一种基于长读长测序的基因单倍型检测方法及其应用与流程
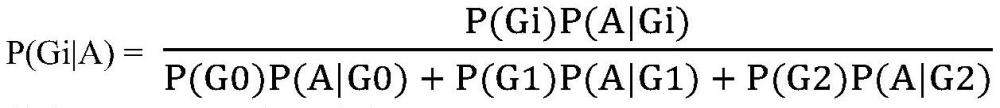
本发明涉及生物,特别涉及一种基于长读长测序的基因单倍型检测方法及其应用。
背景技术:
1、据研究发现,约有25%的药物是由一种单一的在肝脏中高表达的细胞色素p450-2d6(cyp2d6)酶代谢的。cyp2d6基因的多态性使得不同的基因型表现出不同的酶活。p450-2d6的酶活可分为四类:pm(代谢不良型)、im(中间代谢型)、em(正常代谢型)和um(超快代谢型)。不同的酶活对药物代谢的情况存在很大的差异,如8-氨基喹啉类抗疟疾药物伯氨喹(pq),其是一种需要cyp2d6代谢才能产生活性的前药。伯氨喹是预防间日疟原虫复发唯一有效的治疗药物,但对于pm或im表型的患者而言,他们不能将伯氨喹代谢为其活性代谢物,并且此类表型的患者在治愈后间日疟原虫复发的风险也更高。因此,在利用pq控制疟疾时,了解目标人群中pm或im表型的频率至关重要。
2、目前用于检测cyp2d6基因型的方法,如snp-微阵列、qpcr或短读长测序方法的成本已经相对低廉。但是这些检测方法本身存在一定的局限性,如它们只能通过检测已知的突变类型,进而识别常见的cyp2d6基因型,但对新发突变的检出却存在较大的局限性。而对于短读长测序方法,其虽然能发现一些新的变异,但因为cyp2d6和cyp2d7基因序列存在高度相似性,因此,在实际检测中容易导致reads的错配,进而检出错误的变异。此外,这些方法是根据检测到的变异和已知的等位基因频率进行推断,不是直接获得单个等位基因的序列。因此,罕见或新发的等位基因的存在将进一步混淆这些方法的检测结果。
3、长读长测序在一定程度上能够解决目前短读长测序技术所面临的挑战,但由于长读长测序的测序错误率较高(5%-15%),在变异检测中会存在检出错误的变异以及无法检出真实的变异的问题,目前使用的主流变异检测软件仍会产生较多的假阳性变异和遗漏假阴性位点,这会极大的影响cyp2d6基因分型。而且,在利用长读长测序数据研究基因组变异时,snp和indel的检测都是基本检测项目。目前,虽然已有多种不同算法可用于二代测序数据中的snp和indel分析,但由于这些方法是针对二代测序数据开发的,因此在测序错误率较高的长读长测序数据上并不能很好地运行。
4、现有技术中的生信分析方法,目前主流使用的是minimap2+nanopolish软件组合分析方法,对比其他比对软件和变异检测软件的组合分析ppv和sensitivity都是最优的,其中ppv为79.12%,sensitivity为96.43%,f1值为0.8692。但其仍有较多假阳性变异存在(ppv值较低),而这主要是由于长读长测序错误率较高导致的。而这种假阳性变异会对下一步单倍型预测产生偏差,进而影响基因分型。因此,亟需提供一种能够针对cyp2d6基因分型的高准确率的有效长读长测序方法。
技术实现思路
1、本发明旨在至少解决现有技术中存在的上述技术问题之一。为此,本发明的目的在于提供一种基于长读长测序的基因单倍型检测方法及其应用。本发明中的方法通过引物扩增cyp2d6基因序列后用特异性标签index再次扩增cyp2d6基因序列并对不同样本进行标记,然后对不同标签的样本文库pooling后进行nanopore测序分析,通过以特异性标签index作为标记,设置index容错数为1,对样本数据拆分,以有效提高数据拆分有效率,然后通过长读长测序结果推断点突变之间的连锁关系,根据连锁关系分开对单倍型进行预测,最终实现准确地分型单倍型。本发明中的方法相比于现有方法而言,实现了对于nanopore测序中的点突变的矫正,从而极大的提高了f1值,实现了更加准确且有效的cyp2d6基因分型。
2、本发明的第一个方面,提供一种基因单倍型检测方法,包括如下步骤:
3、(1)使用特异性引物对待测样本进行pcr扩增,得到目标片段,然后使用标签引物对目标片段再次pcr扩增,得到带有不同标签的扩增产物;
4、(2)等量混合来源自不同待测样本的带有不同标签的扩增产物,构建nanopore测序库,对nanopore测序库进行长读长测序,测序结果与人类参考基因组进行比对后得到排序后的bam文件,然后进行变异检测,获得vcf文件,使用贝叶斯矫正模型对vcf文件进行矫正,得到矫正后的vcf文件;
5、(3)使用phase命令对矫正后的vcf文件和排序后的bam文件进行分相,然后根据分相结果执行haplotag命令,对数据进行标记,根据标记判断基因单倍型。
6、在本发明的一些实施方式中,所述人类参考基因组为cyp2d6基因参考序列。
7、在本发明的一些实施方式中,所述特异性引物如seq id no:1~2所示。
8、在本发明中,所述特异性引物包括特异性靶向靶序列的结合部分和公共序列部分。其中,公共序列部分用于后续的标签连接。
9、在本发明的一些实施方式中,所述公共序列连接于特异性靶向靶序列的结合部分的5’端。
10、在本发明的一些实施方式中,所述标签引物如seq id no:3~206所示。
11、在本发明中,所述标签引物包括标签部分和公共序列部分。
12、在本发明的一些实施方式中,所述公共序列连接于所述标签部分的3’端。
13、在本发明的一些实施方式中,所述贝叶斯矫正模型为:
14、
15、其中,gi表示目标位点的基因型,以g0、g1和g2分别表示野生、杂合突变和纯合突变;
16、a表示变异等位基因的频率af;
17、p(a|g0)、p(a|g1)、p(a|g2)是分别以对应基因型的先验概率p(g0)、p(g1)、p(g2),经过计算位点样本均值和样本标准差,然后用正态分布拟合得到。
18、在本发明中,基于在生物信息分析方法中加入贝叶斯公式建模,可以对长读长测序检测到的点突变(包括snp和小indel)进行矫正,进而通过长读长测序结果推断点突变之间的连锁关系,并最终实现准确地分型单倍型。
19、在本发明的一些实施方式中,所述基因单倍型检测方法用于cyp2d6基因分型。
20、对于cyp2d6基因分型,长的pcr扩增子的测序不仅可以在不受同源假基因干扰的情况下明确检测变异,还意味着可以直接对长读长reads进行变异分析,从而有效降低了步骤的繁琐性并同时保证了高度的准确性。
21、在本发明的一些实施方式中,在使用贝叶斯矫正模型矫正时,同时使用以下公式进行位点结果校验:
22、
23、其中,a表示变异等位基因的频率af;
24、μ表示位点在野生型样本中的均值;
25、σ表示位点在野生型样本中的标准差;
26、若z值<1.96时,说明待测样本的位点基因型同野生型;
27、若z值≥1.96时,待测样本的位点基因型为上述步骤(2)中得到的vcf文件中的位点基因型。
28、在本发明的一些实施方式中,步骤(3)中,
29、若可进行分相,则使用perl脚本将分相后的vcf文件拆分为两个单倍体的vcf文件,然后用stargezar软件对两个单倍体vcf文件进行基因型检测,最终结合两个单倍体的单倍型作为最终基因型结果;
30、若不可进行分相,用stargezar软件对矫正后的vcf文件进行基因型检测,得到最终基因型结果
31、在本发明的一些实施方式中,所述方法还包括在长读长测序后进行数据处理,包括:
32、用guppy软件提取dna序列信息,并过滤掉q<8的部分,然后通过特异性标签index作为标记,使用python脚本设置index容错数为1对过滤后的数据拆分,去除接头并过滤掉q<9的部分,使用minimap2比对软件进行序列比对,获得sam比对文件,然后用samtools软件进行处理,得到排序后的bam文件,利用bcftools软件的mplieup和call命令,使用multiallelic-caller算法对排序后的bam文件进行变异检测,得到vcf文件。
33、在本发明的一些实施方式中,使用porechop软件去除接头。
34、在本发明的一些实施方式中,使用nanofilt软件过滤掉q<9的部分。
35、在本发明的一些实施方式中,所述minimap2比对软件使用map-ont模式。
36、在本发明的一些实施方式中,在使用samtools软件进行处理时,依次使用view、sort和index命令。
37、在本发明中,所述基因单倍型检测方法的流程图如图1所示。
38、本发明的第二个方面,提供本发明第一个方面所述的基因单倍型检测方法在cyp2d6酶活分型中的应用。
39、在本发明中,通过本发明第一个方面所述的基因单倍型检测方法测定待测样品中的cyp2d6基因型后,可根据本领域中对于cyp2d6酶活对应的基因型进行cyp2d6酶活分型。
40、本发明的有益效果是:
41、1.本发明中的基因单倍型检测方法有效解决了现有技术中cyp2d6基因分型所面临的准确度低的问题,通过简单的pcr扩增结合长读长测序能够更为精准的进行cyp2d6基因分型,且与现有技术的准确度等相比都有着显著的提高。
42、2.本发明中的基因单倍型检测方法引入了贝叶斯矫正模型进行数据矫正,同时还发现minimap2+bcftools相比本领域中出现的minimap2+nanopolish有着更好的检测效果,在实际验证中基本可以实现100%检出的效果。
- 还没有人留言评论。精彩留言会获得点赞!