一种靶向核壳结构载药纳米颗粒及其制备方法与流程
[0001]
本发明涉及纳米生物医药技术领域,尤其涉及一种靶向核壳结构载药纳米颗粒及其制备方法。
背景技术:
[0002]
癌症严重威胁着人类健康,目前临床治疗癌症的方法主要包括手术切除及术后辅以放化疗等多种方法协同治疗,协同治疗策略为克服单一疗法的缺陷提供了有效的解决途径。同时,光疗、基因治疗、免疫治疗等新兴疗法的出现为协同疗法提供了更多可能。为减少化疗药物的毒副作用、实现化疗药物靶向输送、定点可控释放,科学家们已把研究重点聚焦在靶向智能响应型纳米药物载体上,并取得了显著进展。肿瘤组织与正常组织相比,具有明显不同的生理微环境,比如正常组织表现为中性,而肿瘤区域呈弱酸性环境。利用这一显著差别目前已设计合成了多种酸性可降解纳米载体,以便在肿瘤组织精准释放抗肿瘤药物。
[0003]
自从发现rna干扰(rnai)现象以来,小干扰rna(sirna)因为具有靶向沉默特定基因表达的特点,已在生物学和医学研究中得到了广泛研究。人们认为基于sirna的治疗是治疗多种遗传疾病的有效方法。由于sirna分子易受体内核酸酶降解,因此单独使用sirna分子诱导基因沉默的效果很差。另外,sirna分子带负电荷且尺寸较大,几乎无法穿透细胞膜进入细胞质。因此,在利用sirna沉默基因治疗疾病的研究中面临的主要挑战是细胞质内sirna的安全递送。为促进sirna在体内的安全有效递送,科学家们已经做出了巨大的努力发展各种递送载体,主要包括病毒载体、脂质体、聚合物胶束和无机纳米颗粒等。这些纳米载体仍然具有一定的局限性,比如病毒载体存在可能诱导免疫反应和基因突的风险;脂质体、聚合物胶束虽然能通过静电相互作用有效地递送sirna分子,但表面的正电荷通常会增强对细胞膜的损伤,因此这些阳离子递送载体往往会引发严重的细胞毒性问题;无机纳米载体因为长期毒性和药物代谢特性不充分的原因在临床应用也受到了限制。因此,迫切需要寻找新的方案安全高效递送sirna分子。
技术实现要素:
[0004]
有鉴于此,本发明提供了一种靶向核壳结构载药纳米颗粒及其制备方法。本发明提供的靶向核壳结构载药纳米颗粒能够靶向肿瘤组织,有效提高纳米药物在肿瘤组织的富集与滞留,并能够利用在肿瘤组织弱酸性微环境中的响应性可控释放化疗药物和基因药物,且细胞毒性小,安全性高。
[0005]
为了实现上述发明目的,本发明提供以下技术方案:
[0006]
一种靶向核壳结构载药纳米颗粒,包括载药内核和包覆在载药内核表面的载药壳层;
[0007]
所述载药内核包括发光载药核心、修饰在所述发光载药核心表面的细胞穿膜肽以及吸附在所述细胞穿膜肽表面的基因药物;所述发光载药核心的组成成分包括化疗药物、发光共轭聚合物和聚苯乙烯-马来酸酐共聚物;所述细胞穿膜肽为阳离子型细胞穿膜肽;
[0008]
所述载药壳层包括磷酸钙层、掺杂在磷酸钙层中的化疗药物和修饰在磷酸钙层表面的核酸适配体。
[0009]
优选的,所述发光载药核心中,化疗药物、发光共轭聚合物和聚苯乙烯-马来酸酐共聚物的质量比为(10~40):(25~50):(5~20)。
[0010]
优选的,所述发光载药核心的粒径为10~100nm;所述载药内核的粒径为30~200nm;所述靶向核壳结构载药纳米颗粒的粒径为40~300nm。
[0011]
优选的,所述化疗药物包括阿霉素、紫杉醇和喜树碱中的一种或几种;所述基因药物包括sirna、aso和mirna中的一种或几种;所述发光共轭聚合物为聚(5-(2-乙基己氧基)-2-甲氧基-氰基对苯二亚甲基)。
[0012]
本发明还提供了上述方案所述靶向核壳结构载药纳米颗粒的制备方法,包括以下步骤:
[0013]
(1)将化疗药物、发光共轭聚合物和聚苯乙烯-马来酸酐共聚物溶解于有机溶剂中,在超声作用下,将所得混合溶液和水混合,之后将有机溶剂蒸除并将剩余溶液过滤,得到发光载药核心纳米颗粒溶液;
[0014]
(2)将细胞穿膜肽、发光载药核心纳米颗粒溶液和缓冲溶液混合,得到细胞穿膜肽修饰的发光载药核心纳米颗粒溶液;
[0015]
(3)将基因药物和所述细胞穿膜肽修饰的发光载药核心纳米颗粒溶液混合,得到载药内核纳米颗粒溶液;
[0016]
(4)将氯化钙、核酸适配体、化疗药物和载药内核纳米颗粒溶液混合,得到靶向核壳结构载药纳米颗粒原液;
[0017]
(5)将所述载药纳米颗粒原液进行透析处理,得到所述靶向核壳结构载药纳米颗粒。
[0018]
优选的,所述步骤(1)中的有机溶剂包括四氢呋喃、二甲基亚砜和丙酮中的一种或几种;所述混合溶液中化疗药物的浓度为10~40μg/ml,发光共轭聚合物的浓度为25~50μg/ml,聚苯乙烯-马来酸酐共聚物的浓度为5~20μg/ml;
[0019]
所述步骤(1)中将有机溶剂蒸除的方式为加热蒸除,所述加热蒸除的温度为50~200℃,所述加热蒸除在惰性气氛保护下进行。
[0020]
优选的,所述步骤(2)中混合的过程具体为:将细胞穿膜肽溶解于缓冲溶液中,得到细胞穿膜肽溶液,将发光载药核心纳米颗粒溶液和缓冲溶液混合,得到发光载药核心纳米颗粒分散液,然后将所述细胞穿膜肽溶液和所述发光载药核心纳米颗粒分散液混合后避光搅拌;所述细胞穿膜肽溶液中细胞穿膜肽的浓度为5~20mg/ml;所述发光载药核心纳米颗粒分散液中发光载药核心纳米颗粒的浓度为0.01~0.1mg/ml;所述细胞穿膜肽溶液和发光载药核心纳米颗粒分散液的体积比为(5~10):(100~200);
[0021]
所述缓冲溶液为hepes缓冲溶液。
[0022]
优选的,所述步骤(3)中细胞穿膜肽修饰的发光载药核心纳米颗粒溶液的浓度为1~10μg/ml;所述基因药物以基因药物溶液的形式使用,所述基因药物溶液的浓度为10~20μmol/l,所述基因药物溶液和所述细胞穿膜肽修饰的发光载药核心纳米颗粒溶液的用量比为1~5μl:1ml;所述混合的温度为4~37℃。
[0023]
优选的,所述步骤(4)中混合的过程具体为:将氯化钙水溶液和核酸适配体溶液混
合后进行孵育,得到氯化钙和核酸适配体混合溶液;向所述载药内核纳米颗粒溶液中依次加入化疗药物溶液以及氯化钙和核酸适配体混合溶液,混匀后避光搅拌;所述氯化钙和核酸适配体混合溶液中氯化钙的浓度为1~3mol/l,核酸适配体的浓度为10~20μmol/l;所述化疗药物溶液的浓度为1~10mg/ml;所述载药内核纳米颗粒溶液的浓度为10~100μg/ml;所述载药内核纳米颗粒溶液、化疗药物溶液和氯化钙和核酸适配体混合溶液的体积比为1000:(1~5):(1~10)。
[0024]
优选的,所述透析处理用透析袋的截留分子量为100000~1000000。
[0025]
本发明提供了一种靶向核壳结构载药纳米颗粒,包括载药内核和包覆在所述载药内核表面的载药壳层,所述载药内核包括发光载药核心、修饰在所述发光载药核心表面的细胞穿膜肽以及吸附在所述细胞穿膜肽表面的基因药物,载药壳层为磷酸钙壳层,且磷酸钙壳层中掺杂有化疗药物,壳层表面还修饰有核酸适配体。本发明的有益效果如下:
[0026]
(1)本发明提供的靶向核壳结构载药纳米颗粒是一种共载化疗药物和基因药物的纳米颗粒,该纳米颗粒的核心部分具有发光性能,可以通过荧光成像指示纳米载体的分布。
[0027]
(2)本发明在发光载药核心表面修饰阳离子型细胞穿膜肽,阳离子型细胞穿膜肽为正电性,利用静电吸引将带负电性的基因药物吸附,从而实现基因药物和化疗药物的共载;并且本发明的载药内核由于细胞穿膜肽的存在而呈正电性,从而使载药内核具有从溶酶体逃逸进入细胞浆的特点,以达到在细胞浆内释放基因药物和化疗药物的目的。
[0028]
(3)本发明提供的载药纳米颗粒的载药壳层成分为磷酸钙(简称为磷酸钙壳层),磷酸钙壳层在血液循环中能够保持稳定,能够起到保护基因药物和化疗药物的作用,避免基因药物降解和化疗药物泄露,提高基因药物的稳定性和化疗药物的有效性。
[0029]
(4)磷酸钙壳层表面修饰的核酸适配体能够靶向肿瘤组织,可以大大提高载药纳米颗粒识别和结合肿瘤细胞的能力,有效提高纳米药物在肿瘤组织的富集与滞留,解决现有纳米药物因靶向性差引发毒副作用的问题;并且核酸适配体修饰后整个载药纳米颗粒呈负电性,与负电性的细胞膜具有良好的生物相容性。
[0030]
(5)磷酸钙壳层具有酸性可降解的特点,载药纳米颗粒在核酸适配体作用下选择性接触肿瘤细胞,在肿瘤区域富集,载药纳米颗粒进入肿瘤细胞后,首先分布在细胞内的内涵体、溶酶体中,在酸性环境下(ph=5-6)cap壳层逐渐降解,释放出壳层中掺杂的化疗药物,并暴露出纳米颗粒的载药内核,载药内核表面因修饰细胞穿膜肽而呈正电性,能够加速肿瘤细胞摄取并实现溶酶体逃逸,帮助载药内核进入胞浆释放基因药物和化疗药物,显著提高抗肿瘤作用,解决了现有的纳米药物在细胞内经溶酶体途径被免疫清除的问题。
[0031]
综上,本发明提供的靶向核壳结构载药纳米颗粒能够有效提高纳米药物在肿瘤组织的富集与滞留,并能够利用在肿瘤组织弱酸性微环境中的响应性可控释放化疗药物和基因药物,且细胞毒性小,安全性高,实现更高效的化疗-基因联合治疗。
[0032]
本发明还提供了上述方案所述靶向核壳结构载药纳米颗粒的制备方法,本发明的制备过程不涉及高温和强酸、强碱环境,反应温和、操作简单,重复性好,多次重复实验均可得到相同的结果,解决了现有技术中载药纳米颗粒制备条件苛刻、操作复杂的问题。
附图说明
[0033]
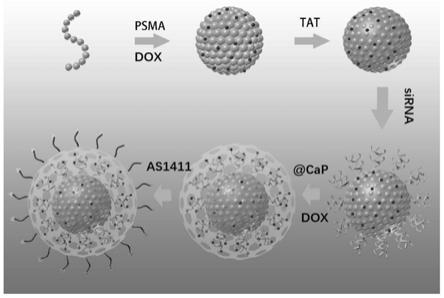
图1为本发明的靶向核壳结构载药纳米颗粒递送化疗药物和基因药物的原理示意
图;
[0034]
图2为本发明制备靶向核壳结构载药纳米颗粒的过程示意图;
[0035]
图3为本发明制备靶向核壳结构载药纳米颗粒的工艺流程图;
[0036]
图4为实施例1制备的d/cpnps的粒径分布统计图;
[0037]
图5为实施例5制备的d/cp
tat
/sirnanps的粒径分布统计图;
[0038]
图6为实施例6制备的t-d/cp
tat
/sirna@capnps的粒径分布统计图;
[0039]
图7为实施例1制备的d/cpnps的zeta电位图;
[0040]
图8为实施例5制备的d/cp
tat
/sirnanps的zeta电位图;
[0041]
图9为实施例6制备的t-d/cp
tat
/sirna@capnps的zeta电位图;
[0042]
图10为d/cpnps和t-d/cp
tat
/sirna@capnps的透射电镜图;
[0043]
图11为实施例6制备的t-d/cp
tat
/sirna@capnps在ph=5和ph=7.2的缓冲溶液中释放dox的曲线图;
[0044]
图12为t-d/cptat/sirna@cap nps在肿瘤细胞hepg-2和正常细胞l02中孵育30min的细胞荧光成像图;
[0045]
图13为t-d/cp
tat
/cy5-sirna@cap nps在肿瘤细胞hepg-2中孵育2h的溶酶体荧光共定位成像图;
[0046]
图14为t-d/cptat/sirna@capnps在肿瘤细胞hepg-2中孵育4h的细胞核荧光共成像图;
[0047]
图15为t-d/cp
tat
/sirna@capnps对肿瘤细胞hepg-2的细胞毒性图;
[0048]
图16为t-d/cp
tat
/sirna@capnps对肿瘤细胞hepg-2的凋亡效果图。
具体实施方式
[0049]
本发明提供了一种靶向核壳结构载药纳米颗粒,包括载药内核和包覆在载药内核表面的载药壳层。
[0050]
在本发明中,所述载药内核包括发光载药核心、修饰在所述发光载药核心表面的细胞穿膜肽以及吸附在所述细胞穿膜肽表面的基因药物;所述发光载药核心的组成成分包括化疗药物、发光共轭聚合物和聚苯乙烯-马来酸酐共聚物(psma)。在本发明中,所述化疗药物优选包括阿霉素(dox)、紫杉醇(taxol)、喜树碱(cpt)中的一种或几种;所述基因药物优选包括sirna、aso、mirna中的一种或几种;所述发光载药核心中化疗药物、发光共轭聚合物和聚苯乙烯-马来酸酐共聚物的质量比优选为(10~40):(25~50):(5~20),更优选为(20~30):(30~40):(10~15);所述发光共轭聚合物优选为具有疏水性质发光共轭聚合物,更优选为聚合物氰基聚苯乙炔衍生物,具体为聚(5-(2-乙基己氧基)-2-甲氧基-氰基对苯二亚甲基)(cn-ppv)(后续简称为cn-ppv),所述cn-ppv的结构式如式i所示;本发明对所述发光共轭聚合物的来源没有特殊要求,采用市售的发光共轭聚合物即可,在本发明的具体实施例中,所述cn-ppv购自加拿大魁北克ads股份有限公司,产品编号为ads110re。
[0051][0052]
本发明对所述聚苯乙烯-马来酸酐共聚物的分子量没有特殊要求,采用本领域技术人员熟知分子量的聚苯乙烯-马来酸酐共聚物即可。在本发明中,所述聚苯乙烯-马来酸酐共聚物具有双亲性,通过物理缠绕作用聚苯乙烯-马来酸酐共聚物和发光共轭聚合物缠绕在一起,形成表面呈负电性的纳米颗粒,并将化疗药物包载在纳米颗粒中,即为本发明的发光载药核心。
[0053]
在本发明中,所述细胞穿膜肽为阳离子型细胞穿膜肽,优选为tat,所述阳离子型细胞穿膜肽具有正电性,通过静电吸附修饰在发光载药核心表面;本发明对所述细胞穿膜肽的来源没有特殊要求,使用市售的细胞穿膜肽即可。
[0054]
在本发明中,所述基因药物呈负电性,通过静电作用吸附在细胞穿膜肽表面。
[0055]
在本发明中,所述载药壳层包括磷酸钙层、掺杂在磷酸钙层中的化疗药物和修饰在磷酸钙层表面的核酸适配体。在本发明中,所述磷酸钙层优选通过生物矿化法在载药内核表面沉积得到,所述磷酸钙层具有孔隙,化疗药物掺杂在磷酸钙层的孔隙中;所述化疗药物的可选种类和上述方案一致,在此不再赘述;所述核酸适配体优选为as1411。本发明对所述核酸适配体的来源没有特殊要求,采用市售的核酸适配体即可。
[0056]
在本发明中,所述发光载药核心的粒径优选为10~100nm,更优选为20~40nm;所述载药内核的粒径优选为30~200nm,更优选为40~80nm;所述靶向核壳结构载药纳米颗粒的粒径优选为40~300nm,更优选为80~120nm。
[0057]
图1为本发明的靶向核壳结构载药纳米颗粒递送化疗药物和基因药物的原理示意图。本发明的载药纳米颗粒在核酸适配体作用下选择性接触肿瘤细胞,进入肿瘤细胞后,首先分布在细胞内的内涵体、溶酶体中,在酸性环境下(ph=5-6)cap壳层逐渐降解,释放出ca
2+
和壳层中掺杂的化疗药物,暴露出纳米颗粒的载药内核,载药内核具有溶酶体逃逸的特性而不会被免疫清除,载药内核进入胞浆后释放基因药物和化疗药物。
[0058]
本发明提供了上述方案所述靶向核壳结构载药纳米颗粒的制备方法,图2为本发明制备靶向核壳结构载药纳米颗粒的过程示意图;图3为本发明制备靶向核壳结构载药纳米颗粒的工艺流程图。下面结合图2~3对本发明的制备方法进行具体说明:
[0059]
本发明提供的制备方法包括以下步骤:
[0060]
(1)将化疗药物、发光共轭聚合物和聚苯乙烯-马来酸酐共聚物溶解于有机溶剂中,在超声作用下,将所得混合溶液和水混合,之后将有机溶剂蒸除并将剩余溶液过滤,得到发光载药核心纳米颗粒溶液;
[0061]
(2)将细胞穿膜肽、发光载药核心纳米颗粒溶液和缓冲溶液混合,得到细胞穿膜肽修饰的发光载药核心纳米颗粒溶液;
[0062]
(3)将基因药物和所述细胞穿膜肽修饰的发光载药核心纳米颗粒溶液混合,得到载药内核纳米颗粒溶液;
[0063]
(4)将氯化钙、核酸适配体、化疗药物和载药内核纳米颗粒溶液混合,得到靶向核
壳结构载药纳米颗粒原液;
[0064]
(5)将所述载药纳米颗粒原液进行透析处理,得到所述靶向核壳结构载药纳米颗粒。
[0065]
本发明将化疗药物、发光共轭聚合物和聚苯乙烯-马来酸酐共聚物溶解于有机溶剂中,得到混合溶液。在本发明中,所述有机溶剂优选包括四氢呋喃、二甲基亚砜和丙酮中的一种或几种;所述混合溶液中化疗药物的浓度优选为10~40μg/ml,更优选为20~30μg/ml,发光共轭聚合物的浓度优选为25~50μg/ml,更优选为30~40μg/ml,聚苯乙烯-马来酸酐共聚物的浓度为5~20μg/ml,更优选为10~15μg/ml。
[0066]
在本发明的具体实施例中,优选先将化疗药物、发光共轭聚合物和聚苯乙烯-马来酸酐共聚物分别溶解于有机溶剂中,得到化疗药物溶液、发光共轭聚合物溶液和聚苯乙烯-马来酸酐共聚物溶液,然后再将化疗药物溶液、发光共轭聚合物溶液和聚苯乙烯-马来酸酐共聚物溶液按比例混合并稀释,得到符合上述浓度要求的混合溶液。
[0067]
得到混合溶液后,本发明在超声作用下,将所得混合溶液和水混合,之后将有机溶剂蒸除并将剩余溶液过滤,得到发光载药核心纳米颗粒溶液。在本发明中,所述水优选为超纯水;本发明优选在超声作用下将混合溶液快速注入装有超纯水的小瓶中,然后继续超声1~2min,以实现混合溶液和水的混合。在本发明中,将所述有机溶剂蒸除的方式优选为加热蒸除,所述加热蒸除的温度优选为50~200℃,更优选为100~150℃,所述加热蒸除优选在惰性气氛保护下进行,更优选在氮气保护下进行;本发明对加热蒸除的时间没有特殊要求,能够将有机溶剂蒸除完全即可。
[0068]
有机溶剂蒸除完全后,本发明优选将剩余溶液冷却至室温,然后过滤。在本发明中,所述过滤用滤头优选为水系滤头,所述水系滤头的孔径优选为0.22μm。
[0069]
采用本发明方法制备的发光载药核心纳米颗粒溶液能够稳定保存12个月以上,不会出现聚沉现象。
[0070]
得到发光载药核心纳米颗粒溶液后,本发明将细胞穿膜肽、发光载药核心纳米颗粒溶液和缓冲溶液混合,得到细胞穿膜肽修饰的发光载药核心纳米颗粒溶液。在本发明中,所述缓冲溶液优选为hepes(羟乙基哌嗪乙磺酸)缓冲溶液;所述hepes缓冲溶液的ph值优选为7.2~7.4。
[0071]
在本发明中,所述混合的过程具体优选为:将细胞穿膜肽溶解于缓冲溶液中,得到细胞穿膜肽溶液,将发光载药核心纳米颗粒溶液和缓冲溶液混合,得到发光载药核心纳米颗粒分散液,然后将所述细胞穿膜肽溶液和所述发光载药核心纳米颗粒分散液混合后避光搅拌。在本发明中,所述细胞穿膜肽溶液中细胞穿膜肽的浓度优选为5~20mg/ml,更优选为10mg/ml;所述发光载药核心纳米颗粒分散液中发光载药核心纳米颗粒的浓度优选为0.01~0.1mg/ml,更优选为0.06mg/ml;所述细胞穿膜肽溶液和发光载药核心纳米颗粒分散液的体积比优选为(5~10):(100~200),更优选为7:200;本发明优选将所得混合液中细胞穿膜肽的浓度控制为0.35mg/ml。在本发明中,所述细胞穿膜肽溶液和发光载药核心纳米颗粒分散液中缓冲物质的浓度均优选为20mmol/l。在本发明的具体实施例中,优选直接将细胞穿膜肽溶解于20mmol/l的hepes缓冲溶液中,得到细胞穿膜肽溶液;发光载药核心纳米颗粒溶液和1mmol/l的hepes缓冲溶液按比例混合,控制所得分散液中hepes的浓度为20mmol/l。
[0072]
在本发明中,所述避光搅拌的温度优选为室温,所述避光搅拌的时间优选为
30min;在避光搅拌过程中,带正电的细胞穿膜肽通过静电相互作用充分结合在带负电的发光载药核心纳米颗粒表面。
[0073]
避光搅拌完成后,本发明优选将所得搅拌液进行离心和超滤,去除多余的细胞穿膜肽,得到细胞穿膜肽修饰的发光载药核心纳米颗粒溶液。在本发明的具体实施例中,优选根据实际需要对所得细胞穿膜肽修饰的发光载药核心纳米颗粒溶液进行稀释。
[0074]
得到细胞穿膜肽修饰的发光载药核心纳米颗粒溶液后,本发明将基因药物和所述细胞穿膜肽修饰的发光载药核心纳米颗粒溶液混合,得到载药内核纳米颗粒溶液。在本发明中,所述细胞穿膜肽修饰的发光载药核心纳米颗粒溶液的浓度优选为1~10μg/ml,更优选为5~10μg/ml;本发明优选将基因药物溶液加入所述细胞穿膜肽修饰的发光载药核心纳米颗粒溶液中,所述基因药物溶液的浓度优选为10~20μmmol/l;所述基因药物溶液和所述细胞穿膜肽修饰的发光载药核心纳米颗粒溶液的用量比优选为(1~5)μl:1ml,更优选为1.25μl:1ml;所述混合的温度优选为4~37℃,更优选为4~10℃。在混合过程中,带有负电的基因药物通过静电相互作用充分结合在细胞穿膜肽表面。
[0075]
得到载药内核纳米颗粒溶液后,本发明将氯化钙、核酸适配体、化疗药物和载药内核纳米颗粒溶液混合,得到靶向核壳结构载药纳米颗粒原液。在本发明中,所述混合的过程具体优选为:将氯化钙水溶液和核酸适配体溶液混合后进行孵育,得到氯化钙和核酸适配体混合溶液;向所述载药内核纳米颗粒溶液中依次加入化疗药物溶液以及氯化钙和核酸适配体混合溶液,混匀后避光搅拌。在本发明中,所述核酸适配体溶液的溶剂优选为ph=7.2~7.4的hepes缓冲液;所述氯化钙和核酸适配体混合溶液中氯化钙的浓度优选为1~3mol/l,更优选为2.5mol/l,核酸适配体的浓度优选为10~20μmol/l,更优选为15~20μmol/l;所述孵育的温度优选为室温,孵育的时间优选为10~60min,更优选为15min;所述化疗药物溶液的浓度优选为1~10mg/ml,所述化疗药物溶液的溶剂优选为去离子水;所述载药内核纳米颗粒溶液的浓度为10μg/ml;所述载药内核纳米颗粒溶液、化疗药物溶液和氯化钙和核酸适配体混合溶液的体积比为1000:(1~5):(1~10),更优选为1000:1:2。
[0076]
在本发明中,所述避光搅拌的转速优选为20rpm,搅拌时间优选为8h。在避光搅拌过程中,表面带正电荷的载药内核纳米颗粒吸附带负电的核酸适配体,使得核酸适配体覆盖在载药内核纳米颗粒表面,在静电吸附作用下ca
2+
与核酸适配体上的po
43-基团逐渐结合沉积在载药内核纳米颗粒表面,形成磷酸钙壳层,在这个过程中实现了核酸适配体的修饰。
[0077]
得到载药纳米颗粒原液后,本发明将所述靶向核壳结构载药纳米颗粒原液原液进行透析处理,得到所述靶向核壳结构载药纳米颗粒。在本发明中,所述透析处理用透析袋的截留分子量优选为100000~1000000,更优选为300000,在本发明的具体实施例中,使用的透析袋优选为md34-300000。
[0078]
在本发明中,最终所得靶向核壳结构载药纳米颗粒分散于hepes缓冲液中,载药纳米颗粒的浓度以及化疗药物、基因药物的载药量可以根据实际所需药量进行调节。在本发明的具体实施例中,载药纳米颗粒的浓度优选为10μg/ml,以载药纳米颗粒的质量计,所述化疗药物的载药量优选为10~100mg/g,更优选为30mg/g,以载药纳米颗粒分散液的体积计,所述基因药物的载药量优选为10~20nmol/l,更优选为15nmol/l。
[0079]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。
[0080]
实施例中的发光共轭聚合物cn-ppv购自cn-ppv购自加拿大魁北克ads股份有限公
司,细胞穿膜肽tat、核酸适配体as1411以及化疗药物购自生工生物工程(上海)股份有限公司。
[0081]
实施例1
[0082]
发光载药核心纳米颗粒(d/cpnps)溶液的制备:
[0083]
利用纳米再沉淀法制备核心发光载药纳米颗粒,步骤如下:
[0084]
(1)将化疗药物阿霉素(dox)、发光共轭聚合物(cn-ppv)和聚苯乙烯马来酸酐共聚物(psma)分别溶解在四氢呋喃中,得到dox溶液、cn-ppv溶液和psma溶液,最终浓度均为1mg/ml,作为原始溶液;
[0085]
(2)将所述dox溶液、cn-ppv溶液和psma溶液混合,并用四氢呋喃稀释,得到混合液,所述混合液中dox、cn-ppv和psma的质量浓度依次为10μg/ml、25μg/ml和5μg/ml。
[0086]
(3)在超声作用下,将所述混合液快速注入超纯水中,得到纳米粒子混合体系,并将纳米粒子混合体系继续超声1min。
[0087]
(4)将纳米粒子混合体系升温至90℃并通入氮气以去除所述溶剂,直至溶剂蒸发完全,将剩余溶液冷却至室温后,用孔径为0.22μm的水系滤头过滤,得到d/cpnps溶液。
[0088]
根据以上方法制备得到的d/cp nps溶液在保存12个月甚至更长时间下仍能保持稳定且不会出现聚沉现象。
[0089]
实施例2
[0090]
发光载药核心纳米颗粒(d/cpnps)溶液的制备
[0091]
利用纳米再沉淀法制备发光载药核心纳米颗粒,步骤如下:
[0092]
(1)将化疗药物阿霉素(dox)、发光共轭聚合物(cn-ppv)和聚苯乙烯马来酸酐共聚物(psma)分别溶解在二甲基亚砜中,得到dox溶液、cn-ppv溶液和psma溶液,最终浓度均为1mg/ml,作为原始溶液;
[0093]
(2)将所述dox溶液、cn-ppv溶液和psma溶液混合后用二甲基亚砜稀释,得到混合液,所述混合液中dox、cn-ppv和psma的质量浓度依次为20μg/ml、50μg/ml和10μg/ml。
[0094]
(3)在超声作用下,将所述混合液快速注入超纯水中,得到纳米粒子混合体系,并将纳米粒子混合体系继续超声2min。
[0095]
(4)将纳米粒子混合体系升温至100℃并通入氮气以去除所述溶剂,直至溶剂蒸发完全,将剩余溶液冷却至室温后,用孔径为0.22μm的水系滤头过滤,得到d/cpnps溶液。
[0096]
根据以上方法制备得到的d/cp nps溶液在保存12个月甚至更长时间下仍能保持稳定且不会出现聚沉现象。
[0097]
实施例3
[0098]
发光载药核心纳米颗粒(d/cpnps)溶液的制备
[0099]
利用纳米再沉淀法制备发光载药核心纳米颗粒,步骤如下:
[0100]
(1)将化疗药物阿霉素(dox)、发光共轭聚合物(cn-ppv)和聚苯乙烯马来酸酐共聚物(psma)分别溶解在丙酮中,得到dox溶液、cn-ppv溶液和psma溶液,最终浓度均为1mg/ml,作为原始溶液;
[0101]
(2)将所述的dox溶液、cn-ppv溶液和psma溶液混合后稀释,得到混合液,所述混合液中dox、cn-ppv和psma的质量浓度依次为40μg/ml、100μg/ml和20μg/ml。
[0102]
(3)在超声作用下,将所述混合液快速注入超纯水中,得到纳米粒子混合体系,并
将纳米粒子混合体系继续超声3min。
[0103]
(4)将纳米粒子混合体系升温至110℃并通入氮气以去除所述溶剂,直至溶剂蒸发完全,将剩余溶液冷却至室温后,用孔径为0.22μm的水系滤头过滤,得到d/cpnps溶液。
[0104]
根据以上方法制备得到的d/cp nps溶液在保存12个月甚至更长时间下仍能保持稳定且不会出现聚沉现象。
[0105]
实施例4
[0106]
tat修饰的发光载药核心纳米颗粒(d/cp
tat
nps)溶液的制备
[0107]
(1)按照实施例1的方法制备d/cp nps溶液;
[0108]
(2)预先将tat溶解在20mmol/l hepes的缓冲溶液(ph=7.2~7.4)中,确定tat的浓度为10mg/ml。向步骤(1)所述d/cpnps溶液中加入1mol/l的hepes的缓冲溶液(ph=7.2~7.4),使d/cpnps分散在20mmol/l的hepes的缓冲溶液(ph=7.2~7.4)中,得到d/cpnps分散液。
[0109]
(3)向d/cpnps分散液中加入tat溶液,使tat浓度为0.35mg/ml,混匀后在室温条件下避光搅拌,使得带正电的tat通过静电相互作用充分结合在带负电的d/cpnps表面。将搅拌后的混合液进行离心超滤,除去多余的tat,得到d/cp
tat
nps溶液。
[0110]
实施例5
[0111]
载药内核纳米颗粒(d/cp
tat
/sirnanps)溶液的制备
[0112]
(1)按照实施例4的方法制备d/cp
tat nps溶液,稀释至纳米颗粒的浓度为10μg/ml。
[0113]
(2)向1ml浓度为10μg/ml的d/cp
tat
nps溶液中加入1.25μl sirna(浓度为20μmol/l),于4℃条件下旋转混合,得到d/cp
tat
/sirnanps溶液。
[0114]
实施例6
[0115]
靶向核壳结构载药纳米颗粒的制备:
[0116]
(1)按照实施例5的方法制备d/cp
tat
/sirnanps纳米颗粒溶液。
[0117]
(2)将cacl2水溶液与as1411溶液混合,室温下孵20min,得到cacl2和as1411混合溶液。
[0118]
(3)向1ml浓度为10μg/ml的d/cptat/sirnanps溶液中加入1μl dox溶液(浓度为10mg/ml)混匀,加入所述cacl2和as1411混合溶液2μl,使用涡轮混匀器混匀,室温条件下置于转盘上以20rpm的转速避光搅拌,得到t-d/cp
tat
/sirna@capnps原液。
[0119]
(4)将所述t-d/cp
tat
/sirna@cap nps原液使用md34-300000透析袋透析,得到t-d/cp
tat
/sirna@capnps。
[0120]
表征和测试:
[0121]
(1)粒径测试
[0122]
使用动态光散射仪对实施例1制备的d/cp nps、实施例5制备的d/cp
tat
/sirnanps和实施例6制备的t-d/cp
tat
/sirna@capnps的粒径进行测试,测试结果如图4~6所示。
[0123]
图4为实施例1制备的d/cp nps的粒径分布统计图,图5为实施例5制备的d/cp
tat
/sirna nps的粒径分布统计图,图6为实施例6制备的t-d/cp
tat
/sirna@capnps的粒径分布统计图;根据图4~6可以看出,d/cp nps的平均粒径为30nm,d/cp
tat
/sirna nps的平均粒径为70nm,t-d/cp
tat
/sirna@capnps的平均粒径为110nm。由此可见,随着逐步修饰,纳米颗粒的尺寸逐渐增大。
[0124]
(2)zeta电位测试
[0125]
使用纳米电位-粒度仪对实施例1制备的d/cp nps、实施例5制备的d/cp
tat
/sirnanps和实施例6制备的t-d/cp
tat
/sirna@capnps的zeta电位进行测试,结果如图7~9所示。
[0126]
图7为实施例1制备的d/cpnps的zeta电位图,图8为实施例5制备的d/cp
tat
/sirna nps的zeta电位图,图9为实施例6制备的t-d/cp
tat
/sirna@capnps的zeta电位图。根据图7~9可以看出,d/cpnps的zeta电位为-25mv,d/cp
tat
/sirna nps的zeta电位为+24mv,t-d/cp
tat
/sirna@capnps的的zeta电位为-6mv。由此可见,tat修饰后,纳米颗粒表面所带电荷由负变正,这一结果使纳米颗粒具备了静电吸引基因药物的性质。
[0127]
(3)形貌测试
[0128]
图10为d/cpnps和t-d/cp
tat
/sirna@capnps的透射电镜图,图10左侧为d/cpnps的透射电镜图,右侧为t-d/cp
tat
/sirna@capnps的透射电镜图。由图10可知,经过逐步修饰后,纳米颗粒尺寸增大,但是形貌没有发生变化。
[0129]
(4)药物释放测试
[0130]
图11为实施例6制备的t-d/cp
tat
/sirna@capnps在ph=5和ph=7.2的缓冲溶液中释放dox的曲线图。图11可知,在ph=7.2缓冲溶液中dox的释放更慢更少,4小时内释放率约为30%;而在ph=5的缓冲溶液中,dox的释放速度快,处理4小时后,纳米颗粒中dox释放率达55%以上,说明弱酸性环境下纳米颗粒的壳层发生降解。
[0131]
(5)荧光成像测试
[0132]
将实施例6制备的t-d/cp
tat
/sirna@capnps在肿瘤细胞hepg中和正常细胞l02中孵育30min,然后进行荧光成像测试,结果如图12所示。
[0133]
图12为t-d/cptat/sirna@cap nps在肿瘤细胞hepg-2和正常细胞l02中孵育30min的细胞荧光成像图,图12左侧为在肿瘤细胞hepg-2中孵育后的荧光成像图,右侧为在正常细胞l02中孵育后的荧光成像图。根据图2可以看出,肿瘤细胞较正常细胞具有明显的荧光亮度,说明t-d/cptat/sirna@capnps具有选择性识别肿瘤细胞的能力。
[0134]
(6)溶酶体荧光共定位成像
[0135]
将实施例6中使用的sirna替换为cy5标记的sirna,制备得到t-d/cp
tat
/cy5-sirna@capnps,将t-d/cp
tat
/cy5-sirna@capnps在肿瘤细胞hepg中孵育2h,进行溶酶体荧光共定位成像测试,使用的荧光染料为hoechst 33342,结果如图13所示。
[0136]
图13为t-d/cp
tat
/cy5-sirna@capnps在肿瘤细胞hepg-2中孵育2h的溶酶体荧光共定位成像图(图13中cnppv表示本发明的载药纳米颗粒)。根据图13可以看出,肿瘤细胞内检测出了明显的红色荧光与绿色荧光,红色荧光来自标记在sirna上的cy5染料,绿色荧光来自溶酶体染料,说明该纳米颗粒成功将sirna递送至细胞质内并实现了溶酶体逃逸。
[0137]
(7)细胞核荧光共成像
[0138]
将实施例6制备得到t-d/cp
tat
/sirna@capnps在肿瘤细胞hepg-2中孵育4h,进行细胞核荧光共成像测试,使用的荧光染料为hoechst 33342,结果如图14所示。
[0139]
图14为t-d/cp
tat
/sirna@cap nps在肿瘤细胞hepg-2中孵育4h的细胞核荧光共成像图。根据图14可以看出,肿瘤细胞内检测出了明显的红色荧光与蓝色荧光,以及细胞核内的红色和蓝色交叠的区域,红色荧光来自标记化疗药物dox,蓝色荧光来自细胞核染料,说
明该纳米药物携带的化疗药物进入到细胞核内。
[0140]
(8)对肿瘤细胞的细胞毒性实验
[0141]
实验步骤:首先将对数生长期细胞用胰蛋白酶消化,制成细胞悬液,并稀释成密度为70000个细胞/ml,取100μl悬液加入96孔板的孔中,使每孔细胞7000个,边缘孔用无菌pbs填充。在5%co2,37℃培养箱中孵育至次日。按实验方案向孔板中加入浓度分别为0μg/ml、2μg/ml、5μg/ml、10μg/ml和20μg/ml的载药纳米颗粒。每种处理设置5个复孔。处理结束后,每个孔中分别加入10μlcck-8溶液,轻轻敲击培养板混匀。按照所述同样的处理方式铺3个96孔板,在培养箱中分别孵育6、12、24小时。使用酶标仪测定450nm光吸收值,按公式计算药物对细胞的抑制率。结果如图15所示。
[0142]
图15为t-d/cp
tat
/sirna@capnps对肿瘤细胞hepg-2的细胞毒性图。根据图15可以看出,在载药纳米颗粒浓度为2μg/ml、5μg/ml、10μg/ml和20μg/ml的条件下,随细胞与纳米药物孵育时间延长,细胞的生存活力明显降低,说明纳米药物对肿瘤细胞具有明显的毒性。
[0143]
(9)细胞凋亡实验
[0144]
实验步骤:
[0145]
仪器参数调节:1、收集1
×
106~3
×
106个细胞,用预冷pbs离心洗涤两次,弃上清。2、加入500μlapoptosis positive control solution重悬,置冰上孵育30分钟。3、用预冷pbs离心洗涤,弃上清。4、加入适量预冷1
×
binding buffer重悬,并加入数量相同且未经处理的活细胞与之混合。加入预冷1
×
binding buffer补充至1.5ml,等分成三管,其中一管为空白对照组、两管为单染组。5、单染管分别加入5μlannexinv-fitc或10μl pi,室温避光孵育5分钟。6、在流式细胞仪上,用空白对照组调节fsc、ssc和荧光通道的电压,并在此电压条件下,用单染组调节荧光通道的补偿。
[0146]
样本检测:1、按实验方案用t-d/cp
tat
/sirna@capnps诱导凋亡。2、用预冷pbs离心洗涤,收集1~10
×
105个细胞(包括培养上清中的细胞)。用双蒸水稀释5
×
binding buffer为1
×
工作液,取500μl 1
×
binding buffer重悬细胞。3、加入5μlannexinv-fitc和10μlpi。4、轻柔涡旋混匀后,室温避光孵育5分钟。
[0147]
实验结果如图16所示。
[0148]
图16为t-d/cp
tat
/sirna@cap nps对肿瘤细胞hepg-2的凋亡效果图;由图16可知,经载药纳米颗粒处理的肿瘤细胞有10%处于早期凋亡阶段,有27%处于晚期凋亡阶段,说明纳米药物对肿瘤细胞的生长有明显的抑制作用。
[0149]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1