一种双靶向近红外仿生给药系统及其制备方法与应用与流程
1.本发明涉及仿生纳米制剂技术领域,尤其涉及一种双靶向近红外仿生给药系统及其制备方法与应用。
背景技术:
2.由乙型肝炎病毒(hbv)引起的乙型肝炎病毒感染导致全球显著的肝脏相关发病率和死亡率。根据世界卫生组织(who)的数据,全世界有近3亿人患有长期的hbv感染,每年有超过78万人死于其并发症(包括肝硬化和原发性肝癌)。预计到2024年,全球乙肝药物市场规模的增长将达到30亿美元,给全球的医疗保健系统带来沉重的负担。目前,由于hbv共价闭路环状dna(cccdna)仍然难以消除,慢性乙型肝炎病毒感染的治疗更倾向于功能性治疗,其目的是持续降低循环中的病毒载量和hbv表面蛋白抗原(hbsag)。在这方面,目前用于临床治疗乙肝的药物,如干扰素(ifn)和核苷类似物(nucs),可以抑制肝炎病毒的复制,减缓肝硬化的进展,适度降低肝细胞癌(hcc)的发病率。然而,这些药物的长时间治疗也伴随着耐药性和不良反应,如骨髓抑制。众所周知,衣壳抑制剂对hbv的生命周期至关重要,并且是直接抗病毒药物减少hbv dna的潜在靶点。最近一份关于新型衣壳组装调节器(cam)的报道显示,一个小分子c39可以通过影响hbv核衣壳的正常组装来阻止hbv dna的合成。最近发表的研究中越来越多的证据表明,反义寡脱氧核苷酸(aso)可以诱导hbv mrna的靶向切割,从而降低病毒蛋白的产生。
3.针对hbv持久性的机制已经取得了重大进展;抗hbv疗法的发展不再局限于直接作用于病毒本身,而是通过多靶点治疗方法靶向病毒生命周期的不同阶段。大量证据证明,这种寻求多药联合治疗复杂疾病的新治疗概念可以有效抑制hbv复制。然而,无论是没有纳米载体的核酸还是高负电荷的小分子药物都很难进入哺乳动物细胞,这给药物治疗带来了重大挑战。此外,向血液中注射不溶性微粒可能引发静脉血栓栓塞,加上药物在体内非特异性分布引起的潜在全身毒性增加,限制了其临床应用。
4.近年来,药物递送纳米颗粒被用来提高药物的给药效率,减少其全身副作用.事实上,大多数纳米颗粒不能区分异常细胞和正常细胞,通常会导致严重的副作用,加上在传递过程中意外释放,因此合理使用给药纳米颗粒仍然是一个巨大的挑战。
5.刺激响应型纳米载体是一种智能纳米载体,有效地减少了药物的意外释放,近年来引起了人们的广泛关注。光刺激响应的纳米颗粒在时间和空间上能确保对装载药物的控制和按需释放。由于他们的可控性和非侵入性,突出了这类纳米载体在药物传递系统和生物医学中的应用潜力。迄今为止报道的光响应纳米载体大都采用紫外光或者可见光来触发药物递送,但是这类光的低穿透率和光毒性限制了它们在体内的应用。由于近红外(nir)光可以穿透组织,对健康组织的损害最小,它在非侵入性治疗中备受关注,并广泛应用于生物医学中的研究。有充分的证据表明,上转换纳米颗粒(ucnps)具有先进的光学特性,如优良的尺寸稳定性和低毒性,可以使低能的近红外光转化为高能的紫外/可见光发射。
6.然而,ucnps表现出较低的载药能力和缺乏精确的靶向能力。此外,纳米颗粒表面
的核酸类药物暴露在复杂的生理和异质环境中很容易被核酸酶降解。因此,迫切需要开发一种具有靶向性高、载药能力强、生物相容性好的新型传递系统。
技术实现要素:
7.针对现有技术中所存在的不足,本发明提供了一种双靶向近红外仿生给药系统及其制备方法与应用,其解决了现有技术中存在的靶向性低、载药能力差、生物相容性低的问题。
8.本发明一方面,提供一种双靶向近红外仿生给药系统,包括红细胞膜和包裹在红细胞膜内的载药纳米载体,所述载药纳米载体包括近红外响应的上转换纳米粒子、抗hbv反义寡核苷酸和抗hbv核衣壳抑制剂,所述上转换纳米粒子表面包覆有介孔二氧化硅,所述抗hbv反义寡核苷酸和抗hbv核衣壳抑制剂分别负载在介孔二氧化硅上。
9.进一步地,所述抗hbv核衣壳抑制剂负载在介孔二氧化硅的介孔通道内,所述抗hbv反义寡核苷酸负载在介孔二氧化硅表面,其中,部分抗hbv反义寡核苷酸负载在介孔通道的孔隙处,用于阻断介孔通道内抗hbv核衣壳抑制剂的释放。
10.进一步地,所述红细胞膜表面修饰有靶向分子,优选地,所述靶向分子为β-d半乳糖糖苷。
11.进一步地,所述近红外响应的上转换纳米粒子为核壳结构,ca
2+
掺杂的nayf4:yb/tm为核,nayf4:yb/nd为壳。
12.进一步地,所述抗hbv反义寡核苷酸为aso,所述抗hbv核衣壳抑制剂为c39。
13.本发明另一方面,提供一种双靶向近红外仿生给药系统的制备方法,包括如下步骤:
14.1)ucnp的合成:热分解法制备nayf4:yb/tm/ca@nayf4:yb/nd;
15.2)ucnp@msn的合成:以ctab为模板剂,将步骤1)制备的nayf4:yb/tm/ca@nayf4:yb/nd依次与硅酸四乙酯、3-氨基丙基三乙氧基硅烷反应,得到介孔二氧化硅包覆的ucnp;
16.3)ucnp@msn-pcaso/c39的合成:在步骤2)得到的ucnp@msn表面修饰pcdna,之后加入抗hbv反义寡核苷酸和抗hbv核衣壳抑制剂,得到载药纳米载体ucnp@msn-pcaso/c39;
17.4)ucnp@msn-pcaso/c39-m-gal的合成:利用gal改性红细胞膜囊包裹步骤3)得到的ucnp@msn-pcaso/c39,得到红细胞膜包裹的载药纳米载体ucnp@msn-pcaso/c39-m-gal。
18.进一步地,步骤2)中,以mg/μl计,nayf4:yb/tm/ca@nayf4:yb/nd和硅酸四乙酯的质量体积比为1:5;硅酸四乙酯和3-氨基丙基三乙氧基硅烷的体积比为10:1。
19.进一步地,步骤3)中,抗hbv反义寡核苷酸的负载量为101.5μmol/g
·
ucnp@msn,按质量百分比算,抗hbv核衣壳抑制剂的负载量为7.3%。
20.进一步地,步骤4)中,所述gal改性红细胞膜囊是由二硬脂酰基磷脂酰乙醇胺-聚乙二醇-半乳糖修饰红细胞膜囊制备得到。
21.本发明又一方面,提供一种双靶向近红外仿生给药系统在制备治疗肝炎药物中的应用。
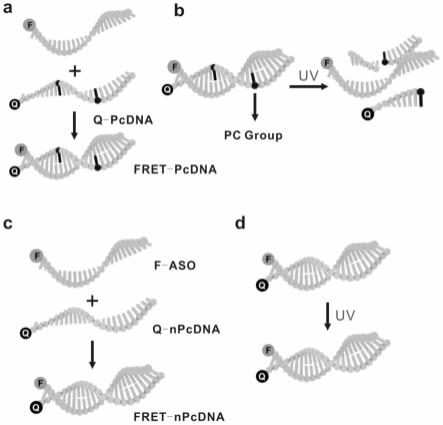
22.术语“npcdna”为与aso互补的单链,“pcdna”为在npcdna中插入了pc linker(ipclink),核苷酸序列如表1所示。pc linker为可光裂解的基团,结构式如式i所示。
aso的荧光光谱;c为紫外照射后pc-aso的琼脂糖凝胶电泳分析;d为紫外线照射后npc-aso的琼脂糖凝胶电泳分析。
32.图3为本发明实施例2、3中三组纳米颗粒的sem和尺寸分布,其中,a为nayf4:yb/tm/ca,b为nayf4:yb/tm/ca@nayf4:yb/nd,c为ucnp@msn。
33.图4为本发明实施例2中nayf4:yb/tm/ca@nayf4:yb/nd的hrtem图像。
34.图5为本发明实施例2中ucnp中镧系离子的多维能量转移路径示意图。
35.图6为本发明实施例3中ucnp@msn的eds图谱。
36.图7为本发明实施例3中ucnp@msn的氮吸附-脱附等温线。
37.图8为本发明实施例3中ucnp@msn在808nm不同功率密度激发下的上转换发光光谱。
38.图9为本发明实施例3、5中ucnp@msn、umac的制备和表征图,其中,a为umac-m-gal合成示意图;b为nayf4:yb/tm/ca;c为nayf4:yb/tm/ca@nayf4:yb/nd;d为ucnp@msn的tem图像(比例尺=20nm);e为ucnp@msn的高角度环形暗场扫描透射电子显微镜(haadf-stem)图像对应的元素(na,f,tm,ca,y,yb,nd,si)映射图像(比例尺=20nm);f为ucnps和ucnps@msn的xrd图谱;g为ucnps@msn-nh2的表面特征(通过chem3d分析的c39的3d结构);h为ucnp@msn-pcaso合成各个阶段的ftir光谱;i为ucnps和ucnps@msn的ucl谱(ucnps@msn的明场和nir荧光图像);j为umac在808nm nir激发下的药物释放曲线;k为在有或没有pc基团和nir激发的情况下从umac释放c39的特异性。
39.图10为本发明实施例4中ucnp@msn、ucnp@msn-pcaso和ucnp@msn-pcaso/c39的紫外/可见吸收光谱。
40.图11为本发明实施例4中c39(a)和fam-aso(b)的标准曲线。
41.图12为本发明实施例5中umc、umac和umac-m-gal中c39的释放动力学。
42.图13为本发明实施例6中umac-m-gal的性能验证,其中,a为umac-m-gal的sem图像;b为umac-m-gal的tem图像;c为sds-page蛋白分析,泳道1和5为标记物、泳道2为rbc膜、泳道3为rbc囊泡和泳道4为umac-m;d为rm、umac和umac-m-gal的流体动力学尺寸分布;e为ucnps、ucnps@msn、uma、uma-m-gal和rbc囊泡的zeta电位;f为各种细胞的细胞裂解物的wb分析;g为用umac-m-gal培养的hepg2.2.15细胞的clsm图像,dapi(蓝色)用于染色细胞核,dil(红色)用于染色rm-gal,fam(绿色)用于标记aso(比例尺为10μm)。
43.图14为本发明实施例6中gal、nhs-peg-dspe、gal-peg-dspe的1h nmr光谱。
44.图15a为本发明实施例6中umac-m-gal在不同生理条件下14天内的稳定性,b为不同时间umac-m-gal在fbs中的水合尺寸,c为聚合物分散指数。
45.图16为本发明实施例7中免疫逃避评估,其中,a为raw264.7巨噬细胞样细胞与各种fam标记的纳米颗粒孵育后的clsm图像(比例尺,50μm);b为raw264.7细胞与各种fam标记的纳米颗粒孵育后的流式细胞术定量;c为对具有不同孵育时间段的仿生纳米颗粒吸收进行定量分析;d为注射后48小时各种仿生纳米颗粒的生物分布;e为注射各种仿生纳米颗粒后48小时的体内药代动力学曲线;(注射后24小时纳米颗粒的血液滞留情况,数据点代表平均值
±
标准差,n=3,ns、*和***分别表示无统计学差异,p《0.05和p《0.001)。
46.图17为本发明实施例7中fret对标记的(pc)umac-m-gal体外近红外光控释放,其中,a为hepg2.2.15细胞与fret对标记的(pc)umac-m-gal孵育后的clsm图像,有nir照射
(nir+)和没有nir照射(nir-)(808nm,2w/cm2),b为流式细胞仪检测结果,c为与fret对标记(pc)umac-m-gal孵育后hepg2.2.15细胞的定量,有和没有nir照射(808nm,2w/cm2)。
47.图18为本发明实施例7中肝脏靶向评估,其中,a为hepg2.2.15细胞与各种纳米颗粒孵育后的clsm图像(比例尺=20μm);b为与fam标记的纳米颗粒孵育3小时后,各种非肝细胞系的流式细胞仪图谱;c为hbv tg小鼠在注射含有用dir染色的umac-m-gal纳米颗粒的pbs后24小时的体内荧光图像;d为注射后24小时处死的hbv-tg小鼠主要器官的离体荧光图像(数据点代表平均值
±
标准差,n=3,ns、*和***分别表示无统计学差异,p《0.05和p《0.001)。
48.图19为本发明实施例7中facs分析不同纳米颗粒在hepg2.2.15细胞中的摄取效率。
49.图20为本发明实施例7中l02细胞摄取umac-m-gal的clsm图像。
50.图21为本发明实施例7中有或没有抗体阻断下umac-m-gal细胞摄取的clsm图像。
51.图22为本发明实施例7中肝靶向评估,其中,a为与fam标记的纳米颗粒孵育3小时后,各种非肝细胞系的clsm图像,b为与fam标记的纳米颗粒孵育3小时后,各种非肝细胞系的流式细胞术定量,d为定量分析在hepg2.2.15细胞中不同孵育时间段的纳米颗粒摄取情况。
52.图23为本发明实施例7中808nm nir照射后um-pcaso的琼脂糖凝胶电泳分析。
53.图24为本发明实施例8中细胞活力评估,其中,a为无nir照射下(对照)hepg2.2.15和l02细胞在不同浓度的umac-m-gal纳米颗粒下孵育的活力,b为通过改变照射时间,hepg2.2.15和l02细胞在808nm nir照射(1.0w/cm2)下的活力,c为通过改变功率密度,hepg2.2.15和l02细胞在808nm nir光下(5分钟)的存活率,d为通过改变纳米粒子浓度,hepg2.2.15和l02细胞在808nm nir光(1.0w/cm2,10分钟)下的活力。
54.图25为本发明实施例8中umac-m-gal在体外的抗病毒作用,其中,a为aso和c39的抗hbv复制作用示意图;b为使用nir照射进行细胞处理的实验程序;为通过elisa测定法检测上清液中的抗原;d-g为有或没有递送系统对hbv的抑制作用的差异。检测参数分别为细胞内hbv dna、hbv 3.5kb rna、hbsag和hbeag;h-k为有或没有nir和pc基团对hbv的抑制作用的差异。检测参数分别为细胞内hbv dna、hbv3.5kbrna、hbsag和hbeag。
55.图26为本发明实施例8中不同合成阶段的纳米颗粒的抗hbv作用。
56.图27为本发明实施例9中不同浓度umac-m-gal的溶血率。
57.图28为本发明实施例9中umac-m-gal在体内的抗病毒作用,其中,a为hbv转基因小鼠实验过程示意图;b-d为不同治疗对hbv抑制作用的差异;e为小鼠肝脏免疫组织分析示意图;f为具有代表性的免疫组织化学图像,显示了处死小鼠肝脏中hbsag和hbeag表达(比例尺=50μm)。
58.图29为本发明实施例9中不同治疗组主要器官的h&e染色图。
59.图30为本发明实施例9中仿生递送系统umac-m-gal的全身毒性评价,其中,a为不同治疗组15天期间的小鼠体重,不同治疗组中的血清生化参数b为bun、c为alt和d为ast水平。
具体实施方式
60.下面结合附图及实施例对本发明中的技术方案进一步说明。若无特殊说明,本发明所用试剂和试验材料均可商业获得。本发明中的dna寡核苷酸序列由sangon biotech co.(中国上海)合成和纯化;hbv转基因小鼠来自重庆医科大学感染性疾病分子生物学教育部重点实验室。
61.实施例1pc-aso的合成
62.为了检查pc-aso的紫外光激活特性,以福斯特共振能量转移(fret)对作为荧光报告物,将携带bhq1(黑洞猝灭基团)的pcdna单链和fam(荧光素)标记的aso链(fam-aso)杂交,形成双链结构。并设立阴性对照,bhq1标记的未携带光响应基团(pc linker)的pcdna单链(npcdna),并与fam(荧光素)标记的aso链(fam-aso)杂交形成双链npc-aso。各序列的核苷酸序列如表1所示。
63.表1 dna寡核苷酸序列
[0064][0065][0066]
可光活化的pc-aso和npc-aso的杂交和光激活特性如图1所示。由图1可知:由于pc基团的破坏,fam-aso从双链结构中释放。实时监测紫外光活化过程,结果如图2a所示。fam的荧光发射明显降低,几乎消失,说明随着双链结构的形成,能量从fam转移到bhq1,证实了pc-aso的形成。在紫外光照射下,pc-aso的荧光发射逐渐恢复,表明fret对结构被破坏。
[0067]
由图2b可知,npc-aso的荧光被fret猝灭,与pc-aso的结果一致。然而,npc-aso的荧光发射并没有随着紫外线照射而增加。琼脂糖凝胶电泳分析pc基团的特异性(图2c,d),表明紫外光激活特性依赖于pc基团。
[0068]
实施例2nayf4:yb/tm/ca@nayf4:yb/nd(ucnps)的合成
[0069]
1、核nayf4:yb/tm/ca的合成
[0070]
采用热分解法在加有12ml油酸(oa),30ml 1-十八碳烯(ode)三口烧瓶中加入1.6mmol lncl3·
6h2o、1.112mmol ycl3·
6h2o、0.32mmol ybcl3·
6h2o、0.008mmol tmcl3·
6h2o和0.16mmol caoa,得到混合物。将混合物在氮气气氛下加热至120℃、15分钟。搅拌条件下将混合物加热至160℃、1小时,然后冷却至室温。加入含有4mmol naoh和7.2mmol nh4f的甲醇溶液10ml,50℃下搅拌30分钟。随后加热至100℃、30分钟除去甲醇。之后在真空下加热至300℃,反应1小时,缓慢冷却至室温。乙醇沉淀,离心,得到nayf4:yb/tm/ca。nayf4:yb/
tm/ca的sem图和尺寸分布如图3a、4b所示,由图3a可知,nayf4:yb/tm/ca的直径约为30nm。
[0071]
2、核壳nayf4:yb/tm/ca@nayf4:yb/nd(ucnp)的合成
[0072]
将0.8mmol lncl3·
6h2o、0.32mmol ycl3·
6h2o、0.4mmol ndcl3·
6h2o加入到含有6ml油酸(oa)、15ml 1-十八碳烯(ode)的三口烧瓶中。在氮气气氛下加热至120℃、15分钟。之后,搅拌条件下加热1小时,然后冷却至室温。逐滴加入2.5ml步骤1制备的核nayf4:yb/tm/ca,室温下搅拌30分钟,然后在80℃下搅拌30分钟。冷却至室温后,加入含有2mmol naoh和3.6mmol nh4f的5ml甲醇溶液,并在50℃搅拌30分钟。随后加热至100℃、30分钟除去甲醇。在真空下加热至300℃,反应1小时,缓慢冷却至室温。用乙醇沉淀,离心,得到核壳nayf4:yb/tm/ca@nayf4:yb/nd。nayf4:yb/tm/ca@nayf4:yb/nd的sem图和尺寸分布如图3b、4c所示,由图3b可知,nayf4:yb/tm/ca@nayf4:yb/nd的平均直径约为42nm。高分辨率透射电镜(hrtem)如图4所示。结果显示,ucnps的晶格间距为0.52nm,ucnps的衍射峰对应于β-nayf4(jcpdscard no.28-1192),说明ucnps具有六方相。
[0073]
如图5所示,与单独的nd
3+
作为敏化剂相比,掺杂nd
3+
的敏化剂降低了生物组织在980nm处的强吸水性,并使ucnp能够在808nm处被激发,从而提高ucnp在生物组织中的渗透深度。
[0074]
实施例3ucnp@msn的合成
[0075]
将实施例2制备的ucnp 20mg溶于2ml氯仿,滴入含有360mg十六烷基三甲基溴化铵(ctab)的10ml水溶液中,使用探头超声波破碎仪进行超声处理400w、1h,得到透明的ucnps/ctab溶液。离心并重复3次。收集沉淀并分散在ddh2o中,加入15μl 2m naoh,搅拌1小时。超声条件下加入100μl四乙氧基硅烷(teos)反应,室温下搅拌12h。加入10μl3-氨基丙基三乙氧基硅烷(aptes)并在室温下搅拌6小时。将产物离心纯化,1%nacl甲醇溶液在40℃条件下提取24h,以除去模板ctab,得到的白色粉末用水和乙醇洗涤3次,真空干燥12h。ucnp@msn的sem图和尺寸分布如图3c所示,由图3c可知,ucnp@msn的平均直径约为57nm。
[0076]
ucnp@msn的介孔特性如图9d所示,显示介孔硅层厚度在10nm左右。ucnp@msn的eds元素分析图和光谱如图9e,图6所示,证实ucnp@msn制备成功。xrd成像如图9f所示,在22
°
处观察到衍射宽峰,这表明ucnp@msn的制备成功。采用自动表面积和孔隙率分析仪测量ucnp@msn的孔隙特性,如图9g,图7所示,结果表明,ucnps@msn的比表面积、孔径和总孔容分别约为2.74nm,373.89m2g-1
,和0.352cm3g-1
,通过chem3d软件分析药物分子c39的大小估计为1.49nm,药物分子c39可以加载到ucnp@msn中。傅里叶变换红外光谱(ftir)(图9h)在3428cm-1
、1089cm-1
和800cm-1
处显示特征峰,证实msn表面改性成功。ucnp和ucnp@msn的上转换发射光谱(图9i,图8)表明,ucnp@msn可以有效地将808nm nir转换为345nm和361nm发射峰的光。
[0077]
实施例4ucnp@msn-pcaso/c39(umac)的合成
[0078]
1、ucnp@msn-pcdna的合成
[0079]
采用edc/nhs化学方法将pcdna修饰在ucnp@msn表面:将pcdna、50mg edc和25mg nhs加入到1ml pbs(ph=5.5)中,37℃孵育1h。之后立即加入ucnp@msn-nh2,调ph值至8。4℃下孵育过夜(pcdna:20μm;ucnp@msn-nh2:2mg
·
ml-1
)。将获得的纳米颗粒ucnp@msn-pcdna离心、洗涤去除未反应的pcdna,并重新分散在dmso中备用(对照样品按照同样的方法制备,不同之处在于将pc替换为npc)。
[0080]
与ucnp@msn相比,还观察到另一个特征峰(nh2≈874cm-1
),表明单链pcdna通过-cooh与ucnp@msn中的-nh2之间通过edc/nhs缩合反应连接。
[0081]
2、ucnp@msn-pcaso/c39的合成
[0082]
将10mg步骤1制备的ucnp@msn-pcdna浸泡在含5mg c-39的dmso溶液中12小时,然后加入aso。随后,搅拌12小时,得到ucnp@msn-pcaso/c39。然后将纳米颗粒ucnp@msn-pcaso/c39离心,pbs(100mm,ph7.4)洗涤去除未反应的c-39。
[0083]
紫外-可见吸收光谱表明制备成功(图10)。通过差异法证实,aso和c39在ucnp@msn上负载成功,负载含量分别为101.5μmol/g
·
ucnp@msn和7.3%(c39)(图11)。zeta电位显示,ucnp@msn-pcaso/c39(或称为umac)的电荷从-28mv下降到-35mv,这说明了pcaso在ucnp@msn表面的成功修饰(图13e)。
[0084]
实施例5umac给药系统控释性试验
[0085]
在808nmnir或无808nmnir条件下,通过nano-500分光光度计和biotech酶标仪测定umac给药系统的紫外吸收值和荧光值,绘制c39药物释放曲线(图9j)。
[0086]
在无808nmnir的umac中,48h时仅释放了约8.1%的c39,表明设计的可光活化pc-aso结构阻断了msn的介孔通道。因此,这些药物被包含在纳米颗粒中。在808nmnir处理组中,c39的释放显著增加,24h时释放了约55.7%的c39,表明nir显著触发了c39的释放(图9k)。结果表明,经连续照射后,umac的药物释放量达到56.8%。通过测定umac在不同溶液中的药物泄漏率来评价umac的药物保留能力。在无照射情况下孵育24h后,观察到很小的药物释放,表明所制备的纳米颗粒具有良好的药物保留能力(图12)。
[0087]
实施例6umac-囊泡(umac-m-gal)的合成
[0088]
1、提取红细胞膜
[0089]
从健康的c57bl/6小鼠采集全血样本,并收集在抗凝管中。用预冷的磷酸盐盐水缓冲液(pbs,ph=7.4)稀释新鲜全血,4℃下4000rpm离心5分钟,并用冷pbs洗涤3次,收集红细胞。将洗涤后的红细胞分散到等渗溶液中,加入4倍体积的低渗溶液混合,在800g 4℃离心10分钟。收集沉淀,用0.25
×
pbs洗涤直至上清液澄清。收集rbc膜并在4℃下储存。
[0090]
2、二硬脂酰基磷脂酰乙醇胺-聚乙二醇-半乳糖(dspe-peg2000-gal)的合成
[0091]
将4-氨基苯基β-d-吡喃半乳糖苷和nhs-peg2000-dspe分别溶解在pbs(ph=7.4)中。将4-氨基苯基β-d-吡喃半乳糖苷溶液滴加到dspe-peg2000-nhs溶液中,摩尔比为5:2,真空下搅拌24小时。反应混合物用透析膜透析24小时,除去过量的gal。
[0092]1h核磁共振(1h nmr)光谱检测游离gal、游离dspe-peg2000-nhs和gal-peg-dspe的结构,结果如图14所示。由结果可知,在gal-peg-dspe光谱中,出现gal(4.85、3.93、6.78和6.97ppm)的化学位移,表明制备成功;nhs条带出现明显改变,表明gal取代n-羟基琥珀酰亚胺基团导致3.18ppm条带消失。在gal-peg-dspe中nhs-peg-dspe的其他ch2谱带分别从3.36到3.14、2.73到2.66、1.261.24ppm,这可能是由于芳族基团的电负性大。
[0093]
3、gal改性红细胞膜囊(rm-gal)的制备
[0094]
将步骤1制备的rbc膜分散在pbs(ph=7.4)中,加入步骤2制备的dspe-peg2000-gal,37℃搅拌1小时。之后,800g离心5分钟,pbs洗涤3次,得到rm-gal。将rm-gal重悬在pbs(ph7.4)中备用。
[0095]
4、ucnp@msn-pcaso/c39-m-gal(umac-m-gal)的制备
[0096]
将实施例4制备的1ml umac(2mg
·
ml-1
)与200nm rm-gal或rm(未进行gal修饰的红细胞膜)混合并通过200nm聚碳酸酯多孔膜多次挤出得到umac-m-gal或umac-m。将纳米颗粒umac-m-gal在13,000rpm离心5分钟,pbs(ph=7.4)洗涤3次,冻干。umac-m-gal的sem图像如图13a所示;tem图像如图13b所示。sds-page蛋白分析和考马斯亮蓝染色如图13c所示,结果显示,红细胞膜蛋白广泛保留在纳米颗粒上,显示红细胞膜成功包裹umac。利用动态光散射(dls)监测红细胞膜包裹前后纳米颗粒的尺寸变化,结果如图13d所示,结果表明umac-m-gal的尺寸约为90nm,与sem和tem的结果一致,最后得到的仿生纳米颗粒的流体动力学直径增加了30nm;ζ电位增加到与rbc囊泡相似的水平(图13e)。umac-m-gal的稳定性如图15a-15c所示,结果显示umac-m-gal在水、磷酸盐缓冲盐水(pbs)、100%胎牛血清(fbs)和含10%胎牛血清的培养基中稳定,甚至在fbs中长达14天。
[0097]
实施例7umac-m-gal的生物学特性
[0098]
1、评估肝源性细胞对umac-m-gal(用fam标记aso链)的细胞摄取。
[0099]
首先,通过免疫印迹分析检测asgpr在肝细胞中的特异性表达,表明与其他组织来源的细胞相比,该受体几乎只在肝细胞中表达(图13f)。然后,将hepg2.2.15细胞和umac-m-gal(用dil标记rm-gal和用fam标记aso)共孵育,检测hepg2.2.15细胞。可观察到dil的大部分信号(红色)与细胞核周围的fam信号(绿色)重叠,这证实了rbc膜成功包裹在umac表面(图13g)。
[0100]
2、免疫逃逸能力检测
[0101]
利用raw264.7小鼠巨噬细胞检测umac-m-gal的抗吞噬能力。细胞分别与umac组、umac-m组和umac-m-gal组纳米颗粒共孵育,使用共聚焦激光扫描显微镜(clsm)成像,结果如图16a所示。使用流式细胞仪定量检测平均荧光强度,结果如图16b所示。结果显示,umac组比umac-m和umac-m-gal组显示出更亮的绿色荧光和平均荧光强度,表明包裹红细胞膜可以有效降低免疫清除。
[0102]
使用icp-ms进行巨噬细胞摄取试验,检测细胞质中si的含量,结果如图16c所示。结果显示,与未包裹rbc膜的纳米颗粒相比,包裹rbc膜的仿生纳米颗粒(umac-m和umac-m-gal)的摄取更低。由此可知,细胞膜的保护可以增强纳米颗粒抵抗免疫清除的能力。
[0103]
在对静脉注射后不同时间点的血液样本进行体内研究,通过icp-ms检测硅含量,表明包裹rbc膜的纳米颗粒在血液中的保留能力明显增强(图16e)。由于网状内皮系统(res)的吞噬细胞主要分布在肝脏和脾脏,从而导致在治疗过程中大量纳米颗粒积累。经检测,umac-m在肝脏和脾脏中的si积累均显著减少,说明细胞膜包裹可以在一定程度上降低res对纳米颗粒的摄取。
[0104]
3、时间和空间控释能力的检测
[0105]
hepg2.2.15细胞与含有fret对标记的umac-m-gal(pcaso)或umac-m-gal(npcaso)共孵育。共培养3h后,用808nm(1.5w/cm2)的激光照射细胞。结果如图17a-c所示,结果显示,在没有激光照射的细胞中几乎没有荧光信号。在近红外(nir)照射下可检测到明显更强的绿色荧光,这表明nir触发了aso-fam链的释放(图17a)。流式细胞仪定量和琼脂糖凝胶电泳显示一致的结果(图17b、图23)。
[0106]
相比之下,用fret对标记的umac-m-gal(npcaso)孵育的hepg2.2.15细胞的绿色荧光强度几乎没有变化(图17a-c),表明pc基团对仿生给药传递系统至关重要。
[0107]
如图18a所示,在与游离aso链孵育时,观察到微弱的绿色荧光信号,这是因为裸dna不能穿透细胞膜。相比之下,与umac共孵育的细胞显示出更强的荧光信号,这表明纳米颗粒可以促进aso链进入细胞。clsm分析和流式细胞术表明,umac-m-gal具有最高的内化效率,这表明gal分子修饰的仿生膜囊泡对改善umac给药系统的内化至关重要(图19)。几乎所有的荧光信号都位于细胞内区域,这表明umac-m-gal是通过内吞途径被吸收。
[0108]
通过共定位研究评估umac-m-gal在l02细胞中的内化,发现孵育2小时后,umac-m-gal与溶酶体红色荧光探针标记的溶酶体具有良好的共定位,也表明umac-m-gal更可能通过内吞途径进入细胞内(图20)。
[0109]
4、评估仿生纳米颗粒的靶向能力
[0110]
使用抗asgpr受体抗体对l02细胞进行了抗体阻断试验。结果显示,umac-m-gal在阻断后的细胞质中具有较弱的荧光信号(图21)。
[0111]
在各种非肝细胞系(a549、hela和mcf7细胞)进行共聚焦分析,表明与l02(正常肝细胞)和hepg2.2.15细胞(将hbv基因组整合到染色体dna的细胞模型)的强荧光信号相比,非肝细胞系中umac-m-gal的荧光积累明显较弱(图22a)。流式细胞术分析显示,在孵育后,与肝细胞结合的umac-m-gal的数量明显高于非肝细胞系(图18b,图22b)。icp-ms结果证实,用gal修饰的仿生纳米颗粒显示出明显更多的硅积累(图22c)。这些结果表明,umac-m-gal具有较高的肝细胞靶向特异性。
[0112]
使用体内荧光成像系统实时跟踪umac-m-gal在体内的生物分布。注射后24小时,用亲脂性荧光染料dir标记的仿生给药系统检测到umac-m-gal在肝脏中大量积累(图18c,d)。为了研究不同纳米颗粒在体内的生物分布,通过检测si含量,分析小鼠主要器官中的纳米颗粒的含量。与体内荧光成像一致,gal修饰的纳米颗粒具有良好的肝脏靶向能力(图16d)。同时,umac-m和umac-m-gal在脾脏中的低积累也证实了红细胞膜的包裹可使纳米颗粒具有免疫逃逸能力。此外,umac-m-gal在肝脏中的高积累可能归因于gal修饰细胞膜的靶向能力。
[0113]
实施例8nir光触发的umac-m-gal体外抗hbv作用
[0114]
1、808nmnir照射含有10%胎牛血清的dmem和umac-m-gal溶液检测温度变化,评估nir激光诱导热效应的风险。结果显示,在浓度为800μg/ml和5w/cm2的近红外照射长达10分钟下,没有检测到高热效应。
[0115]
2、l02细胞和hepg2.2.15细胞用于评估umac-m-gal的体外细胞活力,纳米颗粒的潜在细胞毒性作用可以忽略不计。首先,将细胞分为四组:第一组测定不同浓度的纳米颗粒对细胞活力的影响,第二组用808nm照射30min,第三组用不同功率的808nm照射,第四组评估不同浓度的umac-m-gal在固定功率和照射时间下的细胞毒性。结果显示,纳米颗粒对细胞活力没有影响(图24a)。即使当激光功率达到5w/cm2照射5min或1w/cm2照射10min时,照射组的细胞未表现出明显的细胞毒性作用,说明近红外激光对肝细胞的存活率没有影响(图24b,24c)。与800μg/ml umac-m-gal共孵育和用1.0w/cm2近红外光照射10分钟后,超过90%的hepg2.2.15和l02细胞存活,说明其高生物相容性及其在生物医学应用中的潜在价值(图24d)。
[0116]
3、umac-m-gal对体外hbv复制的抑制作用。
[0117]
hepg2.2.15细胞用于评估umac-m-gal对体外hbv的抑制作用,其作用原理如图25a
所示,操作方法如图25b所示。结果如图25d-g所示,结果表明aso和c39联合使用增强了对hbv的抑制作用。同时,仿生药物给药系统显著提高了抗病毒能力。
[0118]
通过qrt-pcr检测hbv3.5kb rna和细胞内的hbv dna,所用引物序列如表2、seq id no.3-8所示。用酶联免疫吸附法(elisa)检测上清液中hbv病毒抗原(如hbsag和hbeag)的水平,操作方法如图25c所示。
[0119]
表2 rt-qpcr primer
[0120][0121]
由于pc基团的缺失,umac-m-gal在nir照射下对hbv复制没有明显影响,这可能是通过与npcdna杂交抑制药物释放而引起(图25h-i)。相比之下,用pc基团修饰的纳米颗粒组的hbv 3.5kb rna、细胞内hbv dna和hbv病毒抗原在nir照射下显著降低,表明umac-m-gal的hbv抑制活性是由nir介导的激活诱导的(图25j-k)。
[0122]
生物膜和gal对仿生纳米递送系统抑制hbv复制能力的影响。结果表明红细胞膜的包裹和gal的修饰可以促进仿生纳米颗粒的递送。然而,效果并不显著(图26),需要在体内实验中进一步验证。
[0123]
实施例9nir光触发的umac-m-gal在体内抗hbv作用
[0124]
编码hbv基因组的1.2个过长的拷贝(血清型awy)hbv转基因小鼠(hbv-tg c57bl/6),作为动物模型,评估umac-m-gal药物传递系统在体内抑制hbv复制的能力。通过溶血试验,表明纳米颗粒的生物毒性可以忽略不计(图27)。
[0125]
如图28a所示,在使用仿生纳米颗粒和光照处理后的第15天。hbv-tg小鼠血清中与hbv复制相关的参数如图28b-f所示,结果显示相关参数均显著下降。图28b显示,在近红外照射下,aso治疗组的小鼠血清hbv dna仅下降到15%,而aso和c39的nps治疗组的小鼠血清hbv dna下降到1.3%,表明仿生纳米颗粒有功能性治愈的潜力。用仿生纳米颗粒和nir处理后,检测到的hbsag和hbeag的变化水平与hbv dna的变化水平基本一致,但在仅暴露于nir照射的hbv-tg小鼠中没有观察到下降(图28c,28d)。在umac-m-gal纳米颗粒和nir照射后的第15天,采用免疫组化染色研究hbv-tg小鼠肝脏中病毒抗原(hbcag和hbsag)的表达水平(图28f),其表达水平显著下降。此外,各器官的组织病理学分析(h&e染色)显示无器官损伤(图29)。同时,治疗期间的体重(图30a)和治疗后的血液生化参数(图30b-d)均无明显变化。这些实验数据证实,umac-m-gal具有良好的生物相容性和特性,且在体内无全身毒性,在实现慢性hbv感染的功能性治疗方面具有巨大的应用潜力。
[0126]
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1