染色体拷贝数变异程度评估模型、方法及应用与流程
1.本发明涉及基因测序领域,特别是涉及尿路上皮癌患者染色体拷贝数变异(cnv)程度的评估。
背景技术:
2.尿路上皮癌是起源于尿路上皮的一种多源性的恶性肿瘤,包括肾盂癌、输尿管癌、膀胱癌以及尿道癌,是最常见的泌尿系统肿瘤。其中尿路上皮癌可分为非肌层浸润性尿路上皮癌(nmibc)和肌层浸润性尿路上皮癌(mibc)。而10%~15%的肌层浸润性尿路上皮癌患者在确诊时已出现转移。对于t3-t4和(或)n+mo的高危患者,5年生存率仅为25%~35%。吸烟及职业性致癌剂是重要因素。遗传基因缺陷在外因的影响下促发癌变已愈来愈受重视。地区性、种族性发病如“巴尔干肾病”和“马兜铃酸肾病”可能与遗传及环境因素有关。
3.尿脱落细胞fish技术是通过多色的荧光原位杂交检测3号、7号、17号染色体和9p16探针畸变,对尿液标本要求高,适用范围窄,且需有经验丰富的细胞病理学家;同时有低级别尿路上皮癌感性低、假阳性高、常细胞一致性差的问题。尿液dna层面检测单基因/多基因panel突变,如tert启动子c228t和c250t突变、fgfr3突变等,同样存在敏感性和特异性较差的问题。主要原因是尿路上皮癌中基因突变模式多样,包含基因数目较多的大panel在成本收益上较低。
4.苏州宏元生物科技有限公司推出的urocad产品采集10ml尿液中100个以上的细胞(染色体异常肿瘤细胞占比不低于2%),基于一组染色体不稳定区域(3p、3q、5p、7p、7q、8q、9p、 9q、17p、17q),采用低覆盖全基因组测序技术(low-coverage whole-genome sequencing)对样本进行染色体不稳定检测,排查肿瘤相关的染色体变异,分析染色体不稳定性。该检测手段本质上还是基于尿液脱落细胞中的染色体异常肿瘤细胞,其缺点是:1、对尿液取样的要求高(dna提取依赖尿液中的染色体异常肿瘤细胞数目),容易导致ngs实验dna建库失败;2、尿液中可能存在较多的正常脱落细胞,dna提取效率较低,导致背景噪音大;3、阳性率与细胞量有关,细胞量的多少影响结果的准确性;4、早期的尿路上皮癌患者尿脱落细胞少,拷贝数变异程度低;5、算法依赖于10个染色体异常区域,导致灵敏度和特异性降低,容易造成漏检错检;6、以200kb划分窗口计算时容易混入种系的拷贝数变异,产生无法去除的背景噪音。
5.尿液在肾小管中产生,并在每个肾脏的肾盂中聚集,经输尿管流入膀胱,最后尿液储存在膀胱中,直到通过尿道离开身体。由于泌尿生殖道癌的特殊性,尿液中的循环游离dna能够满足特异性突变、结构变异、甲基化检测的需求。
技术实现要素:
6.本发明要解决的技术问题之一是提供一种染色体拷贝数变异程度评估模型,它可以准确判定膀胱癌患者的染色体不稳定性状态。
7.为解决上述技术问题,本发明的染色体拷贝数变异程度评估模型,根据拷贝数变
异负荷评估染色体拷贝数变异程度,所述拷贝数变异负荷为发生拷贝数变异的bin的数目与全基因组bin的数目的比值。
8.bin的划分长度优选设置为1m bp。所述发生拷贝数变异的bin为拷贝数值大于膀胱癌患者拷贝数阈值的bin。所述拷贝数值(logr)的计算公式优选为:;式中,cfdna样本优选为尿液上清cfdna样本;待测cfdna样本每个bin的reads数目优选经过gc、mappability 以及深度差异的校正;健康人cfdna样本每个bin的reads数目优选为健康人尿液上清cfdna参考背景数据库的normal panel中的每个bin的reads数目。
9.本发明要解决的技术问题之二是提供上述染色体拷贝数变异程度评估模型的构建方法,该方法主要包括如下步骤:构建健康人尿液cfdna参考背景数据库;将临床样本尿液cfdna测序数据与参考基因组比对去重后,进行低深度全基因组拷贝数变异和肿瘤分数检测;设置每个bin的长度,去除值为na的bin,获得全基因组bin数目;使用所述参考背景数据库,统计每个bin的拷贝数值logr,所述logr值的计算公式为:;计算膀胱癌患者样本尿液中cfdna染色体区域的拷贝数阈值;将拷贝数值大于所述拷贝数阈值的bin判定为发生拷贝数变异的bin;计算发生拷贝数变异的bin数目占全基因组bin数目的比例,获得各临床样本的拷贝数变异负荷;根据所述拷贝数变异负荷,计算灵敏度、特异度和约登指数,选取最佳临界值作为染色体不稳定性为阳性的判断阈值,完成染色体拷贝数变异程度评估模型的构建。
10.所述参考背景数据库的构建方法,优选包括如下步骤:提取健康人的尿液上清cfdna进行浅层全基因组测序;测序结果与参考基因组比对去重;对人体除y染色体以外的23个染色体进行bin划分,并计算区域覆盖度;通过整个基因组的平均覆盖度标准化每一个bin的覆盖度,根据参考基因组的gc比例、测序深度和比对的偏好性,进行均一化校正,得到所述参考背景数据库。
11.所述膀胱癌患者样本尿液中cfdna染色体区域的拷贝数阈值的计算方法,优选包括如下步骤:分别对正常人样本和膀胱癌患者样本的拷贝数变异值进行皮尔逊相关性检验,剔除弱相关样本;计算每个值非na的bin非na的拷贝数logr值,并按正常人样本和膀胱癌患者样本分别统计取均值,得到logr
normal
和logr
tumor
;对所有的logr
normal
取均值,计算每个logr
tumor
偏离logr
normal
均值的偏离值,并对偏离值取均值,获得膀胱癌患者拷贝数阈值;计算公式
为:;式中,bin个数为去除值为na的bin后的全基因组bin数目。
12.所述膀胱癌患者拷贝数阈值优选为0.9。
13.本发明要解决的技术问题之三是提供基于上述模型的染色体拷贝数变异程度评估方法。该方法用上述染色体拷贝数变异程度评估模型计算样本的拷贝数变异负荷,然后根据拷贝数变异负荷的值评估样本染色体拷贝数变异程度。
14.所述拷贝数变异负荷的具体计算方法,优选包括如下步骤:提取尿液上清cfdna样本,进行浅层全基因组二代测序;对测序数据进行序列比对去重,计算每个bin的reads数目;将每个bin的reads数目和健康人尿液上清cfdna参考背景数据库中每个bin的reads数目相比,计算logr值;将拷贝数logr值大于膀胱癌患者拷贝数阈值的bin判定为发生拷贝数变异的bin;计算发生拷贝数变异的bin的数目与全基因组bin的数目的比值,获得拷贝数变异负荷。
15.染色体拷贝数变异程度评估标准优选为:当拷贝数变异负荷高于4%时,样本染色体不稳定性判定为阳性。
16.本发明要解决的技术问题之四是提供上述染色体拷贝数变异程度评估模型和评估方法的用途。该评估模型和评估方法可用于尿路上皮癌患者的术后监测。
17.本发明构建尿液上清循环游离dna背景库,应用二代测序技术对膀胱癌患者尿液上清中的循环游离dna样本进行低覆盖全基因组测序,检测膀胱癌不同进展时期患者的全基因组染色体不稳定区域,然后通过染色体拷贝数变异负荷模型计算拷贝数变异负荷,判定膀胱癌患者的染色体不稳定性状态,实现对膀胱癌患者的术后监测。与现有技术相比,本发明的染色体拷贝数变异程度评估模型及评估方法具有以下优点和有益效果:1.尿液样本易于获得,尿液cfdna采样侵入性小,可以及时、多次采样,反复检测,实现对患者疾病状态的动态监控,有利于对患者响应状态和预后风险的实时评价;2.相对于基因单个位点的突变,拷贝数变异在肿瘤发生中的贡献率更大,能够解决突变检测敏感性和特异性差的问题;3.相对于fish检测,通量更大,准确性更高;4.对尿液取样要求低,检测使用尿液上清的cfdna,不依赖于尿液中的肿瘤细胞数目即可完成准确的检测,可以克服因尿液中肿瘤细胞数量不足而导致的检测范围局限性,提高检测的准确性和特异性;5.检测所需dna起始量低(仅需1-2ng),尿液采集量只需5-10ml,提升了技术可及性;6.适用范围广,可适用于所有尿路上皮癌,只要肿瘤破碎释放出的游离dna能进入到尿液当中,就可以使用本发明的染色体拷贝数变异程度评估模型及方法对肿瘤患者进行术后监测。
附图说明
18.图1是一个膀胱癌样本的全基因组bin的肿瘤分数检测和异质性评价图。横坐标为染色体位点,纵坐标为拷贝数log2值。处于坐标轴上方的染色体位点代表染色体片段发生扩增;处于坐标轴下方的染色体位点代表染色体片段发生缺失;线段代表该片段发生的是亚克隆事件。其中,样本中肿瘤的含量(tumor fraction)为0.3282;样本肿瘤的倍性(ploidy)为2.5;发生亚克隆的dna占肿瘤dna的比例(subclone fraction)为0.521;发生亚克隆的bin占全基因组bin的比例(fraction genome subclonal)为0.25;发生亚克隆的bin占全基因组发生拷贝数变异的bin的比例(fraction cna subclonal)为0.32。
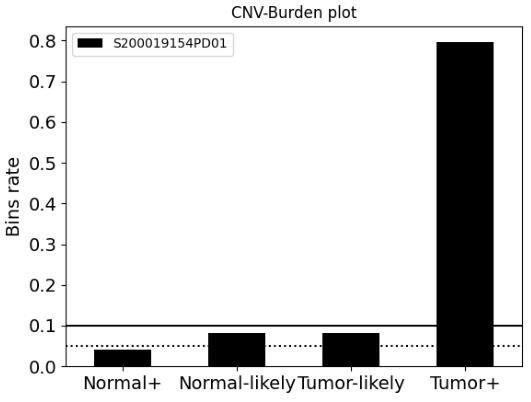
19.图2是一个膀胱癌样本全基因组水平bin的染色体不稳定性分组图。其中tumor+(肿瘤阳性)的bin比例达到79.6%;tumor likely(可能肿瘤)的bin比例为8.2%;normal likely(可能正常)的bin比例为8.1%;normal+(正常)的bin比例为4.1%。
20.图3是去除不合格样本后的数据集的尿液上清cfdna全基因组肿瘤阳性的bin百分比分布图。
21.图4是roc曲线图。
具体实施方式
22.为对本发明的技术内容、特点与功效有更具体的了解,现结合附图及具体实施方式,对本发明的技术方案做进一步详细的说明。
23.实施例1 染色体拷贝数变异程度评估模型的构建1.健康人尿液上清cfdna背景库的生成由于cfdna片段一般在166bp左右,测序读数(reads)长度为150bp,双端测序会产生一定比例的重叠区域。因此,本发明选取1m bp进行bin的划分,克服在100-200bp大小的重叠区域双倍计数的影响。
24.提取30例健康人的尿液上清cfdna进行浅层全基因组测序(swgs),分别与h19参考基因组比对去重后生成bam文件,然后使用readcounter软件对人体除y染色体以外的23个染色体以1m bp步移窗口划分,计算区域覆盖度并生成wig文件。通过整个基因组的平均覆盖度标准化每一个窗口的覆盖度,根据参考基因组的gc比例、测序深度和比对的偏好性,使用hmmcopy进行均一化校正。最后生成健康人尿液cfdna的参考背景数据库,用于纠正由dna建库、测序平台和特异性的cfdna而产生的系统性误差,降低噪音,提高准确性。
25.2.测序数据预处理从ncbi数据库下载18例临床样本(包括9例经过临床诊断为膀胱癌的样本和9例正常人样本)的尿液上清cfdna的swgs测序数据,经与参考基因组比对去重后,使用健康人尿液cfdna的参考背景数据库,运行ichorcna,进行低深度cfdna样本的全基因组cnv(拷贝数变异)和肿瘤分数(tumor fraction)的检测(图1显示了其中一个膀胱癌样本全基因组水平bin的肿瘤分数检测和异质性评价结果),获取软件的运算结果。
26.以1m bp大小设置每个bin的大小(bin的长度为1m bp时,可以更好的摒除种系的拷贝数变异),均匀覆盖整个基因组,总共得到2954个bin,去除值为na的bin,总共有2510个bin入选数据集。统计每个bin的logr值,计算公式为:
。
27.3.cnv的阈值划分(1)正常人尿液中的dna染色体区域的拷贝数计算对9例正常人群样本的cnv值进行皮尔逊相关性检验,剔除弱相关样本1例。
28.对于每个bin非na的logr值进行统计取均值得到logr
normal
,然后对所有的logr
normal
值取均值,计算偏离值范围取均值,获得正常人尿液中循环游离的dna染色体区域的拷贝数阈值为0.19。计算公式如下:;式中,bin个数为2510个。
29.(2)膀胱癌患者尿液中循环游离的dna染色体区域的拷贝数计算对9例膀胱癌患者人群样本的cnv值进行皮尔逊相关性检验,剔除弱相关样本1例。
30.对于每个bin非na的logr值进行统计取均值得到logr
tumor
,然后对所有的logr
normal
值取均值,计算偏离值范围取均值,获得膀胱癌患者尿液中循环游离的dna染色体区域的拷贝数阈值为0.90。计算公式如下:;式中,bin个数为2510个。
31.4.cnv-burden(拷贝数变异负荷)模型构建(1)染色体不稳定性bin的分组根据上述第3步获得的cnv的阈值,对16例临床样本中的每个样本的bin进行分组,拷贝数值为0~0.19:正常(normal+);拷贝数值为0.19~0.545:可能正常(normal likely);拷贝数值为0.545~0.9:可能肿瘤(tumor likely);拷贝数值》0.9:肿瘤(tumor+)。
32.其中,拷贝数值即logr值;normal likely的拷贝数阈值0.545为正常人尿液cfdna染色体区域的拷贝数阈值0.19和膀胱癌患者尿液cfdna染色体区域的拷贝数阈值0.90之和的平均值。
33.图2显示了其中一个膀胱癌样本全基因组水平bin的染色体不稳定性分组。
34.(2)拷贝数变异负荷的计算计算拷贝数变异负荷,即发生拷贝数变异(tumor+分组)的bin数目占全基因组bin数目的比例。计算公式如下:。
35.根据16例临床样本的拷贝数变异负荷分布,计算灵敏度、特异度和约登指数并选取最佳临界值。当拷贝数变异负荷等于4%时,灵敏度和特异度达到最佳。因此,当拷贝数变
异负荷超出4%时,样本染色体不稳定性判定为阳性。
36.实施例2 膀胱癌患者尿液上清cfdna中染色体cnv变异程度评估1.尿液循环游离dna样本的实验室预处理和浅层全基因组二代测序(1)酶切片段化取出 kapa fragmentase buffer 常温融解,混合均匀,置于冰上备用,取出 kapa fragmentase置于冰上,混合均匀,瞬时离心备用。
37.在冰上配制片段化体系:fragmentase buffer 2.5
µ
l,fragmentase 5
µ
l,dna溶液17.5
µ
l,总体积25
µ
l。混合均匀,瞬时离心放置冰上。
38.在 pcr 仪上启动反应程序cycling program i,待温度稳定至4℃时将反应管放进pcr仪进行pcr反应,pcr反应程序如表1所示。
39.表1。
40.(2)末端补平加a上述步骤(1)的反应结束后,根据表2体系,向上述反应pcr管中加入酶反应mix,涡旋混匀离心,设置表3所示程序,在pcr仪上进行反应。
41.表2;表3。
42.将接头连接步骤所需的接头、酶试剂拿出,放2-8℃冰箱或者冰盒上融化。
43.将磁珠从2-8℃冰箱中拿出,置于室温平衡30min以上。配制足量的80%乙醇。
44.准备pcr反应管,最终收集1.5ml离心管,做好标记,准备足量的琼脂糖凝胶用于最
终文库跑胶。
45.(3)接头连接在步骤(2)的pcr程序结束前5-10min,将提前拿出的接头和酶试剂轻弹混匀离心,根据表4体系配制酶试剂混合溶液,涡旋混匀并短暂离心后,加入到步骤(2)的pcr反应后的体系中。然后分别加入相应adapter 1.5
µ
l(10
µ
m),设置表5所示程序在pcr仪上进行反应。
46.表4;表5。
47.(4)磁珠纯化待步骤(3)的pcr程序结束后,将样本取出,将hieff ngs
™ꢀ
dna selection beads磁珠充分涡旋混匀后,加44
µ
l磁珠到上述反应后的pcr管中,充分涡旋混匀,室温孵育5min。将pcr管瞬时离心后放置在磁力架上,等待5min,直到管内溶液完全澄清,小心移除上清。保持pcr管在磁力架上,加入200
µ
l新鲜配制的80%乙醇,室温孵育至少30s。小心吸取并丢掉乙醇,不要碰到磁珠。
48.重复上述步骤一次(总共两次清洗)。
49.将pcr管从磁力架上取下,离心后置于磁力架上,将剩余的乙醇吸取干净,室温下开盖将磁珠晾干,磁珠表面不湿润反光,勿过度干燥开裂,加入21
µ
l h2o,涡旋混匀,静置2min。将pcr管放置在磁力架上,等待2min直到管内溶液完全澄清,小心吸取20
µ
l上清到新的0.2ml pcr管中。
50.(5) pcr扩增配制表6所示试剂,涡旋混匀并短暂离心。设置表7所示程序,在pcr仪上进行反应。
51.表6
;表7。
52.加50
µ
l涡旋混匀的hieff ngs
™ꢀ
dna selection beads磁珠到上述反应后的pcr管中,充分涡旋混匀,室温孵育5 min。将pcr管放置在磁力架上,等待5 min,直到管内溶液完全澄清,小心移除上清。保持pcr管在磁力架上,加入200
µ
l新鲜配制的80%乙醇,室温孵育至少30s。小心吸取并丢掉乙醇,不要碰到磁珠。重复该磁珠纯化步骤一次(总共两次清洗)。
53.将pcr管从磁力架上取下,离心后置于磁力架上将剩余的乙醇吸取干净,室温下开盖将磁珠晾干,勿过干。加入24
µ
l h2o到离心管中,充分涡旋重悬磁珠,室温下放置2 min。置于磁力架上,等待1min,待上清澄清后取23
µ
l至新的pcr管中。
54.(6)文库qc和测序qubit定量:取1
µ
l文库用qubit
®ꢀ
dsdna hs assay kit定量,具体操作参见《thermofish qubit 4.0使用和保养标准操作规程》。
55.qsep100片段分析:文库浓度<15ng/
µ
l时,取1
µ
l文库稀释到0.1-0.5ng/
µ
l,用qsep400检测,具体操作参见《qsep400全自动核酸分析系统标准操作规程》。
56.根据测序仪说明书,使用illumina测序仪进行上机测序。
57.2.测序数据的预处理利用bwa mem算法对dna测序的原始数据进行序列比对,利用picard(2.0.1)算法中的markduplicates功能对比对后的序列进行去重,产生待测尿液样本的去重后测序文件urine_deduped.bam文件。以去重后的测序bam文件为输入文件,运行broad研究所开发的ichorcna(0.1.0)软件。通过计算每个bin的reads数目,然后对每个bin的reads数目进行gc、mappability 以及深度差异的校正,校正后的每个bin的reads数目和构建的30例健康
人尿液上清cfdna参考背景数据库的normal panel中的相应bin的reads数目相比,计算得到logr值,计算公式如下:。
58.3. cnv-burden模型对全基因组染色体稳定性评价根据cnv的阈值,对每个待测样本的bin进行分组,利用cnv-burden模型对待测样本进行全基因组染色体稳定性评价。
59.表8截取了4例样本的染色体稳定性结果。其中,两例样本拷贝数变异负荷远低于4%,染色体稳定性判断为稳定,为健康人样本;两例样本拷贝数变异负荷远高于4%,染色体稳定性判断为不稳定,为膀胱癌患者样本。
60.表8全基因组尿液cfdna样本的染色体稳定性评价。
61.实施例3 cnv-burden算法的准确性评价收集98例临床确诊的尿路上皮癌患者和102例对照组(健康人和非尿路上皮癌的其他尿路疾病患者)的尿液上清cfdna样本,采用实施例2的方法,通过全基因组测序,用实施例1构建的cnv-burden模型,比较尿路上皮癌患者与对照组的染色体异常情况,样本发生染色体不稳定判定为阳性,否则判定为阴性,并将cnv-burden模型的分析结果与金标准病理结果进行比较,去除dna不合格的样本(11例)和病理诊断结果不完整的样本(16例),结果如图3和表9所示,计算本发明的拷贝数变异程度评估方法的灵敏度和特异性,得到roc曲线(参见图4)。发现本发明的评估方法的准确性达89.02%,敏感性86.67%,特异性90.81%,roc曲线的auc面积达0.94。
62.表9 cnv-burden 算法性能评估表。
63.实施例4 一例膀胱癌患者的术后监测一例膀胱癌患者在自2019年10月至2021年8月的接近两年时间内,每3-6个月寄送一次尿液上清进行浅层wgs测序,并运用本发明的染色体cnv变异程度评估模型和方法进行评估。通过cnv-burden模型发现,伴随着治疗的深入,该患者染色体不稳定性正在逐渐缓解,2019年10月至2021年3月染色体不稳定性阳性高风险降低。同时,发现该患者的肿瘤分
数由2019年时的28%逐渐降低至2021年8月的0,表明该患者尿路上皮癌风险降低。参见表10。
64.表10 一例膀胱癌患者术后监测数据。
65.上述实施例仅为本发明的可行或较佳实施例而已,是用来说明本发明的,并非用以限制本发明申请专利的范围,因此,凡依本发明申请专利范围所作的均等变化与修饰,均应属于本发明专利涵盖的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1