一种PS/cPGMA核壳型纳米粒及其制备方法与流程
本发明属于亲和高分子微球制备领域,具体涉及一种PS/cPGMA核壳型纳米粒及其制备方法。
背景技术:
胶乳增强免疫比浊法(LETIA),通过采用物理吸附或共价键合的方式将抗体(或抗原)偶联到纳米颗粒的表面,形成纳米粒-抗体(抗原)复合物,此复合物与样品中的抗原(或抗体),通过抗体抗原反应,形成纳米粒-抗体-抗原聚集颗粒,随着免疫反应的不断发生,聚集的颗粒不断增大,从而导致溶液在一定波长(如600nm)的吸光值发生显著的变化,通过测定免疫反应前后吸光度值的变化可以计算出样品中抗原(抗体)的浓度,从而达到检测的目的。同其它免疫分析方法(如:放射免疫分析法、化学发光免疫分析法、酶联免疫分析法等)相比,LETIA由于具有检测方法简单方便、无放射性污染、可准确定量、稳定性好、同时可在全自动生化仪上实现高通量样品检测等优点,已经越来越多的被应用于临床检测。迄今,LETIA在临床中已经应用于检测β2-微球蛋白、胱抑素C、D-二聚体、脂蛋白a、抗链球菌溶血素“O”、类风湿因子、C-反应蛋白(CRP)、高敏C反应蛋白(hsCRP)、α1-微球蛋白、肌钙蛋白、肌红蛋白、肌酸激酶同工酶、甲胎蛋白、胃蛋白酶原I、前列腺特异性抗原、糖化血红蛋白等多种检测项目,诊断领域已涉及肾功能、风湿、心肌、肿瘤、糖尿病等多个领域,拥有广阔的市场前景。
LETIA中通常使用聚苯乙烯(PS)材料的纳米级胶乳颗粒,抗体可以通过与PS的疏水性相互作用以物理吸附的形式固定于纳米粒表面,然而这种方法制备的抗体-纳米粒复合物稳定性较差。一般通过对PS纳米粒进行表面改性或共聚等方法在纳米粒表面引入活性基团,进而采用化学键和的 方法进行抗体固定化可有效提高试剂的稳定性。
通过与丙烯酸共聚得到的羧基化PS纳米粒在LETIA中最常采用,例如Kyhse-Andersen等(J.Kyhse-Anderson,C.Schmidt.G.Nordin,B.Andersson,et.al.,Serum cystatin C,determined by a rapid,automated particle-enhanced turbidimetric method,is a better marker than serum creatinine for glomerular filtration rate.Clin.Chem.,1994,40(10),1921-1926.)采用羧基化PS纳米粒制备了光抑素C抗体固定化的PS纳米颗粒,并建立了血浆光抑素C的LETIA检测方法。采用化学键合的方法在PS纳米粒表面实现蛋白固定化显著提高了试剂的稳定性,但是由于固定化基质材料(如羧基化PS纳米粒)表面仍有苯环基团暴漏,固定在纳米粒表面的蛋白质仍将有一部分物理吸附(而非化学键合)在纳米粒表面。例如Santos和Forcada(R.M.Santos,J.Forcada.Acetal-functionalized polymer particles useful forimmunoassays.Ⅲ:preparation of latex-protein complexes and their applications.Journal of Materials Science:Materials in Medicine.2001,12:173-180.)报道采用表面醛基修饰的PS纳米粒进行抗体固定化,其中仍有约20%的抗体以物理吸附的形式固定化。
为了进一步的避免蛋白质与胶乳纳米粒的物理吸附作用,使蛋白质完全以化学键合的形式固定到胶乳纳米粒表面,并在此基础上增加蛋白质-纳米粒复合物的稳定性,需对纳米粒的表面进行彻底的亲水性改造,即纳米粒表面覆盖一层亲水性涂层使纳米粒对蛋白质无非特异性吸附作用。早在1984年Litchfield(W.J.Litchfield,A.R.Craig,W.A.Frey,C.C.Leflar,C.E.Looney,M.A.Luddy.Novel shell/core particles for automated turbidimetric immunoassays.Clinical Chemistry,1984,30:1489-1493.)等便利用种子乳液聚合法,制备了外壳亲水的“核壳型”纳米颗粒。该方法中采用PS纳米粒为种子,甲基丙烯酸缩水甘油酯(GMA)为外壳聚合单体,制备了以PS为种子,聚GMA(PGMA)为涂层材料的PS/PGMA“核壳型”纳米粒子,并将其用于制备LETIA检测试剂。采用类似的方法,段殷等(段殷,左锋,柴 宏森,刘建华,王冬梅,徐亮,周晶。单分散PS/PGMA核-壳型高分子微球的制备与表征。天津医科大学学报,2012,18:416-418.)也制备了PS/PGMA纳米粒,并系统的考察了种子纳米粒的粒径、GMA的加入量、乳化剂的加入量、聚合反应时间等因素对核-壳型纳米粒制备的影响。
上述方法虽然通过制备“核壳型”纳米材料实现了纳米粒表面的亲水性改造,但是具有亲水性的外壳聚合物材料是以物理吸附的方式吸附在PS种子纳米粒表面,这易导致“核壳型”纳米粒子的稳定性变差。此外,外壳涂层材料PGMA亲水性较强,在水溶液中长期贮存易溶胀,导致制得的核壳型纳米粒的水力学粒径变大。
技术实现要素:
本发明的目的在于,为了解决现有技术中核壳型PS纳米粒的外壳聚合物材料是以物理吸附的方式吸附在PS纳米粒表面,从而存在稳定性差;并且在水溶液中长期贮存易溶胀,导致纳米粒水力学粒径变大的问题,提供一种PS/cPGMA核壳型纳米粒的制备方法。
为了达到上述目的,本发明的技术方案如下:
一种PS/cPGMA核壳型纳米粒的制备方法,包括步骤如下:
(1)将羧基化PS纳米粒表面进行乙烯基化修饰得到乙烯基化PS纳米粒;
(2)将乙烯基化PS纳米粒通过种子乳液聚合法制备PS/cPGMA核壳型纳米粒。
上述PS/cPGMA核壳型纳米粒的制备方法中,所述羧基化PS纳米粒的平均粒径为60nm-500nm,羧基密度以干球计为0.065mmol/g-0.350mmol/g。
上述PS/cPGMA核壳型纳米粒的制备方法中,所述进行乙烯基化修饰的方法包括以下步骤:将羧基化PS纳米粒分散,经1-(3-二甲氨基丙基)-3- 乙基碳二亚胺盐酸盐活化后,再加入丙烯胺进行反应,得到乙烯基化PS纳米粒。
上述PS/cPGMA核壳型纳米粒的制备方法中,加入的1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐的物质的量为羧基化PS纳米粒上羧基的物质的量的1-5倍。
上述PS/cPGMA核壳型纳米粒的制备方法中,将羧基化PS纳米粒分散于磷酸盐缓冲液中,得到PS纳米粒的水溶液,其中羧基化PS纳米粒的质量为磷酸盐缓冲液质量的2-10%;所述丙烯胺以体积浓度为10%的丙烯胺磷酸盐缓冲液的形式加入,加入量为PS纳米粒水溶液体积的0.5-1.5;所述磷酸盐缓冲液为pH7.4、25mM的磷酸盐缓冲液;所述活化的条件为:室温震荡反应15-60分钟;加入丙烯胺后于4-37℃震荡,反应的时间为3-12小时。
上述PS/cPGMA核壳型纳米粒的制备方法中,所述种子乳液聚合法制备PS/cPGMA核壳型纳米粒的方法包括:
以乙烯基化PS纳米粒为种子,过硫酸钾为引发剂,GMA为单体,碳酸盐缓冲液为分散相,十二烷基磺酸钠为表面活性剂,乙二醇二甲基丙烯酸酯为交联剂。
上述PS/cPGMA核壳型纳米粒的制备方法中,所述种子乳液聚合法制备PS/cPGMA核壳型纳米粒的具体方法包括:
将乙烯基化PS纳米粒水溶液与碳酸盐缓冲溶液按体积比为1:1-1:3混合;然后加入过硫酸钾、GMA、乙二醇二甲基丙烯酸酯和十二烷基磺酸钠搅拌反应,搅拌转速为120转每分钟,反应温度为70-80℃,反应时间为6-10h,制得PS/cPGMA纳米粒;所述乙烯基化PS纳米粒水溶液中的水溶液为pH7.4、25mM的磷酸盐缓冲液,乙烯基化PS纳米粒的质量为磷酸盐缓冲液质量的2-10%。
上述PS/cPGMA核壳型纳米粒的制备方法中,过硫酸钾的质量与乙烯基化PS纳米粒水溶液的体积比为0.1-0.5g/100mL,GMA的质量与乙烯基化PS纳米粒水溶液的体积比为0.2-2g/100mL,十二烷基磺酸钠的质量与乙烯基化PS纳米粒水溶液的体积比为0.01-0.05g/100mL;乙二醇二甲基丙烯酸酯的质量为GMA质量的1-5%。
上述PS/cPGMA核壳型纳米粒的制备方法制备的PS/cPGMA核壳型纳米粒。
上述PS/cPGMA核壳型纳米粒在胶乳增强免疫比浊法中的应用。
与现有技术相比,本发明的优点如下:
1、本发明的PS/cPGMA核壳型纳米粒的制备方法,对羧基化PS纳米粒表面进行乙烯基团活化,使壳层聚合物材料以化学键合的方式固定在羧基化PS种子纳米粒表面,提高制得纳米粒的稳定性。
2、本发明的PS/cPGMA核壳型纳米粒的制备方法,在PGMA壳层材料的制备中,加入了交联剂EDMA,EDMA的投料量占GMA投料量的1-5%,在保证纳米粒表面的亲水性的同时,从而控制壳层材料PGMA在水溶液中的溶胀,进一步提高制得纳米粒的稳定性。
3、本发明的PS/cPGMA核壳型纳米粒的制备方法,选择粒径范围在60nm-500nm的羧基化PS纳米粒为原料,通过EDC活化后易与带有氨基的丙烯胺反应,进而在纳米粒表面引入乙烯基团,为后续种子乳液聚合法制备PS/cPGMA核壳型纳米粒制得一定粒径的乙烯基化PS种子纳米粒。
4、本发明的PS/cPGMA核壳型纳米粒的制备方法制备的PS/cPGMA核壳型纳米粒外壳亲水且在水溶液中稳定性好;外壳亲水可确保蛋白质以化学键合的方式,而非物理吸附固定到纳米粒表面,与蛋白质非特异性吸附作用较弱,可作为LETIA试剂的生产原料使用,与PS纳米粒制备的LETIA试剂相比稳定性显著提高。
附图说明
下面将通过附图和具体实例进一步说明本申请。
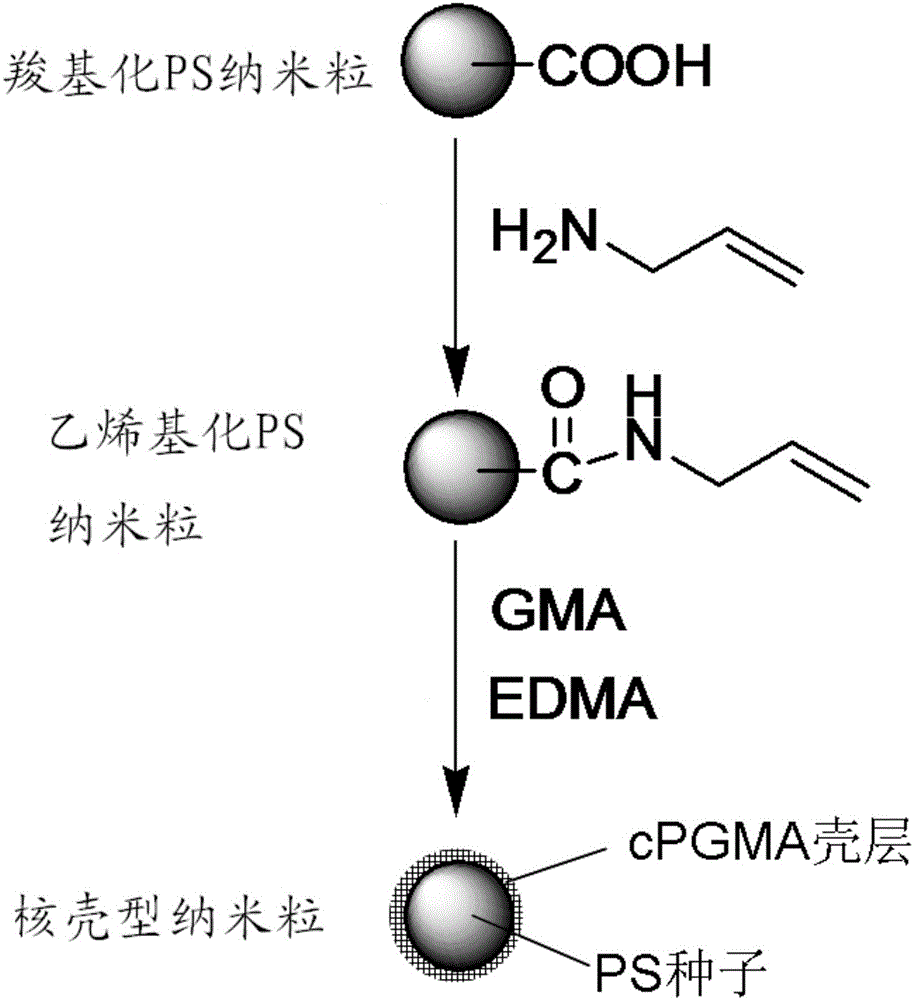
图1为PS/cPGMA纳米粒制备过程示意图。
图2为PS/cPGMA纳米粒的透射电镜照片。
图3为光抑素C检测试剂稳定性测试结果对比;其中,图(A)为PS/cPGMA纳米粒制备的试剂;图(B)为PS纳米粒制备的试剂。
图4为本发明制备的PS/cPGMA核壳型纳米粒与PS/PGMA纳米粒的稳定性结果。
具体实施方式
本发明的所有实施例中的原料、试剂如无特殊说明均为市售商品。下面结合实例对本发明的实施进一步进行说明,但是本发明的实施不仅限于此。
实施例1
根据图1所示的制备过程示意图,制备平均粒径约为120nm的PS/cPGMA纳米粒;
将羧基化PS纳米粒(瑞安市伊普西隆生物科技有限公司)分散于PBS(25mM,pH7.4)中,制备5%(w/w)的PS纳米粒水溶液100mL。羧基化PS纳米粒平均粒径为109.6nm,羧基密度为0.157mM/g(干球),加入150.5mg EDC,室温震荡反应15分钟后,继续加入100mL含10%(v/v)丙烯胺的PBS(25mM,pH7.4)溶液。25℃震荡反应8小时后,用去离子水透析除去未反应的丙烯胺,制得乙烯基化PS纳米粒。用截留分子量为300KD的超滤膜将制得的乙烯基化PS纳米粒浓缩至100mL,5%(w/w)的水溶液。
将上述制备的100mL乙烯基化PS纳米粒水溶液置于一圆底三口烧瓶中,分别加入200mL碳酸盐缓冲液(pH9.0,25mM),0.2g KPS,0.8g GMA,0.016g EDMA,0.02g SDS,在充氮气的条件下,置于75℃水浴中,120转 每分钟搅拌反应8h,制得PS/cPGMA纳米粒。将制得的纳米粒用去离子水透析,并超滤浓缩至5%(w/w)的水溶液。
将制得的PS/cPGMA纳米粒用透射电子显微镜观测,结果参见图2。由图可知,制得的微球具有较好的球形度,粒度分布均匀(图2A),具有明显的核壳型结构(图2B)。通过粒径分析仪测定,制得的PS/cPGMA纳米粒的平均粒径为120.6nm,比种子纳米粒的粒径略有增加(种子纳米粒即羧基化PS纳米粒的平均粒径为109.6nm),粒径增大的部分必然是由于纳米粒壳层的存在而导致,这也与图2显微镜观测的结果一致。
实施例2
根据图1所示的指标过程示意图,制备平均粒径约为65nm的PS/cPGMA纳米粒;
将羧基化PS纳米粒(瑞安市伊普西隆生物科技有限公司)分散于PBS(25mM,pH7.4)中,制备10%(w/w)的PS微球水溶液100mL。羧基化PS纳米粒平均粒径为60.0nm,羧基密度为0.065mM/g(干球),加入124.6mg EDC,室温震荡反应15分钟后,继续加入50mL含10%(v/v)丙烯胺的PBS(25mM,pH7.4)溶液。4℃震荡反应12小时后,用去离子水透析除去未反应的丙烯胺,制得乙烯基化PS纳米粒。用截留分子量为300KD的超滤膜将制得的乙烯基化PS纳米粒浓缩至100mL,10%(w/w)的水溶液。
将上述制备的100mL乙烯基化PS纳米粒水溶液置于一圆底三口烧瓶中,分别加入300mL碳酸盐缓冲液(pH9.0,25mM),0.5g KPS,2g GMA,0.02g EDMA,0.05g SDS,在充氮气的条件下,置于70℃水浴中,120转每分钟搅拌反应10h,制得PS/cPGMA纳米粒。将制得的纳米粒用去离子水透析,并超滤浓缩至5%(w/w)的水溶液。用粒径分析仪测定制得的PS/cGMA纳米粒的平均粒径为65.0nm。
实施例3
根据图1所示的指标过程示意图,制备平均粒径约为520nm的PS/cPGMA纳米粒;
将羧基化PS纳米粒(瑞安市伊普西隆生物科技有限公司)分散于PBS(25mM,pH7.4)中,制备2%(w/w)的PS微球水溶液100mL。羧基化PS纳米粒的平均粒径为500nm,羧基密度为0.350mM/g(干球),加入134.2mg EDC,室温震荡反应15分钟后,继续加入150mL含10%(v/v)丙烯胺的PBS(25mM,pH7.4)溶液。37℃震荡反应3小时后,用去离子水透析除去未反应的丙烯胺,制得乙烯基化PS微球。用截留分子量为300KD的超滤膜将制得的乙烯基化PS纳米粒浓缩至100mL,2%(w/w)的水溶液。
将上述制备的100mL乙烯基化PS纳米粒水溶液置于一圆底三口烧瓶中,分别加入100mL碳酸盐缓冲液(pH9.0,25mM),0.1g KPS,0.2g GMA,0.01g EDMA,0.01g SDS,在充氮气的条件下,置于80℃水浴中,120转每分钟搅拌反应6h,制得PS/cPGMA纳米粒。将制得的纳米粒用去离子水透析,并超滤浓缩至5%(w/w)的水溶液。用粒径分析仪测定制得的PS/cGMA纳米粒的平均粒径为520.1nm。
实施例4
PS/cPGMA纳米粒制备的光抑素C(Cys-C)检测试剂的稳定性考察。
取实施例1制备的PS/cPGMA微球水溶液(5%w/w)20mL,加入20mL碳酸盐缓冲液(pH9.0,25mM)混合后,加入1.25g甘氨酸,室温震荡反应17h。而后加入1g NaBH4终止反应,将制得的微球用去离子水透析,并超滤浓缩至5%(w/w)的水溶液。制得羧基化PS/cPGMA微球,经电位滴定法测定羧基密度为0.150mM/g(干球)。
分别取上述制得的羧基化PS/cPGMA微球与粒径及羧基密度相同的羧基化PS微球各1mL,制备Cys-C LETIA检测试剂。R1与R2的配方及制备方法如下:
R1:60mL,其中含有50mM PBS(pH7.4),0.5%BSA,0.05%NaN3,150mM NaCl。
R2:15mL,制备方法如下:取羧基化微球溶液或羧基化PS/cPGMA微球1mL,与1mL 50mM 2-吗啉代乙磺酸(MES)缓冲液(pH6.0)混合后,加入EDC与N-羟基硫代琥珀酰亚胺各10mg后,室温震荡反应20min。加入10mg Cys-C多克隆抗体(产自瑞安市伊普西隆生物科技有限公司),37℃水浴震荡反应3h。随后,加入0.5g甘氨酸终止反应。将上述反应液用截留分子量为500kD的膜包切向流过滤除去未偶联的抗体,所用的洗液为50mM PBS(pH7.4),切向流过滤后收集微球溶液15mL。最后向微球溶液中加入BSA 0.3g,制得R2。
Cys-C标准品稳定性的测定:对8mg/L,4mg/L,1mg/L标准品进行测试,采用加速试验的方法进行稳定性测试。即:将上述制备的试剂均分为14份,完全密封后放入37℃水浴中,每天取出1份对标准品进行测试,连续测试14天。测试结果参见图3。
由图可知,采用PS/cPGMA纳米粒制备的试剂,考察期间内,测试结果无明显变化趋势,标准品的信号值相对标准偏差(RSD)小于2.3%(图3A);采用PS纳米粒制备的试剂,考察期间内,测试结果有显著下降趋势,标准品的信号值相对标准偏差(RSD)显著增大(图3A)。这充分说明PS/cPGMA制备的LETIA检测试剂比PS纳米粒制备的检测试剂具有更好的稳定性。
实施例5
将实施例1制备的PS/cPGMA纳米粒和PS/PGMA纳米粒(根据段殷等,单分散PS/PGMA核-壳型高分子微球的制备与表征。天津医科大学学报,2012,18:416-418.中的方法制备)的溶胀性进行比较。
将两种纳米粒均用50mM PBS(pH7.4,含0.05%NaN3)溶液配制成5%(w/w)的微球水溶液,并将其密封后放置在37℃水域中,测定21天 内的粒径变化情况,对比结果参见图4。
由图4可知,PS/PGMA纳米粒粒径显著增大(从第一天测定的120.4nm增大到第21天的167.8nm),而PS/cGMA纳米粒的粒径并未见明显变化(第一天测得的粒径为120.2nm,而到第21天的为120.0nm)。这说明本专利制备的纳米粒稳定性得到了显著提高。
- 还没有人留言评论。精彩留言会获得点赞!
- 磁性胶体核壳结构γ‑Fe2O3及Fe3O4的制备方法与流程
- 一种核壳结构Au@SiO2负载Pt纳米球的制备方法与流程
- 一种核壳结构Ag@TiO2@Pt纳米复合材料的制造方法与工艺
- 一种基于Ag@In核壳结构的高温钎料及其制备方法与流程
- 一种核壳结构Au@Co(OH)2纳米微球的制备方法与流程
- 一种制备核‑壳结构的高介低损钛酸铜钙基陶瓷的方法与流程
- 一种具有核壳结构的微球复合负极材料及其制备方法和应用与制造工艺
- 一种防潮耐水含橡胶籽油的核壳结构纳米改性变压器油及其制备方法与制造工艺
- 一种耐盐雾含茶籽油的核壳结构纳米改性变压器油及其制备方法与制造工艺
- 一种含锭子油的高流动性核壳结构纳米改性变压器油及其制备方法与制造工艺
- 一种核壳结构Au@SiO2负载Pt纳米球的制备方法与流程
- 一种核壳结构Ag@TiO2@Pt纳米复合材料的制造方法与工艺
- 一种基于Ag@In核壳结构的高温钎料及其制备方法与流程
- 一种核壳结构Au@Co(OH)2纳米微球的制备方法与流程
- 一种制备核‑壳结构的高介低损钛酸铜钙基陶瓷的方法与流程
- 一种具有核壳结构的微球复合负极材料及其制备方法和应用与制造工艺
- 一种防潮耐水含橡胶籽油的核壳结构纳米改性变压器油及其制备方法与制造工艺
- 一种耐盐雾含茶籽油的核壳结构纳米改性变压器油及其制备方法与制造工艺
- 一种含锭子油的高流动性核壳结构纳米改性变压器油及其制备方法与制造工艺
- 一种含椰子油的抗氧化核壳结构纳米改性变压器油及其制备方法与制造工艺