一种诊断待测者是否是乳腺癌的系统以及生物标志物的制作方法
1.本发明涉及医学领域,具体而言,涉及利用蛋白组学筛选乳腺癌的生物标志物并用于乳腺癌的诊断,尤其涉及一种预测乳腺癌发生风险的生物标志物及其应用。
背景技术:
2.蛋白质组学(proteomics)是研究细胞、组织或生物体中蛋白质组成、定位、变化及其相互作用规律的科学,包括对蛋白质表达模式和蛋白质组功能模式的研究。随着质谱技术的发展,液相色谱与质谱联用技术(lc-ms/ms)已成为蛋白质组学研究中最主要的工具。蛋白质组学的发展对寻找疾病的诊断标志、筛选药物靶点、毒理学研究等有重要意义,也因此被广泛应用于医学研究。
[0003][0004]
越来越多的证据表明,各种因素(即基因和环境因素)可能与乳腺癌的发生和发展有关。乳腺癌患者的早期诊断是乳腺癌治疗的重要方面之一。在各种诊断平台中,影像学技术是主要的诊断手段,可以为乳腺癌患者提供有价值的数据。此外,利用生化生物标志物,如蛋白质、dna、mrna 和 microrna,可以作为乳腺癌患者的新诊断和治疗工具。随着免疫学和分子生物学的发展,肿瘤相关蛋白标志物在乳腺癌的诊治中显示出越来越重要的临床价值,已成为必不可少的辅助诊断、观察疗效和判断预后的生物学指标。
[0005]
临床上已发现多种可以用于乳腺癌诊断、病理分型和临床分期、判断预后和疗效的肿瘤标志物,但目前常用的乳腺癌标志物的(ca153、ca125、cea)诊断效能均不理想,尚未发现一种特异的肿瘤标志物对乳腺癌诊断有较高的敏感度以及特异性。
[0006]
因此,寻找新的乳腺癌诊断相关标志物,多种标志物相结合,采用合适的乳腺癌诊断预测模型,具有重要的临床价值。
技术实现要素:
[0007]
针对现有技术中存在的问题,本发明提供了一种用于乳腺癌检测的生物标志物,利用蛋白质组学的方法,通过分析乳腺癌患者和正常人的血液中具有显著性差异的蛋白质,筛选出系列全新的能早期预示乳腺癌发生风险的生物标记物,并从中进一步筛选出一组生物标志物构建乳腺癌的诊断模型,可用于便捷、无创、高效地预测个体是否患乳腺癌,满足临床所需。
[0008]
一方面,本发明提供了一种生物标志物在制备预测个体是否是乳腺癌的试剂中的用途,所述生物标志物选自如下的一种或多种或者由下列标志物组成:dcd、plin1、fabp4、sfrp1、s100b、cryab、pgm5、cd36和adh1b。
[0009]
本发明通过tmt标记定量蛋白质组学研究,用lc-ms/ms超高效液相色谱-串联质谱联用方法分析健康组和乳腺癌病人组两组血液样品,再通过正交偏最小二乘法判别在乳腺癌样品和对照样品之间有显著差异的蛋白质,最终得到9种的与乳腺癌关联的蛋白质,作为生物标志物,可用于高效准确地预测个体是否乳腺癌。
[0010]
在一些方式中,所述可用于预测个体是否是乳腺癌的生物标志物,可以生物标志物为检测目标制备检测试剂,例如样品前处理试剂、抗原或抗体等适用于所述生物标志物检测的生物试剂及试剂盒;也可以开发成适用于所述生物标志物lc-uv或lc-ms检测的标准化试剂或试剂盒等。
[0011]
在一些方式中,所述的标志物如下:皮敌菌素(dcd)为uniprot数据库编号为p81605的蛋白或者氨基酸序列;脂溶素-1(plin1)为uniprot数据库编号为o60240的蛋白或者氨基酸序列;脂肪酸结合蛋白,脂肪细胞(fabp4)为uniprot数据库编号为p15090的蛋白或者氨基酸序列;分泌的卷曲相关蛋白1(sfrp1)为uniprot数据库编号为q8n474的蛋白或者氨基酸序列;s100钙结合蛋白b (s100b)为uniprot数据库编号为p04271的蛋白或者氨基酸序列;α-晶状体蛋白 b 链(cryab)为uniprot数据库编号为p02511的蛋白或者氨基酸序列;磷酸葡萄糖变位酶样蛋白 5(pgm5)为uniprot数据库编号为q15124的蛋白或者氨基酸序列;血小板糖蛋白 4(cd36)为uniprot数据库编号为p16671的蛋白或者氨基酸序列;全反式视黄醇脱氢酶 [nad(+)] (adh1b) 为uniprot数据库编号为p00325的蛋白或者氨基酸序。
[0012]
进一步地,所述生物标志物包括dcd、plin1、fabp4、sfrp1、s100b、cryab、pgm5、cd36和adh1b。
[0013]
在一些方式中,所述的生物标志物包括皮敌菌素(dcd);脂溶素-1(plin1);脂肪酸结合蛋白,脂肪细胞(fabp4);分泌的卷曲相关蛋白1(sfrp1);s100钙结合蛋白b (s100b);α-晶状体蛋白 b 链(cryab);磷酸葡萄糖变位酶样蛋白 5(pgm5);血小板糖蛋白 4(cd36);全反式视黄醇脱氢酶 [nad(+)] (adh1b)。
[0014]
进一步地,所述试剂用于检测体液样本中的生物标志物,所述体液样本包括血液、尿液、唾液、汗液中的任意一种。
[0015]
在一些方式中,本发明的所述生物标志物是通过血液样本筛选获得的,尤其适于开发成用于乳腺癌预测的血液检测试剂或试剂盒等。
[0016]
本发明从血液筛选到乳腺癌的生物标志物,这些生物标志物在乳腺癌患者和非乳腺癌患者的血液中存在显著性差异,通过收集血液样本,即可通过检测个体血液中这些生物标志物来预测或辅助诊断该个体是否有乳腺癌或患有乳腺癌的可能性,或者可以检测某一群体血液中的这些生物标志物,进而将该群体分为乳腺癌组或非乳腺癌组。
[0017]
进一步地,所述检测体液样本中的标志物,为检测个体的体液样本中生物标志物的有无或相对丰度或浓度。
[0018]
在一些方式中,优选采用相对丰度来表示,所述相对丰度为高效液相色谱-串联质谱获得的检测图谱中该生物标志物的峰面积。比如某个生物标志物在对照样品(未患乳腺癌的个体)里测出的平均峰面积是500,在乳腺癌样品里测出的平均峰面积是3000,那么就认为该生物标志物在乳腺癌样本中的丰度是对照样本中的6倍。
[0019]
另一方面,本发明提供了一种预测个体是否是乳腺癌的生物标志物组合,所述生物标志物选由下列标志物组成:dcd、plin1、fabp4、sfrp1、s100b、cryab、pgm5、cd36和adh1b。
[0020]
通过检测临床乳腺癌样本的数据显示,仅仅采用这9种生物标志物预测乳腺癌,其auc值就能达到0.964,其效果明显好于现有生物标志物联合预测乳腺癌模型的效果。
[0021]
另一方面,本发明提供了一种预测个体是否是乳腺癌的试剂盒,所述试剂盒包括如上所述的用于检测上述9生物标志物,或如上所述的生物标志物组合的检测试剂。
[0022]
在一些方式中,所述检测试剂为如上所述生物标志物的抗体,所述抗体为单克隆抗体。
[0023]
再一方面,本发明提供了一种预测待测者是否是乳腺癌的系统,所述系统包括数据分析模块,所述数据分析模块用于分析待测者的生物标志物的检测值,所述生物标志物为如下生物标志物的组合:dcd、plin1、fabp4、sfrp1、s100b、cryab、pgm5、cd36和adh1b;或。
[0024]
进一步地,所述数据分析模块通过将待测者生物标志物的检测值代入如下方程,计算预测个体是否是乳腺癌的预测值,从而评估个体是否是乳腺癌,所述方程为:
[0025]
其中,y为预测值,i表示第i个生物标志物,n表示生物标志物的个数,其中n=9,xi表示第i个生物标志物的检测值,单位为μg/ml;c表示预测类型:阴性、阳性,p(yc)表示预测所诉预测类型为c的先验概率,其中,所述的阴性和阳性样本的先验概率均为0.5;σci和μci分布表示在类别y为c时,标志物xi对应的标准差和平均值,所诉xi对应的标准差和平均值如下表:
。
[0026]
进一步地,当诊断模型预测值y》0.513时,认为待测者为乳腺癌患者;当模型预测值y≤0.513时,认为待测者不为乳腺癌患者。
[0027]
进一步地,还包括数据检测系统,数据输入、输出界面;所述数据检测系统用于检测样本中的生物标志物,获得检测值;所述数据输入、输出界面中的输入界面用于输入生物标志物的检测值,经所述的数据分析模块分析检测值后,输出界面用于输出个体是否是乳腺癌的分析结果。
[0028]
再一方面,本发明提供了如上所述的系统用于构建预测个体是否是乳腺癌的概率值的检测模型的用途。
[0029]
本发明的有益效果为:1、 筛选到9种全新的能早期预示乳腺癌发生风险的生物标记物dcd、plin1、fabp4、sfrp1、s100b、cryab、pgm5、cd36和adh1b;2、 分别采用9种生物标志物构建乳腺癌的不同的诊断模型,发现采用包括dcd、plin1、fabp4、sfrp1、s100b、cryab、pgm5、cd36和adh1b的9种生物标志物构建的乳腺癌诊断模型最优,可用于更高效地预测个体是否患乳腺癌,auc值达到0.984,其效果明显好于现有的乳腺癌诊断模型。
[0030]
3、 对于同样的标记物物质,不同的模型获得不同的诊断效果,筛选出最有的诊断预测模型。
附图说明
[0031]
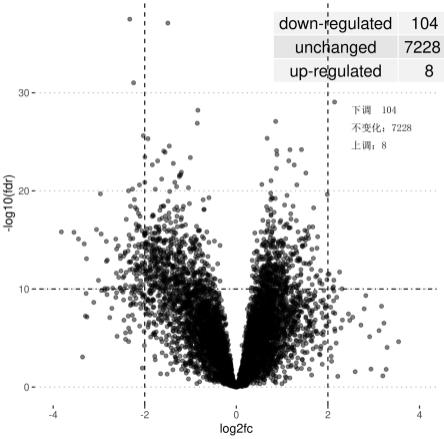
图1为实施例1中的tcga数据库中健康对照与乳腺癌两组的wilcoxon结果图。
[0032]
图2为实施例1中的cptac数据库中健康对照与乳腺癌两组的wilcoxon结果图。
[0033]
图3为实施例1中来源于tcga和cptac数据库的差异基因与血液蛋白的韦恩图。
[0034]
图4为实施例1中的健康对照与乳腺癌两组的roc和boruta分析结果图。
[0035]
图5为实施例3中的基于9种不同算法构建的最优模型性能评估结果图。
[0036]
图6为实施例3中构建的乳腺癌联合诊断模型在模型组中的roc曲线。
[0037]
图7为实施例3中构建的乳腺癌联合诊断模型在测试组中的roc曲线。
[0038]
图8为实施例3中构建的乳腺癌联合诊断模型在测试组中的性能评估结果图。
[0039]
详细说明(1)诊断或者检测这里的诊断或者检测是指对于样本中的生物标志物进行检测或者化验,或者目的生物标志物的含量,例如绝对含量或者相对含量,然后通过目标标志物是否存在或者数量的多少来说明提供样本的个体是否可能具有或患某种疾病,或者具有某种疾病的可能性。这里的诊断与检测的含义可以互换。这种检测的结果或者诊断的结果是不能直接作为患病的直接结果,而是一种中间结果,如果获得直接的结果,还需通过病理学或者解剖学等其它辅助手段才能确认患有某种疾病。例如,本发明提供了多种与乳腺癌具有关联性的新的生物标志物,这些标志物的含量的变化与是否患有乳腺癌具有直接的关联性。
[0040]
(2)标志物或生物标志物与乳腺癌的联系标志物和生物标志物在本发明中具有相同的含义。这里的联系是指某种生物标志物在样本中出现或者含量的变化与特定疾病具有直接的关联性,例如含量的相对升高或者降低,表示这种患有这种疾病的可能性相对健康人员更高。
[0041]
如果样本中多个不同的标志物同时出现或者含量的相对变化,表示这种患有这种疾病的可能性相对健康人员也更高。也就是说标志物种类中,某一些标志物与患病的关联性强,有些标志物与患病的关联性弱,或者有些甚至与某种特定的疾病无关联。对于那些关联性强的标志物中的一种或者多种,可以作为诊断疾病的标志物,与那些关联性弱的标志物可以与强的标志物组合来诊断某种疾病,增加检测结果的准确性。
[0042]
针对本发明发现的血清中的众多生物标志物,这些标志物都可以用来进行区分乳腺癌与健康人群。这里的标志物可以单独作为单个的标志物来进行直接的检测或者诊断,选择这样的标志物表示该标志物的含量的相对变化与乳腺癌具有强的关联性。当然,可以理解的是,可以选择与乳腺癌关联性强的一种或者多种标志物的同时检测。正常的理解是,在一些方式中,选择关联性强的生物标志物来进行检测或者诊断可以达到一定标准的准确性,例如60%,65%,70%,80%,85%,90%或者95%的准确性,则可以说明,这些标志物可以获得诊断某种疾病的中间值,但并不表示就能直接确认患有某种疾病。
[0043]
当然,也可以选择roc值越大的差异蛋白质来作为诊断的标志物。所谓的强,弱一般通过一些算法来计算确认,例如标志物与乳腺癌贡献率或者权重分析。这样的计算方法可以是显著性分析(p值或fdr值)和倍数变化(fold change),多元统计分析主要包括主成分分析(pca)、偏最小二乘判别分析(pls-da)和正交偏最小二乘判别分析(opls-da),当然还包括其他的方法,例如roc分析等。当然,其它的模型预测方法也是可以的,在具体选择生物标志物的时候,可以选择本发明所公开的差异蛋白质,也可以选择或者结合其它现有公知的标志物组合通过模型方法进行预测。
具体实施方式
[0044]
下面结合附图和实施例对本发明作进一步详细描述,需要指出的是,以下所述实施例旨在便于对本发明的理解,而对其不起任何限定作用。本实施例中使用的试剂均为已知产品,通过购买市售产品获得。
[0045]
实施例1 利用公共数据库筛选乳腺癌的生物标志物1. 数据收集利用tcga数据库下载乳腺癌组织样本的rna测序数据(https://portal.gdc.cancer.gov/),组织样本筛选标准为:经病理确诊为原发性乳腺癌,且具有完整的临床资料和随访结果,最终获得1097个原发性乳腺癌组织样本和114个正常组织(健康)样本用于后续分析。从cptac数据库(https://proteomics.cancer.gov/programs/cptac)下载乳腺癌组织样本的蛋白质组学数据,包含143个原发性乳腺癌组织样本和114个正常组织(健康)样本用于后续分析。
[0046]
2. 数据预处理通过tcga数据库下载的rna测序数据首先采用tpm方法对原始reads count数据进行归一化处理。然后对归一化后的tpm值采用log2进行对数转换,使其定量值成正态分布。为了获得高质量的定量结果,将在所有样本中的平均表达值小6的基因(rna)进行了剔除。最终获得了12427个基因的rna定量结果。通过cptac数据库下载的蛋白质组学数据,首先筛选出采用特异性肽段计算得到相对定量ratio值,然后采用log2进行对数转换,使其定量值成正态分布。为了获得高质量的定量结果,将在小于10个样本中有定量信息的蛋白进行了剔除。最终获得了7340个蛋白的rna定量结果。
[0047]
3. 差异分析采用单变量分析和多元统计分析结合的方式进行差异蛋白质和转录本的筛选,其中单变量分析主要包括特征分子在不同分组中的显著性分析(p值或fdr值)和倍数变化(fold change),多元统计分析主要包括接受者操作特性曲线(roc)分析和基于随机森林算法的boruta特征筛选。所有统计分析均使用r完成,具体的r相关信息见表2。
[0048]
表2:本发明所用的r及其相关信息
[0049]
计算变量投影重要度(variable importance for the projection,vip)以衡量各蛋白质的表达模式对各组样本分类判别的影响强度和解释能力,进一步进行wilcoxon秩和检验得到校正后的p值(fdr)。按照fdr《1e-10和fold change》2的条件筛选,在cptac来源的蛋白质组学数据中获得104个下调蛋白和8个上调蛋白(详见图1),在tcga来源的rna测序数据中获得359个下调和232上调基因(详见图2)。
[0050]
为了更加精准定位潜在的乳腺癌候选标志物,我们将cptac数据库和tcga数据库筛选的得到的差异表达基因与the human protein atlas数据库中的血液蛋白进行了比较分析。我们将三者交集部分的23个基因定义为潜在的乳腺癌候选标志物,(详见图3)。
[0051]
为了评估每个标志物在乳腺癌诊断预测中的作用,我们采用roc和boruta分析方法对每个标志物进行了评估,结果图见图4,横坐标为roc分析得到的auc,纵坐标为wilcoxon检验计算得到的-log10(fdr),点的大小代表boruta分析得到的vip值。
[0052]
在23个候选标志物中,按照vip》5和auc》0.9进一步筛选,共计找到9种更为显著的候选标志物,详见见表3。
[0053]
表3:乳腺癌与正常健康的差异标志物
[0054]
表3中fdr值越小和/或vip值越大,在一定程度上说明该差异化合物在两组间的差异性越显著,同时也说明该差异化合物可能具有更高的诊断价值。
[0055]
全新发现的乳腺癌差异生物标志物,可作为乳腺癌与健康鉴别诊断的候选生物标志物,选择其中的一种或多种的组合,可用于乳腺癌的辅助诊断,或者用于区分乳腺癌与健康人群。
[0056]
实施例2:9种单一生物标志物预测乳腺癌本实施例利用实施例1中筛选出的单个生物标志物建立乳腺癌的预测或诊断模型,用于区分乳腺癌和非乳腺癌,或者从群体中筛选出乳腺癌患者,或者用于预测个体是否是乳腺癌患者或个体或者待测者得乳腺癌的可能性。
[0057]
建立实施例1提供的9种的每个蛋白质的roc曲线,通过曲线下面积(auc)的大小来判断实验结果优劣。auc为0.5表示单个蛋白质无诊断价值;auc大于0.5,说明单个蛋白质具有诊断价值;auc越大,说明单个蛋白质的诊断价值越高,结果如表4所示。
[0058]
表4:roc分析乳腺癌与正常健康样本各差异蛋白质的roc值及相关信息
[0059]
9种生物标志物的浓度变化与是否患乳腺癌的关联性的高低,可以通过表4中的auc值、敏感性、特异性等来区分,其中auc值最为直观和明显。auc值越高,表示该生物标志物越能准确区分乳腺癌人群和非乳腺癌人群。
[0060]
由表4可以看出,9种生物标志物的浓度变化与是否患乳腺癌都具有明显的关联性,单独采用9种生物标志物中的任意一种,其浓度变化用于区分乳腺癌人群和非乳腺癌人群,auc值都能达到0.7以上,都具有较高的准确性,其中s100钙结合蛋白b(s100b)的关联性最高,auc值达到0.768。
[0061]
实施例3:9种差异蛋白质联合鉴别乳腺癌与健康正常人群的分类模型及其建立利用单一的生物标志物虽然也能区分乳腺癌与非乳腺癌血清样本或进行乳腺癌的预测,但一般来说将多种生物标志物进行组合,其区分或预测的准确性更高。
[0062]
但是,预测乳腺癌准确性更高的单一生物标志物,在与其他一种或多种生物标志物组合后,其在该组合中起的作用不一定越大,同时也并非生物标志物的个数越多,其组合的预测准确性(auc值)就越高,因此还需要进行大量验证实验。
[0063]
本实施例对由血清中皮敌菌素(dcd);脂溶素-1(plin1);脂肪酸结合蛋白,脂肪细胞(fabp4);分泌的卷曲相关蛋白1(sfrp1);s100钙结合蛋白b (s100b);α-晶状体蛋白 b 链(cryab);磷酸葡萄糖变位酶样蛋白 5(pgm5);血小板糖蛋白 4(cd36);全反式视黄醇脱氢酶 [nad(+)](adh1b),9种蛋白标志物构建的模型进行研究。
[0064]
1. 获取数据研究人群:从2019.8-2019.12收集了500例乳腺癌和500例健康对照,所有入组的患者签署知情同意书。乳腺癌患者均为活体组织经病理学确认结果,健康对照为常规体检正常(不含有结节,或者是不是乳腺癌的人群)。将入组人员按照8:2的比例分为模型组(乳腺癌n=400,
健康对照n=400)和测试组(乳腺癌n=100,健康对照n=100)。数据信息如表5:表5:建模样本信息
[0065]
乳腺癌患者的纳入标准:(a)无其他恶性肿瘤病史,(b)采血后一个月内进行手术治疗,且经术后病理证实为乳腺癌。对照组的健康人选自体检中心;通过胸部x射线或薄切片计算机断层扫描证实这些个体没有乳腺结节,也没有恶性肿瘤病史。在知情同意后,将收集的所有血清样品储存在-80℃的血清库中。
[0066]
本实施例对采集到的血清样本进行酶联免疫吸附剂检测(elisa),获得血清中皮敌菌素(dcd);脂溶素-1(plin1);脂肪酸结合蛋白,脂肪细胞(fabp4);分泌的卷曲相关蛋白1(sfrp1);s100钙结合蛋白b (s100b);α-晶状体蛋白 b 链(cryab);磷酸葡萄糖变位酶样蛋白 5(pgm5);血小板糖蛋白 4(cd36);全反式视黄醇脱氢酶 [nad(+)] (adh1b),9种蛋白标志物的浓度,单位:μg/ml。
[0067]
2. 实验数据统计分析shapiro wilk的测试用于评估正态分布,并且使用非参数检验wilcoxon测试分别分析模型组和测试组中乳腺癌患者和健康对照之间的血液标志物浓度的差异。在模型组中,采用多种机器学习方法相结合的方法构建8种乳腺癌标志物的联合诊断模型。使用预测概率值以95%置信区间(ci)估计接收器操作员特征(roc)曲线下面积(auc),以评估多变量诊断模型的辨别能力。使用测试组,计算youden指数(yi)以确定用于区分乳腺癌患者与正常对照的预测概率cut-off值。此外,构建并比较了单个标志物和不同亚组的roc。计算标准描述性统计数据,例如频率,平均值,中位数,阳性预测值(ppv),阴性预测值(npv)和标准偏差(sd)以描述研究群体的实验结果。使用r3.6.1进行统计学分析,p值小于0.05被认为是统计学上显著的。
[0068]
3. 乳腺癌联合诊断模型(9mp)构建步骤s101,将模型组中样本的皮敌菌素(dcd)、脂溶素-1(plin1)、脂肪酸结合蛋白脂肪细胞(fabp4)、分泌的卷曲相关蛋白1(sfrp1)、s100钙结合蛋白b (s100b)、α-晶状体蛋白 b 链(cryab)、磷酸葡萄糖变位酶样蛋白 5(pgm5)、血小板糖蛋白 4(cd36)、全反式视黄醇脱氢酶 [nad(+)] (adh1b), 9种蛋白标志物的浓度矩阵作为原始训练数据集。
[0069]
s102,设定用于构建预测模型的监督分类算法,以及算法的超参数优化过程中网格搜索范围。监督分类算法包含:神经网络、梯度提升、广义线性模型、随机森林、逻辑回归、支持向量机、朴素贝叶斯和混合判别分析8种算法。该步骤中,对每种算法设定模型的超参数优化的网格搜索范围如下表6所示。
[0070]
表6:8种算法的参数网格搜索范围
[0071]
s103,根据步骤s102设定的算法和超参数设定范围,选择其中一种算法和对应的超参数组合方式,作为预测模型构建的参数。
[0072]
s104,将原始数据集按k折交叉验证机制,分割成k个子集。为确保每一折子集中,多数类样本和少数类样本比例与原始数据集相同,需采用分层k折交叉验证(stratified k-folds cross validation)机制来进行数据分割。
[0073]
s105,根据步骤s104分割得到的k个训练数据子集,选择其中一个子集作为验证集ddev。
[0074]
s106,将步骤s105中未选择的训练数据子集合并形成训练数据池dtrainl。
[0075]
s107, 根据步骤s106得到的训练数据集d.train, 基于所选择的有监督分类算法和超参数构建预测模型。
[0076]
s108,根据步骤s107得到的预测模型,在验证集d.dev进行评估得到auc值,并将当前预后预测模型与相应的auc值存储在预测模型池pool中。步骤s108为根据步骤s107得到
的预测模型,在当前迭代中确定的验证集上进行评估,并将模型和评估结果都存储到预测模型池中,供以后预测模型选择使用。该步骤中提到的评估,可以是auc值,也可以是其他合理的对模型性能进行评估的指标。
[0077]
s109,判断是否每个子集全部做过验证集。步骤s109为判断步骤s104得到的k个子集是否都已作为验证集,进行过模型的训练。如果所有的子集均作为验证集并完成了训练,则执行步骤s110;若有子集并未作为验证集,则执行步骤s105。该步骤确保原始数据集中,每一个样本均做过验证集,提高模型稳定性,防止模型过拟合于某个子集。
[0078]
s110,将得到预测模型池pool所有模型的auc平均值作为本次组合方式模型的最终性能评估值。并将模型参数和最终性能评估auc值存入最优模型池pool.best。
[0079]
s111,判断每种算法和对应的所有超参数组合方式是否全部构建预测模型。步骤s111为判断步骤s102得到所有算法和对应的超参数组合方式是否都进行过预测模型的构建。如果所有组合方式均作完成了模型的构建,则执行步骤s112;若有组合方式未完成模型的构建,则执行步骤s103。
[0080]
s112,从步骤s111迭代结束后得到的最优模型池pool.best中,对于每种算法选择auc值最高的预测模型,存入乳腺癌诊断的候选预测模型集m.set。
[0081]
s113,从步骤s112获得的模型集m.set,在测试组d.test中进行评估得到auc值。将auc值最大的模型作为乳腺癌诊断的最终预测模型4. 乳腺癌联合诊断模型(9mp)参数优化结果通过上述模型构建步骤执行,我们得到了9种不同算法下最优模型。建模过程中采用10倍交叉验证方法,通过auc、灵敏度和特异性三个方面对模型进行了性能评估。如表7和图5所示:朴素贝叶斯(naive_bayes)算法在auc、灵敏度和特异性三个方法的性能评估得分均为最大(auc为0.977;灵敏度为0.905;特异性为0.925)。
[0082]
表7:glmnet算法不同超参数组合下构建模型的auc
[0083]
基于上述分析结果,选择朴素贝叶斯(naive_bayes)算法构建的最优模型作为乳腺癌诊断的最终预测模型,其构建模型的方程为:
[0084]
其中,y为预测值,i表示第i个生物标志物,n表示生物标志物的个数(n=9),xi表示第i个生物标志物的检测值(μg/ml);c表示预测类型(阴性、阳性),p(yc)表示预测预测类型为c的先验概率,这里阴性和阳性样本先验概率均为0.5;σci和μci分布表示在类别y为c时,标志物xi对应的标准差和平均值,详细数值见下表8。
[0085]
表8:模型中9种生物标志物在不同类型样本中浓度的平均值和标准差
[0086]
从以上可以看出,对于同样的标志物组合,采用不同的算法获得不同模型对于诊断或者预测结果差异大,所以,当标志物相同的时候,不同的算法也是非常关键的。
[0087]
5. 乳腺癌联合诊断模型(9mp)诊断阈值确定以模型组中的预测值绘制roc曲线,并根据约登(youden)指数值设置最佳诊断截断值为0.513。即当诊断模型预测值≤0.513时,认为待测者不为乳腺癌患者;当模型预测值>0.513时,认为待测者为乳腺癌患者。结果如图6所示:模型在模型组中auc为0.978,灵敏度为90.5%,特异性为93.5%。
[0088]
6. 乳腺癌联合诊断模型(9mp)验证以测试组中的预测值绘制roc曲线,如图7所示,auc为0.984。并根据约登(youden)指数值设置最佳诊断截断值为0. 513。即当诊断模型预测值≤0. 513时,认为待测者不为乳腺癌患者或者健康个体;当模型预测值>0. 513时,认为待测者为乳腺癌患者。结果如图8所示:模型在测试组中的准确率为93.5%,kappa值为0.87,灵敏度为91.4%,特异性为95.8%,阳性预测率为96%,阴性预测率为91%。
[0089]
虽然本发明披露如上,但本发明并非限定于此。任何本领域技术人员,在不脱离本发明的精神和范围内,均可作各种更动与修改,因此本发明的保护范围应当以权利要求所限定的范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1