一种归一化并比较水平基因转移网络的方法与流程
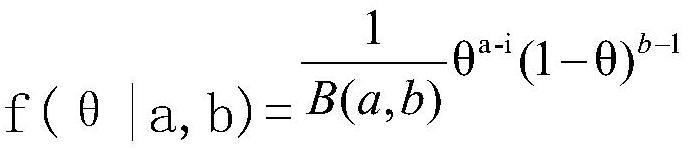
本发明涉及基因转移网络,尤其涉及一种归一化并比较水平基因转移网络的方法。
背景技术:
1、人体微生物和人类有着复杂的相互作用。研究发现,人体微生物和多种疾病都相关。人体微生物是个复杂的生态系统,以肠道微生物组为例,ferretti等人利用菌株水平的宏基因组图谱来跟踪母婴间的细菌传播;他们发现母婴对中共有的菌株;在最近关于肠道微生物组的研究中,不少研究者建立了宏基因组和人类表型之间的联系,但这些工作未能从系统的角度对肠道微生物组进行建模。近年,研究人员应用hgt网络分析宏基因组,提取微生物组的特征,并探寻微生物组和表型的关联。
2、存在的缺点或不足:
3、虽然目前研究者建立了宏基因组和人类表型之间的联系,但是这些工作无法从系统的角度对肠道微生物区系进行建模。hgt网络是一种系统性分析宏基因组的工具,然而由于hgt网络会受到测序数据量的影响,可能会导致一些假阳性的宏基因组和表型的关联,因此需要归一化并比较水平基因转移网络。本发明涉及一种归一化hgt网络,并计算hgt网络拓扑属性的方法。可以标准化的比较试验组和对照组的宏基因组,从而系统性地建模并分析宏基因组。
技术实现思路
1、本发明的目的是为了解决现有技术中存在的缺陷,而提出的归一化并比较水平基因转移网络的方法。
2、为了实现上述目的,本发明采用了如下技术方案:
3、一种归一化并比较水平基因转移网络的方法,包括以下步骤:
4、s1:采用贝叶斯概率模型将hgt根据测序量进行归一化;
5、s2:在hgt网络中,将每个基因组被表示为一个顶点,其中,如果hgt发生在两个基因组之间,则在两个顶点之间有一条边;
6、s3:采用不同的分类级别对基因组序列进行注释,得到了不同分类等级的hgt网络;
7、s4:通过图的密度、传递性、同配性和代数连通性几种拓扑性质对hgt网络进行分析。
8、进一步地,在步骤s1中:使用localhgt来检测每个样本中的hgt,并且得到支持每个hgt断点对的reads数量,其中,当所有样本中的平均reads数目为q时,根据模型推断;
9、当总reads数目为q时,则支持每个hgt断点对的reads数目的概率分布,根据这个概率分布,过滤hgt断点;
10、过滤标准为:在总reads数目为q的情况下,预期的reads数目大于x(默认为2)的概率高于90%。
11、进一步地,在步骤s4中:图密度表示图中存在的边的数目除以图可能具有的最大边数;
12、传递性为通过观察到的三角形数与图中可能的最大三角形数之比计算获得,传递性度量顶点的相邻顶点相互连接的总体概率;
13、同配性为图中的节点与具有相似度的节点相连接的趋势,通过关联度与关联度的皮尔逊相关系数来量化关联度;
14、代数连通度是图的拉普拉斯矩阵的第二小特征值;
15、拓扑属性是使用python模块networkx计算。
16、相比于现有技术,本发明的有益效果在于:
17、由于hgt事件会受到测序数据量的影响,因此在所有样本中使用贝叶斯模型根据测序量对hgt事件进行归一化处理,大大减少了hgt网络受测序数据量的影响所导致一些假阳性的宏基因组和表型关联的情况发生,并且使试验组和对照组的宏基因组比较更加标准化。
18、对基因组在不同的分类水平上进行注释,我们得到不同水平的hgt网络。使用图密度、传递性、同配性和代数连通性来描述网络,并在不同组别间的样本中比较hgt网络,以对宏基因组和人体表型关联提供新的分析方向。通过计算多种拓扑属性来分析hgt网络,也能更加系统性地建模并分析宏基因组。
技术特征:
1.一种归一化并比较水平基因转移网络的方法,其特征在于,包括以下步骤:
2.根据权利要求1所述的归一化并比较水平基因转移网络的方法,其特征在于,在步骤s1中:使用localhgt来检测每个样本中的hgt,并且得到支持每个hgt断点对的reads数量,其中,当所有样本中的平均reads数目为q时,根据模型推断;
3.根据权利要求2所述的归一化并比较水平基因转移网络的方法,其特征在于,过滤hgt断点的过滤标准为:在总reads数目为q的情况下,预期的reads数目大于x(默认为2)的概率高于90%。
4.根据权利要求1所述的归一化并比较水平基因转移网络的方法,其特征在于,在步骤s4中:图密度表示图中存在的边的数目除以图可能具有的最大边数;
技术总结
本发明公开了一种归一化并比较水平基因转移网络的方法,包括以下步骤:S1:采用贝叶斯概率模型将HGT根据测序量进行归一化;S2:在HGT网络中,将每个基因组被表示为一个顶点,其中,如果HGT发生在两个基因组之间,则在两个顶点之间有一条边;S3:采用不同的分类级别对基因组序列进行注释,得到了不同分类等级的HGT网络;S4:通过图的密度、传递性、同配性和代数连通性几种拓扑性质对HGT网络进行分析。使用图密度、传递性、同配性和代数连通性来描述网络,并在不同组别间的样本中比较HGT网络,以对宏基因组和人体表型关联提供新的分析方向,通过计算多种拓扑属性来分析HGT网络,也能更加系统性地建模并分析宏基因组。
技术研发人员:陈银,赵梓丞,李静
受保护的技术使用者:深圳百人科技有限公司
技术研发日:
技术公布日:2024/1/13
- 还没有人留言评论。精彩留言会获得点赞!