个处理器或处理器实现的模块可以分布在多个地理位置上。即使在说明书或权利要求书中可能提到要由处理器执行的某些处理,也应将其解释为包括多个分布式处理器的联合操作。
计算机系统700可以包括被配置为经由总线708彼此通信的主存储器704和静态存储器706。计算机系统700可以还包括图形显示单元710(例如等离子体显示面板(pdp)、液晶显示器(lcd)、投影仪或阴极射线管(crt))。由处理器702控制的图形显示单元710显示图形用户界面(gui)以显示一个或多个结果以及由本文描述的过程生成的数据。计算机系统700还可以包括字母数字输入设备712(例如键盘)、光标控制设备714(例如鼠标、轨迹球、操纵杆、运动传感器或其他指向工具)、存储单元716(硬盘驱动器、固态驱动器、混合驱动器、存储磁盘等)、信号生成设备718(例如扬声器)和网络接口设备720,它们也被配置为经由总线708进行通信。
存储单元716包括计算机可读介质722,在其上存储了体现本文描述的方法或功能中的任意一个或多个的指令724。在计算机系统700执行指令724期间,指令724也可以全部或至少部分地驻留在主存储器704内或处理器702内(例如在处理器的高速缓存存储器内),主存储器704和处理器702也构成计算机可读介质。指令724可以经由网络接口设备720在网络726上被被发送或接收。
尽管在示例实施例中将计算机可读介质722示出为单个介质,但是术语“计算机可读介质”应被认为包括能够存储指令(例如指令724)的单个非暂时性介质或多个介质(例如集中式或分布式数据库、或相关联的高速缓存和服务器)。计算机可读介质可以包括能够存储指令(例如指令724)的任意介质,指令由处理器(例如处理器702)执行并且使得处理器执行本文公开的方法中的任意一个或多个。该计算机可读介质可以包括但不限于以固态存储器、光学介质和磁性介质形式的数据存储库。
8.其他注意事项
实施例的前述详细描述参考附图,其示出了本公开的特定实施例。具有不同结构和操作的其他实施例不脱离本公开的范围。参考在本说明书中阐述的申请人发明的许多替代方面或实施例的某些特定示例来使用术语“发明”等,并且其使用或不存在均不旨在限制申请人的发明或权利要求的范围。仅为了方便读者,本规范分为几部分。标题不应解释为对本公开范围的限制。该定义旨在作为本公开的描述的一部分。将理解的是,在不脱离本公开的范围的情况下,可以改变本公开的各种细节。此外,前述描述仅出于说明的目的,而非出于限制的目的。
- 还没有人留言评论。精彩留言会获得点赞!