用于高分辨率图案化的含硅烷醇有机-无机杂化涂层的制作方法
本发明涉及使用聚氢基倍半硅氧烷树脂涂料组合物在基材上进行图案化步骤的基于辐射的方法。
本发明还涉及可沉积为聚氢基倍半硅氧烷树脂涂层的经水解的前体溶液,通过施加经掩蔽的辐射,所述涂层可以非常高的分辨率图案化;并涉及经涂覆基材和用所述前体溶液在图案化前后形成的涂层。
背景技术:
传统上已通过光刻工艺的进展实现了微电子器件中复杂电路的微型化,其中可定义特征的极限受到光刻工艺中使用的光的波长的限制。通过一般曝光工具,该行业已从436nm(g-线)过渡到405nm(h-线)、365nm(i-线)、248nm(krf准分子)和最终的193nm(arf准分子)的限定波长(okoroanyanwu2015;gangnaik等人2017;desimone等人2014)。
在248nm和193nm波长下使用的光刻胶(resist)材料为化学放大光刻胶(car),其组成和微结构已随着在基材上印刷更精细特征的需求而演变。作为这种趋势的延续,λ=13.5nm(92ev)的极紫外(euv)波长是下一代光刻的主要候选波长(desimone等人2017)。
在过去的十年里,人们已在用于大规模生产集成电路的euv光刻(euvl)的开发方面进行大量投资。提出了各种纳米光刻技术,包括电子束光刻(ebl)、软光刻、纳米压印光刻(nil)、光子束光刻(pbl)或扫描探针光刻,来扩展可在二维表面上印刷的特征的按比例缩小。
euvl的成功需要新的光刻胶材料,这些材料都有着某些优点和缺点。功能性光刻胶应提供rls特性,即分辨率(r)、线边缘粗糙度(ler)和敏感性(s),如higgins等人2011所述。
已开发出众多不同的材料途径来使用euvl形成图案。在一般层面上,这些材料可分为不同的三类:有机、硅基和含金属光刻胶。
有机光刻胶(常被称为非-car光刻胶)可表现出足够的ler,但由于不良的euv吸收特性而需要高的剂量,因此处于很大的劣势中。为了提高光刻胶的敏感性,已采用car材料。在另一方面,car材料受患于不良的ler,这主要归因于car中光子散粒噪声和光酸产生剂分布的统计学影响。
为了应对两种有机类型光刻胶的挑战,已制备了在光刻胶材料中包含辐射敏感组分的制剂。具有13.5nmeuv光子的高摩尔吸收率的元素为金属。因此,人们对含金属的光刻胶越来越感兴趣,其中向其他光刻胶材料添加金属以改善敏感性,或者以分子光刻胶、金属氧化物纳米颗粒、有机金属前体的形式或以金属-有机骨架的形式采用金属。
这样的光刻胶已显示出高的敏感性、分辨率,并因此被认为是未来euvl光刻胶的关键候选材料。然而,它们的主要缺点在于,金属在集成电路的制造中极度不利,因为它们可破坏晶体管的功能,并且ler特性需要改善。此外,desimone等人和watanabe指出,在euv扫描仪中,含金属光刻胶中的金属物种会与原子h或自由基h*相互作用形成金属氢化物(mxhy),这对光学器件的寿命构成严重威胁。另外,在金属基光刻胶和化学放大光刻胶两者中,高的ler都是问题(desimone等人2017)。
car的另一个缺点在于低的抗蚀刻性和图案的不稳定性(grigorescu和hagen2009)。因此,为了向基材转移图案,car既需要富硅的中间层又需要富碳的下层。基于金属和硅的无机树脂的优点在于,由于无机中间层通过euvl直接图案化,所以将简化光刻堆叠。
氢倍半硅氧烷(hsq)是一种广泛使用的负型电子束和euvl光刻胶材料。由于其实现高密度图案即小于10nm节距结构的潜力,已进行了深入的研究以找到最佳工艺。然而,其患于敏感性非常低,需要浓缩显影剂,并已知在加工过程中相对不稳定,从而使其工业采纳受到限制,如(yang等人2009)所述。
存在若干可促进适合于euvl的工艺的开发的因素,如显影剂温度、显影剂浓度和显影剂组成。尽管现有技术已有进展,但仍在寻找提供rls特性而又无过度金属污染的功能性光刻胶。
技术实现要素:
本发明的一个目的是提供新型氢倍半硅氧烷树脂涂料组合物。
本发明的另一个目的是提供制备新型氢倍半硅氧烷树脂的方法。
本发明的第三个目的在于提供用光辐射将经聚氢基倍半硅氧烷涂覆基材图案化的方法。
本发明的又一个目的在于提供经涂覆基材,所述经涂覆基材包含适于通过光辐射图案化的辐射敏感涂层。
常规的hsq树脂不含硅烷醇官能团。本发明中令人惊奇地发现,与hsq树脂相比,hsq树脂中硅烷醇部分的存在大大增强涂层的图案形成能力。结果,与迄今文献中描述的那些相比,本类型的含硅烷醇的聚氢基倍半硅氧烷在euv光刻中表现出更高的敏感性。
看来膜中存在的树脂的硅烷醇基团会与由euv从sih生成的中间反应性硅物种反应,但这只是一种可能的解释。
此外,已发现,官能化基团的添加进一步增强了聚氢倍半硅氧烷树脂的敏感性。含有官能团的含硅前体的添加,将增强树脂在水性显影剂中的溶解性并还改善树脂的敏感性。
还进一步发现,在一些实施方案中,使用金属氧化物化合物作为水解混合物的一部分或者单独地作为添加剂或作为纳米颗粒添加将改善树脂的敏感性。
因此,本发明提供了一种氢倍半硅氧烷树脂的溶液,其包含有机液体和与具有硅-碳键和硅-氢的硅-氧网络或硅-氧-金属中的si-h或si-r相比约0.001m至约1m的硅烷醇。
可通过使三官能硅烷与其他含硅或金属的前体进行水解/缩合反应以获得含金属和硅烷醇的聚氢基倍半硅氧烷,来制备本类型的含金属和硅烷醇的聚氢基倍半硅氧烷树脂溶液。
这些溶液可用于在半导体基材上产生流延的含硅烷醇有机-无机杂化涂层,以在其上形成图案。
本发明还提供了一种经涂覆基材,所述经涂覆基材包含辐射敏感涂层,所述辐射敏感涂层包含具有硅-碳键和硅-氢和/或具有硅烷醇键的硅-氧网络或硅-氧-金属网络。
本发明还提供了一种用特定波长的光辐射将经含金属和硅烷醇的聚氢基倍半硅氧烷涂覆基材图案化的方法,所述方法包括以下步骤:沿选定的图案辐照经涂覆基材以形成具有经辐照涂层的区域和有未经辐照涂层的区域的经辐照结构,和选择性地显影所述经辐照结构以去除实质部分的未经辐照涂层而形成经图案化基材。
此外,本发明提供了一种用特定波长的光辐射将经含金属和硅烷醇的聚氢基倍半硅氧烷涂覆基材图案化的方法,其中所述波长为13.5nm或更小。
此外,本发明提供了一种经图案化基材,所述经图案化基材包含具有表面和在沿所述表面的选定区域处存在而在沿所述表面的其他区域处不存在的第一涂层的基材。通常,所述第一涂层包含具有硅-碳键和硅-氢和/或具有硅烷醇键的硅-氧网络或硅-氧-金属网络。或者,所述第一涂层可溶于至少一些有机液体中,或所述第一涂层可溶于碱水溶液中。
更具体地,本发明的特征在于独立权利要求的特征部分中陈述的内容。
通过本发明获得了相当大的优势。
因此,本溶液可用于在半导体基材上流延涂层,以通过随后的烘烤、辐照和显影步骤形成图案。特别地,本发明允许以工业上可行的方式控制树脂的微观结构,并解决现有技术的缺点。例如,可调节含金属和硅烷醇的聚氢基倍半硅氧烷树脂中的硅烷醇含量,这对于获得在euvl中应用的高度敏感的涂料是非常期望的。
此外,可通过前体的共聚,限制或促进涂层在显影剂中的溶解性,来改变包含具有硅-碳键和硅-氢和/或具有硅烷醇键的硅-氧网络或硅-氧-金属网络的涂层的溶解性。
本文描述的发明解决了关键的rls挑战,并另外提供高的抗氧等离子体蚀刻性,从而使得光刻堆叠的简化成为可能。
另外,包含具有硅-碳键和硅-氢和/或具有硅烷醇键的硅-氧网络或硅-氧-金属网络的涂层的使用允许使用行业标准的2.38重量%tmah溶液来显影图案,和采取措施设计经辐照涂层在显影剂中的溶解度特性。
在一个重要的方面,本发明提供了可通过辐照图案化的硅氧烷。
从以下对实施方案的详细讨论,本技术的其他特征和优点将显现。
附图说明
图1以侧视图示意性地示出了三层光刻堆叠的组件;
图2以侧视图示意性地示出了四层光刻堆叠的组件;
图3以侧视图示意性地示出了根据一个实施方案用含金属和羟基的聚氢倍半硅氧烷树脂图案化;
图4示出了含硅烷醇的聚氢基倍半硅氧烷的euv交联机理;
图5为sem图像,示出了使用实施例1中的材料在施加65.4mj/cm2的euv剂量下通过euvl获得的ler=1.5nm的22nm半节距图案化线;
图6a为使用对比例1中制备的2%聚合物在5000µc/cm2的剂量下获得的具有50nmhp的线的sem图像;
图6b为使用实施例1中制备的2%聚合物在450µc/cm2的剂量下获得的具有50nmhp的线的sem图像;
图7示出了由实施例1、3和5及对比例1和2中的聚合物制得的膜的ftir光谱,表明了对于不同的合成,硅烷醇含量的差异;
图8包括三个sem图像,展示了a)实施例1、b)实施例7和c)实施例10在辐射之后的电子束结果;从图8可以明显看出,在实施例7和10中,要获得具有改善的ler的类似图案,所需的剂量超出实施例1中40%;
图9含三个sem图像,展示了a)实施例1、b)实施例14和c)实施例17的电子束结果;从图9可以明显看出,获得图案所需的剂量在b)中降低了17%,在c)中降低了33%;和
图10含三个sem图像,展示了a)实施例1、b)实施例20、c)实施例21的电子束结果,示出了其在相同剂量下的性能。
具体实施方式
本发明的实施方案涉及制备含金属和硅烷醇的聚氢基倍半硅氧烷树脂溶液的方法,其中使三官能硅烷与合适的其他含硅或金属的前体进行水解/缩合反应以获得含金属和硅烷醇的聚氢基倍半硅氧烷。
本材料可表征为“含硅烷醇的有机-无机杂化材料”。这样的材料含源自有机化合物以及源自无机化合物的残基,这将在下文说明。此外,本材料含硅烷醇基团,即通常沿其主链、特别是沿其主硅氧烷链,显示出si-o-h连接性的基团。
树脂或涂料的硅含量通常为至少30重量%,特别是35重量%或更多。通常,硅的最大含量为约52.9重量%。
通常,聚氢基倍半硅氧烷树脂主链的每个重复单元有约1个硅烷醇基团。在一个实施方案中,聚氢基倍半硅氧烷树脂主链的每个单元平均有约0.8至1.2个硅烷醇基团。
因此,在一个实施方案中,提供了一种氢倍半硅氧烷树脂涂料组合物,其在液相中包含树脂,所述树脂在硅-氧网络中显示出硅烷醇(si-oh)和硅-氢(si-h)及任选地硅-碳键、或在硅-氧-金属网络中显示出硅-氢(si-h)和任选地硅-碳键,si-h与si-oh的峰高比为约2:1至100:1。
在一个实施方案中,提供了一种氢倍半硅氧烷树脂涂料组合物,其在液相中包含与具有硅-碳键和硅-氢的硅-氧网络或硅-氧-金属中的si-h或si-r相比约0.001m至约1m的硅烷醇。
当与本发明组合物中任选存在的组分结合使用时,术语“潜”是指在组合物或树脂的加工过程中(例如由光)激活的性质。因此,“潜”催化剂为特别是由光激活的物质,例如当树脂或组合物暴露于光时。类似地,组合物中的潜酸和潜碱可在暴露于光时激活。
根据一个实施方案,本发明的技术涉及一种适于在基材上形成可辐照固化的硅氧烷层的组合物,所述组合物包含硅氧烷聚合物,所述硅氧烷聚合物含有sio部分、多个沿聚合物分布的反应性位点、和第一sih部分、第二sioh部分、和中间芳族和非芳族部分、含金属-氧键的第四部分,其中所述聚合物的分子量为500至50000g/mol,并且所述组合物优选还包含酸和/或碱催化剂和溶剂。
根据一个优选的实施方案,本发明的技术涉及一种适于制备可在基材上流延的涂料制剂的组合物,并且其中基材上的涂层可通过辐照可图案化物而图案化,并且其中所述涂料由通式(i)表示:
在式(i)中,符号a、b、c和d各自表示独立地选自1至1000的整数,z代表官能团,m代表金属原子,r1至r8代表烃基原子团,a、b、m、o、y、z、p、q和x各自独立地代表0至3的整数。
上述组合物通过水解如下物质来获得:
具有至少两个或三个连接至硅的可水解基团的第一单体含氢硅化合物(“前体a”)与
任选的第二单体硅化合物,其具有至少零个、一个、两个或三个烃基原子团和至少一个连接至该化合物的硅原子的可水解基团(“前体b”)、
任选的第三单体硅化合物,其具有至少一个官能团和至少一个连接至该化合物的硅原子的可水解基团以形成硅氧烷材料,其中所述官能团增强在显影剂中的溶解(即,“前体c”)、和
任选地基于一种或多种可水解金属氧化物前体的第四前体,
将硅氧烷材料配制成在合适的溶剂体系中的稳定组合物(即,“前体d”)。
在一个实施方案中,前体b与任选地前体a、c和d中的一个或多个一起使用。
本发明中使用的前体的比率可变化。前体a的用量可为0-100mol%,前体b的用量可为0-50mol%,前体c的用量可为0-20mol%,前体d的用量可为0-50mol%。前体1和2一起构成组合物的至少50mol%,特别是至少60mol%,例如至少70mol%。
硅氧烷组合物可通过在同一反应容器中、或分别在指定的部分中、或每种前体独立地进行水解和缩合来获得。
通过选择前体的相对量,可调节聚氢基倍半硅氧烷树脂中的硅烷醇含量。
本发明特别适合于制备包含通过水解具有通式ii的第一硅化合物而获得的聚(有机硅氧烷)的组合物
r1a-si-r2b (ii)
其中
a为1或2的整数,
b为2或3的整数,
r1表示氢原子,和
r2表示可水解基团,其可独立地选自羟基、烷氧基、酰氧基和卤素。
在“卤素”的含义中,每个可水解基团优选且独立地代表氯或溴。
在“烷氧基”的含义中,每个可水解基团优选且独立地代表具有1至6个、特别是1至4个碳原子的烷氧基基团,如甲氧基、乙氧基、正丙氧基、异丙氧基、丁氧基、仲丁氧基或叔丁氧基。在“酰氧基”的含义中,酰基基团具有1至4个碳原子。因此,作为可水解基团,每个酰氧基优选且独立地代表乙酰氧基、丙烯酰氧基或甲基丙烯酰氧基。
这样的前体的具体实例有三乙氧基硅烷(hteos)、三甲氧基硅烷(htmos)、二乙氧基硅烷和三氯硅烷。
本发明还涉及包含通过水解具有通式ii的第一硅化合物与具有通式iii的前体而获得的共聚(有机硅氧烷)的组合物
r3m-sir2n-r4o(iii)
其中
r3和r4可独立地选自烷基基团、芳基基团、芳烷基基团、卤代烷基基团、卤代芳基基团、卤代芳烷基基团、具有环氧基团的有机基团、巯基基团、烷氧基芳基基团、酰氧基芳基基团、异氰脲酸酯基团、羟基基团、环状氨基基团、或氰基基团或它们的组合,
r2和r3也可以是烷氧基基团、酰氧基基团或卤素基团,
m为0至1的整数,
n为2至4的整数,和
o为0至1的整数,
其中m+n+o的总值不可超过4。
本发明还涉及包含通过水解具有通式ii的第一硅化合物与具有通式iv的前体而获得的共聚(有机硅氧烷)的组合物
zx-r5y-sir2n-r6z(iv)
其中
z为基团如羟基、羧酸、巯基、胺或其盐、或季铵盐,其促进在水性显影剂中的溶解性,和
r5为共价结合到z和si两者的间隔基基团,并可独立地选自衍生自烷基基团、芳基基团、芳烷基基团、卤代烷基基团、卤代芳基基团、卤代芳烷基基团、烯基基团、烷氧基芳基基团、酰氧基芳基基团的二价基团或它们的组合,
r6可独立地选自烷基基团、芳基基团、芳烷基基团、卤代烷基基团、卤代芳基基团、卤代芳烷基基团、具有环氧基团的有机基团、巯基基团、烷氧基芳基基团、酰氧基芳基基团、羟基基团、环状氨基基团、或氰基基团或它们的组合。
r2可以是烷氧基基团、酰氧基基团或卤素基团,
y为0至2的整数,
n为1至3的整数,和
z为0至11的整数,
其中y+n+z的总值不可超过4。
在式iv的结构中,x表示间隔基基团r3可涵盖的值,并必须等于或大于1。
在一个实施方案中,式iv包括具有游离或受保护的脂族或酚类羟基基团的硅物种。
在一个实施方案中,式iv包括具有游离或受保护的脂族或酚类羧酸基团的硅物种。
本发明最后涉及包含通过水解具有通式ii的第一硅化合物与具有通式v的前体而获得的共聚(有机硅氧烷)的组合物
r7p-mr8q(v)
其中
r7可独立地选自烷基基团、芳基基团、芳烷基基团、卤代烷基基团、卤代芳基基团、卤代芳烷基基团、烯基基团、烷氧基芳基基团、酰氧基芳基基团或它们的组合,
m可独立地选自能够形成可被水解和缩合成金属氧化物或金属-氧代氢氧化物(oxohydroxide)的前体的任何金属,
p为0至1的整数并且要求m能够形成稳定的碳金属键,
q为从mox-p获得的数的整数,其中mox为金属前体的氧化态,p为共价连接到金属前体的碳原子团r7的数目,
r8可以是起与另一金属原子的键合的作用的烷氧基基团、酰氧基基团、卤素基团、羟基基团或氧原子;或者r8也可由金属的二齿配体或配位配体组成。
在本发明的上下文中,
“烷基基团”代表c1-10烷基基团,特别是c1-4烷基基团;
“烷氧基基团”代表c1-10烷氧基基团,特别是c1-4烷氧基基团;
“芳基基团”代表具有1至5个稠合或共轭的环并具有4至30个碳原子的芳族环基团。
“芳烷基基团”代表具有1至5个环和4至30个碳原子并带有1至6个、特别是1至3个各具有1至10个、特别是1至4个碳原子的烷基基团作为环取代基的芳族环基团,
“烷氧基芳基”代表具有1至5个环和4至30个碳原子并带有1至6个烷氧基基团作为环取代基的芳基基团,其中所述烷氧基基团具有1至10个、优选1至4个碳原子,和
“酰氧基芳基”代表具有1至5个环和4至30个碳原子并带有1至6个酰氧基基团作为环取代基的芳基基团,其中所述酰氧基基团含有1至4个碳原子。
卤素取代基的实例包括氟、氯、溴和碘基团。
通常,式v的金属化合物为金属β-二酮或β-酮酯或其组合。
在“m”的含义中,金属通常选自过渡金属和稀土金属。金属的实例包括如下:具有各种氧化程度的锑、铋、锗、铪、铁、钼、铈、镧、钇、锡、钛、锆和钨,例如锆、铪、铝、钛和锡及其组合。
特别地,金属可选自锑(iii)、铋(iii)、锗、铪(iv)、铁(iii)、钼(v)、铈(iv)、镧(iii)、钇(iii)、锡(iv)、钛(iv)、锆(iv)和钨(v)。
前体ii的具体实例包括但不限于四甲氧基硅烷、四氯硅烷、四乙酰氧基硅烷、四乙氧基硅烷、四正丙氧基硅烷、四异丙氧基硅烷、四正丁氧基硅烷、甲基三甲氧基硅烷、甲基三乙氧基硅烷、甲基三氯硅烷、甲基三乙酰氧基硅烷、甲基三丙氧基硅烷、甲基三丁氧基硅烷、甲基三苯氧基硅烷、甲基三苄氧基硅烷、乙基三甲氧基硅烷、乙基三乙氧基硅烷、苯基三甲氧基硅烷、苯基三氯硅烷、苯基三乙酰氧基硅烷、苯基三乙氧基硅烷、γ-氯丙基三甲氧基硅烷、γ-氯丙基三乙氧基硅烷、γ-氯丙基三乙酰氧基硅烷、3,3,3-三氟丙基三甲氧基硅烷、γ-甲基丙烯酰氧基丙基三甲氧基硅烷、γ-巯基丙基三甲氧基硅烷、γ-巯基丙基三乙氧基硅烷、β-氰基乙基三乙氧基硅烷、氯甲基三甲氧基硅烷、氯甲基三乙氧基硅烷、二甲基二甲氧基硅烷、苯基甲基二甲氧基硅烷、二甲基二乙氧基硅烷、苯基甲基二乙氧基硅烷、γ-氯丙基甲基二甲氧基硅烷、γ-氯丙基甲基二乙氧基硅烷、二甲基二乙酰氧基硅烷、γ-缩水甘油氧基甲基三甲氧基硅烷、缩水甘油氧基甲基三乙氧基硅烷、α-缩水甘油氧基乙基三甲氧基硅烷、α-缩水甘油氧基乙基三乙氧基硅烷、β-缩水甘油氧基乙基三甲氧基硅烷、β-缩水甘油氧基乙基三乙氧基硅烷、α-缩水甘油氧基丙基三甲氧基硅烷、α-缩水甘油氧基丙基三乙氧基硅烷、β-缩水甘油氧基丙基三甲氧基硅烷、β-缩水甘油氧基丙基三乙氧基硅烷、γ-缩水甘油氧基丙基三甲氧基硅烷、γ-缩水甘油氧基丙基三乙氧基硅烷、γ-缩水甘油氧基丙基三丙氧基硅烷、γ-缩水甘油氧基丙基三丁氧基硅烷、γ-缩水甘油氧基丙基三苯氧基硅烷、α-缩水甘油氧基丁基三甲氧基硅烷、α-缩水甘油氧基丁基三乙氧基硅烷、β-缩水甘油氧基丁基三乙氧基硅烷、γ-缩水甘油氧基丁基三甲氧基硅烷、γ-缩水甘油氧基丁基三乙氧基硅烷、δ-缩水甘油氧基丁基三甲氧基硅烷、δ-缩水甘油氧基丁基三乙氧基硅烷、(3,4-环氧环己基)甲基三甲氧基硅烷、(3,4-环氧环己基)甲基三乙氧基硅烷、β-(3,4-环氧环己基)乙基三甲氧基硅烷、β-(3,4-环氧环己基)乙基三乙氧基硅烷、β-(3,4-环氧环己基)乙基三丙氧基硅烷、β-(3,4-环氧环己基)乙基三丁氧基硅烷、β-(3,4-环氧环己基)乙基三苯氧基硅烷、γ-(3,4-环氧环己基)丙基三甲氧基硅烷、γ-(3,4-环氧环己基)丙基三乙氧基硅烷、δ-(3,4-环氧环己基)丁基三甲氧基硅烷、δ-(3,4-环氧环己基)丁基三乙氧基硅烷、缩水甘油氧基甲基甲基二甲氧基硅烷、缩水甘油氧基甲基甲基二乙氧基硅烷、α-缩水甘油氧基乙基甲基二甲氧基硅烷、α-缩水甘油氧基乙基甲基二乙氧基硅烷、β-缩水甘油氧基乙基甲基二甲氧基硅烷、β-缩水甘油氧基乙基乙基二甲氧基硅烷、α-缩水甘油氧基丙基甲基二甲氧基硅烷、α-缩水甘油氧基丙基甲基二乙氧基硅烷、β-缩水甘油氧基丙基甲基二甲氧基硅烷、β-缩水甘油氧基丙基乙基二甲氧基硅烷、γ-缩水甘油氧基丙基甲基二甲氧基硅烷、γ-缩水甘油氧基丙基甲基二乙氧基硅烷、γ-缩水甘油氧基丙基甲基二丙氧基硅烷、γ-缩水甘油氧基丙基甲基二丁氧基硅烷、γ-缩水甘油氧基丙基甲基二苯氧基硅烷、γ-缩水甘油氧基丙基乙基二甲氧基硅烷、γ-缩水甘油氧基丙基乙基二乙氧基硅烷、γ-缩水甘油氧基丙基乙烯基二甲氧基硅烷、γ-缩水甘油氧基丙基乙烯基二乙氧基硅烷和苯基磺酰氨基丙基三乙氧基硅烷。
前体iii的具体实例包括但不限于4-乙酰氧基苯基乙基三氯硅烷、4-乙酰氧基苯基乙基三甲氧基硅烷、4-乙酰氧基苯基乙基三乙氧基硅烷、4--(乙酰氧基苯基乙基)甲基二氯硅烷、4--(乙酰氧基苯基乙基)甲基二甲氧基硅烷、4-(乙酰氧基苯基乙基)甲基二乙氧基硅烷、三乙氧基甲硅烷基丙基氨基甲酸酯、三乙氧基甲硅烷基丙基马来酰胺酸、n-(3-三乙氧基甲硅烷基丙基)-4-羟基丁酰胺、n-(3-三乙氧基甲硅烷基丙基)葡糖酰胺、(3-三乙氧基甲硅烷基)丙基琥珀酸酐、脲基丙基三乙氧基硅烷、脲基丙基三甲氧基硅烷、3-羟基-3,3-双(三氟甲基)丙基三乙氧基硅烷、4-(甲氧基甲氧基)三甲氧基甲硅烷基苯和6-(甲氧基甲氧基)-2-(三甲氧基甲硅烷基)萘。
前体iv的具体实例包括但不限于锑(iii)醇盐如乙醇锑(iii)、铋(iii)醇盐如异丙醇铋(iii)、锗醇盐如四乙氧基锗、铪(iv)醇盐如丁醇铪(iv)、铁(iii)醇盐如乙醇铁(iii)、钼(v)醇盐如乙醇钼(v)、铈(iv)醇盐如异丙醇铈(iv)、镧(iii)醇盐如异丙醇镧(iii)、钇(iii)醇盐如异丙醇钇(iii)、锡(iv)醇盐、烷基和芳基取代的锡(iv)醇盐如四叔丁氧基锡、四异丙氧基锡、钛(iv)醇盐如丁醇钛(iv)和异丙醇钛(iv)、锆(iv)醇盐如丁醇锆(iv)、钨(v)醇盐和钨(vi)醇盐如乙醇钨(v)和乙醇钨(vi)。
在一个实施方案中,制备部分缩合的聚氢基倍半硅氧烷树脂的方法包括使用hteos(hsi(oc2h5)3)或hteos与其他三官能硅烷的混合物,对其进行受控的水解/缩合反应。
在这样的方法中,三官能硅烷通常选自甲基三甲氧基硅烷(mtmos)、甲基三乙氧基硅烷(mteos)、三甲氧基乙基硅烷(etmos)或二乙氧基二甲基硅烷(dmdeos)或三甲氧基苯基硅烷(phtmos)。
在一个实施方案中,将单体溶解在meothf中,meothf用作该合成的溶剂。或者,使用其他溶剂如etothf或meothf与etothf的混合物,或meothf、etothf或r-othf的合成溶剂,其中r由3至12个碳组成。
在一个实施方案中,用摩尔比在0.0001m至1m的范围内的酸溶液获得单体的受控水解。
根据一个实施方案,水解和聚合在完全没有溶剂的情况下进行,或者在有机溶剂中如在醇、酯、酮和醚中进行。
具体的合适溶剂有丙酮、乙基甲基酮、甲醇、乙醇、异丙醇、丁醇、乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸丁酯和四氢呋喃。特别合适的溶剂有酮和醚,特别是当水解存在的过程中通过与硅原子配位而具有一定的稳定化效应时。这样的实例有乙基甲基酮、甲基四氢糠基醚和乙基四氢糠基醚。
通过添加摩尔比在0.0001m至1m的范围内的酸或碱溶液来获得单体的受控水解。
该合成中可使用有机或无机酸。
可使用无机酸如硝酸、硫酸、盐酸、氢碘酸、氢溴酸、氢氟酸、硼酸、高氯酸、碳酸和磷酸。优选地,硝酸或盐酸由于它们的低沸点而使用,这使得产物的纯化简单。
在其他选择中,可使用各种有机酸代替无机酸。有机酸有羧酸、磺酸、醇、硫醇、烯醇和酚组。实例有甲磺酸、乙酸、乙磺酸、甲苯磺酸、甲酸或草酸。
合成中使用的碱可类似地是无机或有机的。典型的无机碱和金属氢氧化物、碳酸盐、碳酸氢盐及其他将产生碱性水溶液的盐。这样的材料的实例有氢氧化钠、氢氧化钾、氢氧化铯、氢氧化钙、碳酸钠和碳酸氢钠。另一方面,有机碱包括较大的一组,包括有机酸的金属盐(如乙酸钠、乙酸钾、丙烯酸钠、甲基丙烯酸钠、苯甲酸钠)、直链、支链或环状烷基胺(如二氨基乙烷、腐胺(purtescine)、尸胺、三乙胺、丁胺、二丁胺、三丁胺、哌啶)、脒和胍(如8-二氮杂双环(5.4.0)十一碳-7-烯、1,1,3,3-四甲基胍、1,5,7-三氮杂双环[4.4.0]-癸-5-烯)、磷氮烷(如p1-t-bu、p2-t-bu、p4-t-bu)和季铵化合物(如四甲基氢氧化铵、四乙基氢氧化铵、四丁基氢氧化铵)。
水解和缩合过程中反应混合物可保持在-30℃至170℃的范围内的温度下。
本领域技术人员知道,较低的反应温度将提供对反应改进的控制,但以长的反应时间为代价,而过高的温度可能会使过程过快而无法适当控制。
在0-100℃的温度下,优选1至48h或2-24h的反应时间。
根据本发明的方法在有机溶剂体系中产生部分交联的、任选地含金属的有机硅氧烷聚合物,所述聚合物相对于聚苯乙烯标准物测得的分子量为约5,00至100,000g/mol,特别是约1,000至50,000g/mol。
本发明的另一个实施方案是,把在其中进行水解和聚合反应的溶剂在聚合反应后换为将通过某种形式的稳定化而给材料提供更好的涂覆性能和产品储存性的溶剂。
稳定化有机溶剂体系例如由有机醚任选地与其他一种或多种共溶剂混合形成。
在一个实施方案中,有机醚为通常包含4至26个碳原子并任选地含有其他官能团如羟基基团的直链、支链或环状醚。
特别合适的实例有五元和六元环状醚,其任选地在环上带有取代基。
其他合适的醚有例如(c1-20)链烷二醇(c1-6)烷基醚。所述链烷二醇烷基醚的实例有丙二醇单甲醚、丙二醇二甲醚、丙二醇正丁醚、二丙二醇单甲醚、二丙二醇二甲醚、二丙二醇正丁醚、三丙二醇单甲醚及其混合物。
本发明的醚的特别优选的实例有甲基四氢糠基醚、四氢糠醇、丙二醇正丙醚、二丙二醇二甲醚、丙二醇正甲醚、丙二醇正乙醚及其混合物。
稳定化溶剂体系仅由包含上述种类的醚的溶剂组成,或由这样的醚与水解反应的典型反应介质或其他溶剂如丙二醇单甲醚乙酸酯的混合物组成。在这样的情况下,醚的比例为溶剂总量的约10至90重量%,特别是约20至80重量%。
由选定的溶剂和树脂材料组成的辐射敏感制剂的固体含量在0.1%至不超过50%的范围内,优选在0.5%至10%的范围内。
在一个实施方案中,在缩合的聚氢基倍半硅氧烷树脂中,将制剂与不同的光酸产生剂和光碱产生剂及敏化剂组合以用作euvl负性光刻胶。
涂覆过程中,使用固体含量(或聚合物含量)来调节所得的膜厚度。
为了改善涂层均匀性方面的涂层性能,可使用不同的表面活性剂如有机硅或含氟表面活性剂来降低含硅烷醇的聚氢基倍半硅氧烷制剂涂层的表面张力。如果需要,使用这样的表面活性剂可改善涂层品质。与含硅烷醇的聚氢基倍半硅氧烷的量相比,表面活性剂的量在0.001质量%至不超过10质量%的范围内。
组合物还可含有盐,特别是无机盐。
在一个实施方案中,硅氧烷预聚物溶液的粘度为约0.5厘泊(cp)至约150cp。有机液体的闪点可为至少10℃并且在20℃下的蒸气压小于约10kpa。
如上文所讨论,本文描述的辐射敏感的含金属和硅烷醇的聚氢基倍半硅氧烷组合物可用于在器件内直接形成所需的无机材料结构和/或用作为有机光刻胶的替代品的可辐射图案化的无机光刻胶。任一情况下都可利用显著的加工改进,并还可改进经图案化材料的结构。
与hsq树脂相比,hsq树脂中硅烷醇部分的存在大大改善涂层的图案形成能力。
通常在非水溶剂中的含金属和硅烷醇的聚氢基倍半硅氧烷溶液将提供高分辨率图案化,其中所述溶液对于具有良好辐射敏感性的可辐射图案化的涂层的形成具有高度稳定性。
用含金属和硅烷醇的聚氢基倍半硅氧烷溶液形成的涂层为经图案化氧化硅涂层的形成提供优异的直接图案化。在感兴趣的实施方案中,暴露于辐射把经辐照的涂层材料转化为抵抗用显影剂组合物去除的材料,使得其可被选择性地去除。
因此,涂层可被负性图案化。选择性去除至少一部分涂层材料可留下图案,其中涂层的一些区域已被去除而暴露出下面的基材。在辐照后涂层的显影之后,经图案化的氧化硅材料可用来促进器件形成中以优异的图案分辨率加工。涂层材料可设计为对选定的辐射如极紫外光、紫外光和/或电子束敏感。此外,可将前体溶液配制为稳定的,具有适宜的货架寿命以便商业销售。
所述辐射敏感涂层材料可用作负性辐射图案化涂层。在负性图案化中,暴露于辐射将把经辐照的涂层材料转化为相对于未经辐照的涂层材料更抵抗用显影剂组合物去除的材料。选择性去除至少一部分涂层材料将留下图案,其中一些区域已被去除而暴露出下面的基材。
集成电子器件等的形成通常涉及材料的图案化,以在结构内形成单独的元件或部件。该图案化可涉及覆盖堆叠层的选定部分的不同组合物,堆叠层垂直和/或水平地彼此接合以引起期望的功能。
各种材料可包括半导体(其可具有选定的掺杂剂)、电介质、电导体和/或其他类型的材料。为了形成高分辨率图案,可使用辐射敏感的有机组合物来引入图案,该组合物可被称为光刻胶,因为该组合物的一些部分被加工成抵抗显影/蚀刻,使得可使用选择性的材料去除来引入选定的图案。
可用随同选定图案或图案负片的辐射来将光刻胶暴露,并形成具有显影剂抵抗性区域和显影剂可溶解区域的图案或潜像。
在euv或类似的辐照方法如电子束过程中,sih物种将反应形成中间体——反应性硅物种(图4)。该中间体需要大气水分或增大的剂量来进行后续反应。事实上,针对euvl提出的许多金属光刻胶都表现出类似的挑战,其中反应性的中间体需要大气水分来进行使材料交联产生负性光刻胶涂层材料的后续反应。因此,我们的发现是在膜中引入适量的硅烷醇基团,这些硅烷醇基团将与由euv从sih生成的中间体反应性硅物种直接反应。
结果,与迄今文献中描述的那些相比,含硅烷醇的聚氢基倍半硅氧烷在euv光刻中展现出更高的敏感性。
基于上文,本发明的技术提供了光刻胶膜的形成,做法是:
向半导体基材上施加如上所述的组合物以形成光刻胶下层膜,并烘烤所述组合物。
在一个实施方案中,提供了一种制造半导体器件的方法,其包括:
-向半导体基材上施加光刻胶下层膜或若干下层膜并烘烤该组合物,以形成一个或多个光刻胶下层膜;
-向一个或多个光刻胶下层膜上施加根据权利要求1的组合物作为光刻胶,以形成光刻胶膜;
-将光刻胶膜暴露于光;
-在光暴露之后,对光刻胶膜显影以形成光刻胶图案;
-使用光刻胶图案蚀刻光刻胶下层膜;和
-使用如此图案化的光刻胶膜和如此图案化的光刻胶下层膜制造半导体基材。
在一个实施方案中,提供了一种制造半导体器件的方法,其包括:
-在半导体基材上形成有机下层膜;
-向有机下层膜上施加用于形成光刻胶膜的组合物并烘烤该组合物,以形成光刻胶膜;
-将光刻胶膜暴露于光;
-在光暴露之后,对光刻胶膜显影以形成光刻胶图案;
-使用光刻胶图案蚀刻光刻胶下层膜;
-使用如此图案化的光刻胶下层膜蚀刻有机下层膜;和
-使用如此图案化的有机下层膜制造半导体基材。
所述技术还提供了一种制造半导体器件的方法,所述方法包括:
-向半导体基材上施加光刻胶下层膜或若干下层膜并烘烤该组合物,以形成一个或多个光刻胶下层膜;
-向一个或多个光刻胶下层膜上施加根据权利要求1的组合物作为光刻胶,以形成光刻胶膜;
-将光刻胶膜暴露于光;
-在光暴露之后,对光刻胶膜显影以形成光刻胶图案;
-使用光刻胶图案蚀刻光刻胶下层膜;和
-使用如此图案化的光刻胶膜和如此图案化的光刻胶下层膜制造半导体基材。
此外,一种制造半导体器件的方法包括以下步骤:
-在半导体基材上形成有机下层膜;
-向有机下层膜上施加用于形成光刻胶膜的组合物并烘烤该组合物,以形成光刻胶膜;
-将光刻胶膜暴露于光;
-在光暴露之后,对光刻胶膜显影以形成光刻胶图案;
-使用光刻胶图案蚀刻光刻胶下层膜;
-使用如此图案化的光刻胶下层膜蚀刻有机下层膜;和
-使用如此图案化的有机下层膜制造半导体基材。
涂料的组成也对euvl光刻胶材料所需的其他属性有着显著影响。出于实际原因,必须有足够长的涂覆后延迟(pcd)。与在辐照过程中使用环境或氮气覆盖层的先前使用的光刻步进机和扫描仪不同,euvl扫描仪辐照在真空中进行。从接合的或独立的光刻胶轨道转移通过euvl扫描仪中的各个腔室可能要花很长时间,因此,在这些步骤中,涂料组合物应在气体抽空腔室中保持稳定。
因此,材料应表现出足够的pcd稳定性,这意味着可使用溶剂或显影剂轻松地去除涂层而不在基材上留下任何残留物。令人惊奇的是,已发现膜中的si-h和si-oh之间的相关性对pcd稳定性有着影响。如果图案化过程中的固化时间过长,则膜中过高的si-oh含量会使材料过早交联,从而使未暴露的区域的显影变得不可能。ftir是一种评估si-h与si-oh比率的快速简便的工具,并且我们观察到,在某些比率下,材料会产生高分辨率图案、低的ler和良好的pcd稳定性。
更具体地说,已记录了ftir光谱以验证合成的含硅烷醇的聚氢基倍半硅氧烷的化学结构,其在图7中呈现。
从该图中可以看出,含硅烷醇的聚氢基倍半硅氧烷显示出强且宽的si-oh峰,范围从3200至3700cm-1,这归因于氢键合的硅烷醇网络。另外,si-h官能作为以约2248cm-1为中心的尖峰观察到。因此,优选地,si-h与si-oh峰高比的比率应至少为或优选大于4:1,但至多为或小于100:1,或更优选大于3:1但小于50:1。
本发明涉及所描述的组合物在半导体基材上的图案形成中的用途。
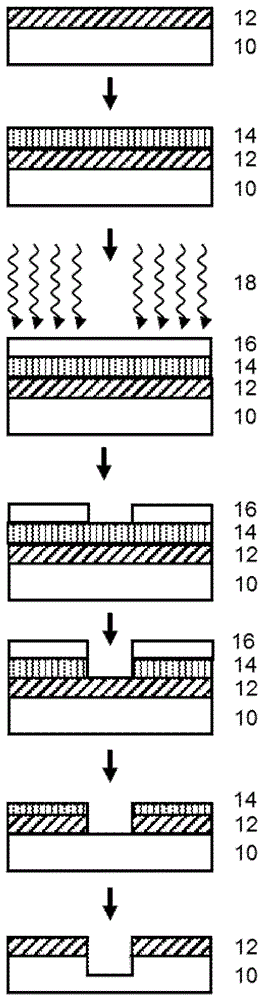
接下来转到附图,图1示出了当前使用的典型工艺,其中在基材表面10上涂覆有机下层12,然后涂覆和固化无机中间层14,该无机中间层14通常由高含硅材料组成。在此之上涂覆光刻胶16。
在使用193nm波长进行公知的辐照18之后,形成图案并显影。这样的层的组合在文献中经常被描述为“三层”堆叠,这指的是层数。随后可应用公知并使用的图案转移蚀刻工艺,来将形成在光刻胶上的图案转移到基材(图1)。
值得指出的是,光刻中使用的各个层的堆叠可高于3层。
层的另一常用组合是四层堆叠(参见图2),其中首先施加有机下层(或基材)12,然后涂覆和固化通常由高含硅材料组成的无机中间层14。在此之上施加有机底部抗反射层18,然后施加光刻胶16,从而完成图2的四层堆叠。
熟悉光刻胶图案化中所用下层材料领域的技术人员知道,所有描述的下层不仅可涂覆和固化,而且可通过气相沉积工艺如化学气相沉积(cvd)、原子层沉积(ald)及类似方法来施加。
对于熟悉本领域的技术人员而言,相同的程序可应用于13.5nm的euvl。
然而,由于吸收挑战、光酸和光子辐照二者的随机分布,因此工艺中使用的常见car面临着严峻的挑战,特别是与ler和lwr有关。
因此,大量的兴趣已转向无机光刻胶的使用,无机光刻胶有望简化堆叠,其中既减少层数又减少ler和lwr挑战。在大多数情况下,无机光刻胶由一种或多种呈易水解或已水解的金属氧化物前体形式的化合物的金属氧化物和氢倍半硅氧烷组成。这些无机光刻胶的优点在于:金属氧化物光刻胶产生前所未有的euvl剂量敏感性,而氢倍半硅氧烷聚合物则赋予优异的分辨率及低的ler/lwr和非常弱的euvl敏感性。
因此,两种方法都具有使其难以在euvl图案化工艺中使用的属性。
本发明使用含金属-硅烷醇的硅氧烷作为优选的方法来实现堆叠简化和图案化工艺中使用的金属的减少两者,参见图3。
在本发明的一个实施方案中,在euv中对基材20涂覆呈旋涂碳或无定形碳层形式的主要有机的涂层22。根据本发明的可图案化薄膜22基于的是含金属和硅烷醇的聚氢基倍半硅氧烷树脂24。
与文献中的hsq树脂相比,该涂层易于通过euvl图案化,具有高敏感性,且与文献中所述的car和无机树脂相比,ler/lwr显著较低。
特别地,本发明提供了在不存在金属的情况下用双层结构高分辨率图案化的措施,这在无显著产量损失地生产功能全面的晶体管管芯中可能是显著的优势。众所周知,含硅材料对euv的吸收相对较低。另一方面,已经确定euv会从图4的hsq树脂中的si-h部分生成不稳定的硅烷醇。
基于本文给出的实施例,很显然,与文献中描述的方法相比,将促进硅烷醇缩合或sih氧化的也含金属的添加剂会对获得较低的敏感性有利。
所述改进可通过以下获得:
1.与仅含硅的树脂相比提高树脂的敏感性,或
2.与不含金属的制剂相比加速缩合过程
本发明通过在集成电路制造中使用的表面20上沉积富碳下层22来实现。该下层通过气相工艺如cvd或通过旋涂并固化来沉积。然后施加按照本发明的组合物24,产生主要是硅并富含金属的氧化物涂层,该涂层可通过新型图案化技术如euvl直接图案化。
在通过旋涂或其他合适的液体沉积方法施加后,应用预烘烤步骤以去除涂层材料中包含的溶剂和挥发性化合物。为了避免制造设备中的污染,这是必要的。
预烘烤可根据溶剂和挥发性组分的沸腾温度在不同的温度和时间下进行。通常,预烘烤可在60℃至120℃下进行30秒到至多不超过30分钟的时间。
在干燥或预烘烤步骤之后,通过任选使用的掩模将涂层暴露于euv光、电子束或类似的暴露方法(26)来在基材上生成图案。生成图案所需的剂量最小为10mj/cm2,最大为200mj/cm2。
如前所述,由于各种原因,包括工业应用中的合理产量和解决rls折衷等,优选较低的剂量。
在euv暴露之后任选进行暴露后烘烤。该步骤可改善经暴露区域的交联,这将导致光刻胶的更高对比度。暴露后烘烤可在80℃到至多不超过150℃的温度下进行30秒到至多不超过30分钟的时间。
进行显影以去除未经暴露区域,从而获得所设计的图案。在显影步骤中,光刻胶24的未经暴露区域将溶解在显影剂中。
可有效地使用该行业中常用的基于碱如四甲基氢氧化铵、氢氧化钾、氢氧化钠的具有0.1%至25%的不同质量比的碱水溶液显影剂。显影通过使用喷涂或搅炼显影剂溶液法来施加显影剂的公知方法进行。或者,可将基材浸没在由显影剂组成的浴中。
可任选地进行固化步骤以使经暴露并显影的图案致密化而获得进一步交联的材料,或通过让材料表现出一定的回流的工艺来改变光刻胶图案的形状。与在无固化步骤的情况下制得的材料相比,该步骤可产生抗蚀刻性更高的材料。固化可在120℃至400℃下进行2分钟到至多不超过30分钟。可通过加热放置在热板上或烘箱中的涂层来进行固化。
可在辐射敏感的涂料制剂中添加不同的光酸产生剂(pag)、光碱产生剂(pbg)和敏化剂。这样的添加剂是car中的关键组分,并且其随机分布是光刻胶制剂中ler挑战的常见根本原因。
通常,光酸产生剂和光碱产生剂可选自非离子型酸产生剂、和非离子碱类型或离子类型的光碱产生剂。光碱产生剂可与敏化剂结合使用。
令人惊奇的是,未观察到ler随pag而增加,并且pbg既没有减少所需的剂量也没有因暴露期间的随机分布挑战和散粒噪声而增加ler。不过,可向涂料制剂中添加pag或pbg来增强辐射敏感涂料的酸或碱催化缩合,以促进膜的经暴露区域的缩合,从而产生潜在地更致密的图案。
pag、pbg和敏化剂可利用的种类繁多。仅举几个例子来说,pag包括离子型光酸产生剂如芳基重氮鎓盐、二芳基卤鎓盐、三芳基硫鎓盐等。与含硅烷醇的聚氢基倍半硅氧烷的量相比,离子型光酸产生剂的量在0.001质量%至不超过10质量%的范围内。
不同的光酸产生剂包括非离子型酸产生剂如硝基苄基酯、砜、磷酸盐、n-羟基酰亚胺磺酸盐、苯酚的磺酸酯、重氮萘醌、含卤素的化合物、亚氨基砜。与含硅烷醇的聚氢基倍半硅氧烷的量相比,非离子型光酸产生剂的量在0.001质量%至不超过10质量%的范围内。
光碱产生剂可以是co(iii)-胺,并可添加烷基胺盐、o-酰基肟、苄氧基羰基衍生物、甲酰胺。与含硅烷醇的聚氢基倍半硅氧烷的量相比,光碱产生剂的量在0.001质量%至不超过10质量%的范围内。
我们通过euv光刻证实了这一理论并在图8中呈现了图案的扫描电子显微镜(sem)图像。
如实施例1中所提及,使用具有2%固体含量的聚合物1来制备膜,用euv辐照,最后用2.38%的tmah显影以获得图案化的图像。如图8中可见,已分别在例如65.4mj/cm2、86.5mj/cm2和82mj/cm2的剂量下获得了22nm、30nm和50nm的不同半节距的高分辨率sem图像。
此外,我们使用电子束光刻进一步证实了我们的理论。我们使用了不同含量即1%、2%和3%的实施例1中制得的聚合物下的三种制剂。图9中的sem图像显示,在600µc/cm2、400µc/cm2和200µc/cm2下已获得50nm的图案线。如图可见,对于具有更高固体含量的样品观察到更高的敏感性。这是因为膜中硅烷醇的含量较高,从而增加了交联的密度和反应性。
然而,如样品3和5中所见,当sih与sioh的比率小于4时,膜中硅烷醇的量变得太大。高的硅烷醇含量导致材料不表现出适合于euv的pcd(图5-6和表1),因为涂层相对较快地自交联而不再可能用显影剂去除。另一方面,如果sih与sioh的比率大于100,则膜的敏感性将显著降低并因此需要大量的euv剂量。
因此,我们广泛研究了合成途径来调节树脂中sih/sioh比率的量,做法是使用不同的水解时间和/或在不同的溶剂中和/或使用不同的单体比率进行反应。ftir结果呈现在图7中。
在实际的euv光刻应用中,在软烘烤后euv光刻胶膜的涂覆后延迟(pcd)需要至少为1小时。因此,我们对我们开发的聚合物和制剂广泛地进行了pcd测试。程序汇总在表1中。
x为对其中pcd对于euv光刻来说良好的样品给出的标记,而y是实施例中仅在80℃的低软烘烤温度下进行1min才良好,z则在高到足以完全去除膜中的溶剂的任何软烘烤温度下都不可接受。
为了进一步改善敏感性,可向待用作光刻胶的聚合物溶液中添加金属氧化物纳米颗粒。可添加不同的金属氧化物纳米颗粒如氧化钛纳米颗粒、杂化钛铝氧化物纳米颗粒、或铪纳米颗粒。
图10a、10c和10d中呈现了使用电子束光刻的结果。纳米颗粒的添加可经由对硅烷醇和硅烷醇与si-h的交联反应的催化活性来增加涂膜的敏感性。另一个好处在于,膜中包含的金属氧化物可改善微电子制造过程中等离子体蚀刻中的抗蚀刻性或蚀刻选择性。另外,已发现向聚合物1中添加溶解性增强剂如仅2.5重量%的hfipteos显著增加了线边缘粗糙度(图8c中的sem图像)。
基于上述内容,提供了以下实施方案:
一种前体溶液,其包含有机液体和与具有硅-碳键和硅-氢的硅-氧网络或硅-氧-金属中的si-h或si-r相比约0.001m至约1m的硅烷醇,并且所述前体溶液的粘度为约0.5厘泊(cp)至约150cp。有机液体的闪点可为至少10℃并且在20℃下的蒸气压小于约10kpa。
一种用特定波长的光辐射将经含金属和硅烷醇的聚氢基倍半硅氧烷涂覆基材图案化的方法,所述方法包括以下步骤:沿选定的图案辐照经涂覆基材以形成具有经辐照涂层的区域和有未经辐照涂层的区域的经辐照结构,和选择性地显影所述经辐照结构以去除实质部分的未经辐照涂层来形成经图案化基材。
一种用特定波长的光辐射将经含金属和硅烷醇的聚氢基倍半硅氧烷涂覆基材图案化的方法,其中所述波长为13.5nm或更小。
一种经涂覆基材,其包含平均厚度不超过约5微米并且在沿涂层的任何点处相对于平均值的厚度变化不超过约50%的辐射敏感涂层,所述涂层包含具有硅-碳键和硅-氢和/或具有硅烷醇键的硅-氧网络或硅-氧-金属网络。
一种经图案化基材,其包含具有表面和在沿所述表面的选定区域处存在而在沿所述表面的其他区域处不存在的第一涂层的基材。通常,所述第一涂层包含具有硅-碳键和硅-氢和/或具有硅烷醇键的硅-氧网络或硅-氧-金属网络。或者,所述第一涂层可溶于至少一些有机液体中,或所述第一涂层可溶于碱水溶液中。
一种经图案化基材,包含具有表面和在沿所述表面的选定区域处存在而在沿所述表面的其他区域处不存在的第一涂层的基材。通常,所述第一涂层包含具有硅-碳键和硅-氢和/或具有硅烷醇键的硅-氧网络或硅-氧-金属网络,并且在氧等离子体或灰化过程中的蚀刻速率低于50nm/min。
以下非限制性实施例示意了实施方案。
实施例
凝胶渗透色谱数据在配备有串联连接的shodexkf柱(kf-g;kf-803l;kf-802;kf-801)的agilent1260infinitylc上收集。检测器和柱温保持在40℃下。thf洗脱液的流速为1.0ml/min。
使用brukervertex70用傅立叶变换红外光谱(ft-ir)来研究化学结构。使用膜样品来进行分析。
涂覆后延迟(pcd)测试:用pgmea稀释来自实施例的聚合物溶液以获得具有2%固体含量的溶液。通过在硅晶片上以1500rpm的速度旋涂该聚合物溶液30秒来制备膜样品。然后在热板上用80℃至150℃的范围内的温度进行1分钟的软烘烤。将膜在室温下保存1h,然后用2.38%的tmah显影,最后用去离子水重复洗涤三次并用氮气干燥。
测量显影前后的膜厚度以评价pcd。如果在软烘烤后膜完全被tmah显影剂去除,则其在pcd方面良好。结果汇总在表1中。
在vistec工具上进行电子束光刻。经由旋涂机以1500rpm的旋转速度将2%固体的制剂在硅晶片上旋涂30秒。然后在热板上于80℃的温度下进行1分钟的软烘烤。电子束剂量在100kv和1na电流下从100到900µc/cm2不等。通过将膜在2.38%的tmah溶液中浸没1分钟来进行显影步骤。然后将膜用去离子水重复洗涤三次并最后用氮气干燥。
euv光刻:经由旋涂机以1500rpm的旋转速度将2%固体含量的聚合物制剂在硅晶片上旋涂30秒。然后在热板上于80℃的温度下进行1分钟的软烘烤。软烘烤后的膜样品在paulscherrer研究所(瑞士菲利根)用xil-iieuv工具辐照。剂量从2到150mj/cm2不等。通过将膜在2.38%的tmah溶液中浸没1分钟来进行显影步骤。然后将膜用去离子水重复洗涤三次并最后用氮气干燥。
实施例1
在一个烧瓶中,将121.55克的量的三乙氧基硅烷(hteos)溶解在234.60克甲基四氢糠基醚(meothf)中。将58.65克meothf、25.62克去离子水(diw)和11.71克0.01摩尔硝酸的混合物混合,并分三等份以1.5小时加到上述的hteos溶液中。加完后,让反应继续混合另外20小时。进行蒸发以获得21-25%的固体含量。加入四倍于聚合物重量的甲基乙基酮(mek)。用0.2微米的过滤器过滤所获得的聚合物溶液。凝胶渗透色谱数据显示,mw为5001,mn为2239,多分散性为2.23。
实施例1的聚合物的蚀刻数据在表1中给出。所使用的旋涂碳(soc)为pibond制造的soc300。
表1:实施例1中制得的聚合物的蚀刻数据。
实施例2
在一个烧瓶中,将121.55克的量的hteos溶解在234.60克乙基四氢糠基醚(etothf)中。将58.65克etothf、25.62克diw和11.71克0.01摩尔硝酸的混合物混合,并分三等份以1.5小时加到上述的hteos溶液中。加完后,让反应继续混合另外20小时。进行蒸发以获得21-25%的固体含量。加入四倍于聚合物重量的mek。用0.2微米的过滤器过滤所获得的聚合物溶液。凝胶渗透色谱数据显示,mw为7396,mn为2903,多分散性为2.54。
实施例3
在一个烧瓶中,将5.510克的量的hteos和0.567克四甲氧基硅烷(tmos)(摩尔比为90:10)溶解在11.730克meothf中。将2.932克meothf、1.290克diw和0.590克0.01摩尔硝酸的混合物混合,并分三等份以1.5小时加到上述的hteos和tmos溶液中。加完后,让反应继续混合另外20小时。进行蒸发以获得21-25%的固体含量。加入四倍于聚合物重量的mek。用0.2微米的过滤器过滤所获得的聚合物溶液。凝胶渗透色谱数据显示,mw为4497,mn为2059,多分散性为2.18。
实施例4
在一个烧瓶中,将5.223克的量的hteos和0.854克tmos(摩尔比为85:15)溶解在11.729克meothf中。将2.930克meothf、1.290克diw和0.590克0.01摩尔硝酸的混合物混合,并分三等份以1.5小时加到上述的hteos和tmos溶液中。加完后,让反应继续混合另外20小时。进行蒸发以获得21-25%的固体含量。加入四倍于聚合物重量的mek。用0.2微米的过滤器过滤所获得的聚合物溶液。凝胶渗透色谱数据显示,mw为2022,mn为1191,多分散性为1.69。
实施例5
在一个烧瓶中,将3.154克的量的hteos和2.293克tmos(摩尔比为50:50)溶解在11.729克meothf中。将2.930克meothf、1.330克diw和0.610克0.01摩尔硝酸的混合物混合,并分三等份以1.5小时加到上述的hteos和tmos溶液中。加完后,让反应继续混合另外20小时。进行蒸发以获得21-25%的固体含量。加入四倍于聚合物重量的mek。用0.2微米的过滤器过滤所获得的聚合物溶液。凝胶渗透色谱数据显示,mw为3359,mn为1669,多分散性为2.01。
对比例1
在一个烧瓶中,将60.77克的量的hteos溶解在117.25克乙醇中。将29.37克乙醇、6.40克diw和2.93克0.01摩尔硝酸的混合物混合,并分三等份以1.5小时加到上述的hteos溶液中。加完后,让反应继续混合另外20小时。通过加入250克pgmea进行溶剂交换,并进行溶剂蒸发以获得21-25%的固体含量。加入四倍于聚合物重量的mek。用0.2微米的过滤器过滤所获得的聚合物溶液。凝胶渗透色谱数据显示,mw为16598,mn为8432,多分散性为1.97。
对比例2
在一个烧瓶中,将60.77克的量的hteos溶解在117.25克乙醇中。将146.60克乙醇、6.40克diw和2.93克0.01摩尔硝酸的混合物混合,并分三等份以1.5小时加到上述的hteos溶液中。加完后,让反应继续混合另外20小时。通过加入300克1-buoh进行溶剂交换,并进行溶剂蒸发以获得21-25%的固体含量。加入四倍于聚合物重量的甲基乙基酮(mek)。用0.2微米的过滤器过滤所获得的聚合物溶液。凝胶渗透色谱数据显示,mw为3188,mn为1967,多分散性为1.62。
聚合物测试
用gpc测定实施例14至18的聚合物的分子量,并与对比例1和2的材料比较。然后通过以1500rpm的旋转速度旋涂2%的聚合物溶液30秒,来从各种聚合物制备膜样品。然后用80℃至150℃的范围内的温度进行30秒至2分钟的软烘烤。
表2中给出了材料的gpc数据和涂覆后延迟数据。
表2:用不同的组合物产生的材料的gpc数据和涂覆后延迟。
图例(对于pcd结果):
x=对于在80℃/1h和150℃/1h下烘烤后至少1h的pcd良好,
y=在80℃/1h后良好但在150℃/1h下不良好,和
z=在80℃/1h和150℃/1h下均差
注:对于euvl,pcd时间必须至少为1小时,以便可安全地进行暴露。
结果评价:实施例1展示了具有低ler的高分辨率图案化(图5)。与用较低硅烷醇含量制备的类似材料(对比例)相比,实施例1类似地显示出(图6)对辐射的高敏感性。另一方面,太高的硅烷醇含量导致材料具有不良的pcd性能(实施例3-5)。因此,优选地,si-h与si-oh峰高比(图7)的比率应大于4但小于100,或更优选大于3但小于50。
实施例6
用hteos:hfipteos=99:1的摩尔比制备聚合物溶液
在一个烧瓶中,将5.941克的量的hteos和0.136克3-羟基-3,3-双(三氟-甲基)丙基三乙氧基硅烷(hfipteos)(摩尔比为99:1)溶解在11.729克meothf中。将2.930克etothf、1.300克diw和0.590克0.01摩尔硝酸的混合物混合,并分三等份以1.5小时加到上述的hteos和hfipteos溶液中。加完后,让反应继续混合另外20小时。进行蒸发以获得21-25%的固体含量。加入四倍于聚合物重量的mek。用0.2微米的过滤器过滤所获得的聚合物溶液。凝胶渗透色谱数据显示,mw为3970,mn为2023,多分散性为1.96。
实施例7
hteos:hfipteos=97.5:2.5的摩尔比
在一个烧瓶中,将5.744克的量的hteos和0.334克hfipteos(摩尔比为97.5:2.5)溶解在11.729克meothf中。将2.930克etothf、1.290克diw和0.590克0.01摩尔硝酸的混合物混合,并分三等份以1.5小时加到上述的hteos和hfipteos溶液中。加完后,让反应继续混合另外20小时。进行蒸发以获得21-25%的固体含量。加入四倍于聚合物重量的mek。用0.2微米的过滤器过滤所获得的聚合物溶液。凝胶渗透色谱数据显示,mw为3519,mn为1766,多分散性为1.99。
实施例8
hteos:hfipteos=95:5的摩尔比
在一个烧瓶中,将5.430克的量的hteos和0.648克hfipteos(摩尔比为95:5)溶解在11.729克meothf中。将2.930克etothf、1.290克diw和0.590克0.01摩尔硝酸的混合物混合,并分三等份以1.5小时加到上述的hteos和hfipteos溶液中。加完后,让反应继续混合另外20小时。进行蒸发以获得21-25%的固体含量。加入四倍于聚合物重量的mek。用0.2微米的过滤器过滤所获得的聚合物溶液。凝胶渗透色谱数据显示,mw为3069,mn为1569,多分散性为1.95。
实施例9
hteos:hfipteos=90:10的摩尔比
在一个烧瓶中,将4.855克的量的hteos和1.223克hfipteos(摩尔比为90:10)溶解在11.729克meothf中。将2.930克etothf、1.290克diw和0.590克0.01摩尔硝酸的混合物混合,并分三等份以1.5小时加到上述的hteos和hfipteos溶液中。加完后,让反应继续混合另外20小时。进行蒸发以获得21-25%的固体含量。加入四倍于聚合物重量的mek。用0.2微米的过滤器过滤所获得的聚合物溶液。凝胶渗透色谱数据显示,mw为2661,mn为1524,多分散性为1.74。
实施例10
聚合物1+hfipteos=97.5:2.5摩尔
用pgmea稀释实施例1中制得的聚合物以获得2%固体含量的溶液。以聚合物1:hfipteos=97.5:2.5摩尔比的摩尔比加入一定量的hfipteos。将两个溶液在室温下混合30分钟。然后将最终溶液用0.2微米的过滤器过滤。
聚合物测试
用gpc测定实施例1和6至9的聚合物的分子量。通过以1500rpm的旋转速度旋涂2%的聚合物溶液30秒来制备各种聚合物的膜样品。然后用80℃至150℃的范围内的温度进行30秒至2分钟的软烘烤。
表3中给出了材料的gpc数据和涂覆后延迟数据。
表3:用不同的组合物产生的材料的gpc数据和涂覆后延迟。
图例(对于pcd结果):
x=对于在80℃/1h和150℃/1h下烘烤后至少1h的pcd良好,
y=在80℃/1h后良好但在150℃/1h下不良好,和
z=在80℃/1h和150℃/1h下均差
注:对于euvl,pcd时间必须至少为1小时,以便可安全地进行暴露。
结果评价。实施例6-9证实了官能团增强在显影剂中的溶解性的作用。官能团的添加还降低了获得如图8中所示的图案所需的剂量。另外,这样的官能团的使用允许使用较低浓度的显影剂溶液,这将降低工艺成本并减少环境和健康问题。实施例10显示,树脂组合物可通过将前体分开或同时水解来获得(实施例6-9)。
实施例11
hteos:mteos=90:10的摩尔比
在一个烧瓶中,将5.423克的量的hteos和0.654克甲基三乙氧基硅烷(mteos)(摩尔比为90:10)溶解在11.729克meothf中。将2.930克etothf、1.290克diw和0.590克0.01摩尔硝酸的混合物混合,并分三等份以1.5小时加到上述的hteos和mteos溶液中。加完后,让反应继续混合另外20小时。进行蒸发以获得21-25%的固体含量。加入四倍于聚合物重量的mek。用0.2微米的过滤器过滤所获得的聚合物溶液。凝胶渗透色谱数据显示,mw为2525,mn为1277,多分散性为1.97。
实施例12
hteos:dmdeos=90:10的摩尔比
在一个烧瓶中,将5.524克的量的hteos和0.554克二甲基二乙氧基硅烷(dmdeos)(摩尔比为90:10)溶解在11.729克meothf中。将2.930克etothf、1.290克diw和0.590克0.01摩尔硝酸的混合物混合,并分三等份以1.5小时加到上述的hteos和dmdeos溶液中。加完后,让反应继续混合另外20小时。进行蒸发以获得21-25%的固体含量。加入四倍于聚合物重量的mek。用0.2微米的过滤器过滤所获得的聚合物溶液。凝胶渗透色谱数据显示,mw为2207,mn为1148,多分散性为1.92。
实施例13
hteos:phtmos=90:10的摩尔比
在一个烧瓶中,将5.359克的量的hteos和0.719克phtmos(摩尔比为90:10)溶解在11.729克meothf中。将2.930克etothf、1.250克diw和0.570克0.01摩尔硝酸的混合物混合,并分三等份以1.5小时加到上述的hteos和phtmos溶液中。加完后,让反应继续混合另外20小时。进行蒸发以获得21-25%的固体含量。加入四倍于聚合物重量的甲基乙基酮(mek)。用0.2微米的过滤器过滤所获得的聚合物溶液。凝胶渗透色谱数据显示,mw为1930,mn为746,多分散性为2.59。
结果评价。实施例11-13证实了可用来与hteos或其他含氢前体共聚的几种其他单体的作用。
实施例14
聚合物1+氧化钛=98:2(重量%)
通过将异丙醇钛(iv)(tiip,15.887克)溶解在异丙醇(200g)中来合成氧化钛纳米颗粒。分别制备硝酸0.01m(5.033克)、乙酸(3.357克)和ipa(200g)水溶液的混合物并逐滴加到tiip溶液中。反应在室温下进行6h,然后向反应混合物中加入pgme(500克)并经由旋转蒸发器从溶液去除ipa。继续蒸发溶剂以获得2重量%固体的溶液。通过0.2微米的过滤器过滤溶液后最终获得产物。然后,用pgmea稀释实施例1中制得的聚合物以获得2%固体含量的溶液。以聚合物1:tiox=98:2(重量%)的质量比加入一定量的氧化钛纳米颗粒。将两个溶液在室温下混合30分钟。然后将溶液用0.2微米的过滤器过滤。
实施例15
聚合物1+氧化钛=95:5(重量%)
制备样品的步骤与实施例17相似,不同的是添加的氧化钛的量为5重量%。
实施例16
聚合物1+氧化钛=90:10(重量%)
制备样品的步骤与实施例15中相似,不同的是添加的氧化钛的量为10重量%。
实施例17
聚合物1+氧化钛铝=97:3(重量%)
用pgmea稀释实施例1中制得的聚合物以获得2%固体含量的溶液。以聚合物1:氧化钛铝=97:3的质量比加入一定量的氧化钛铝纳米颗粒。在室温下混合两个溶液30分钟。然后将溶液用0.2微米的过滤器过滤。
实施例18
聚合物1+氧化钛铝=90:10(重量%)
氧化钛铝纳米颗粒的合成通过tiip和异丙醇铝(aloipr)的受控水解/缩合进行。在配备有磁棒的圆底烧瓶中,将tiip(27.831克)和aloipr(5克)的混合物溶解在ipa(320克)溶剂中。ti∶al的摩尔比为8∶2。在另一个烧瓶中,将8.377克水、7.350克乙酸和200克ipa的混合物混合。将第二个烧瓶中的溶液逐滴加到单体溶液中。加完后,向反应混合物中加入200克pgme并在室温下继续搅拌16小时。然后蒸发ipa和水以得到2%固体的溶液。通过0.2微米的过滤器过滤溶液后最终取得反应产物。
样品制备步骤与实施例17中相似,不同的是添加的氧化钛铝的量为10重量%。
实施例19
聚合物1+氧化铪纳米颗粒=90:10(重量%)
氧化铪纳米颗粒合成:通过正丁醇铪(iv)的受控水解/缩合来合成纳米颗粒。将20克的量的正丁醇铪(iv)(95%)溶解在正丁醇溶剂(800克)中。以铪:maa=1:1.5的摩尔比加入一定量的甲基丙烯酸(maa,5.26克)并混合1小时。在室温下通过向反应介质中逐滴加入水(2.91克)和n-buoh(600克)的混合物来进行水解步骤。加完后,让反应继续20小时。然后蒸发正丁醇直至获得3%固体的溶液。在用0.2微米的过滤器过滤溶液后取得反应产物。然后,用pgmea稀释实施例1中制得的聚合物以获得2%固体含量的溶液。以聚合物1:hfox=90:10质量比的质量比加入一定量的氧化铪纳米颗粒。将两个溶液在室温下混合30分钟。然后将溶液用0.2微米的塑料micron过滤。
聚合物测试
用gpc测定实施例14至18的聚合物的分子量。通过以1500rpm的旋转速度旋涂2%的聚合物溶液30秒来制备膜样品。然后用80℃至150℃的范围内的温度进行30秒至2分钟的软烘烤。
表4中给出了材料的gpc数据和涂覆后延迟数据。
表4:用不同的组合物产生的材料的涂覆后延迟。
图例(对于pcd结果):
x=对于在80℃/1h和150℃/1h下烘烤后至少1h的pcd良好,
y=在80℃/1h后良好但在150℃/1h下不良好,和
z=在80℃/1h和150℃/1h下均差
注:对于euvl,pcd时间必须至少为1小时,以便可安全地进行暴露。
结果评价。实施例14-19证实了添加的金属氧化物材料的有益效果。仅添加很少的重量百分数的金属氧化物材料就可显著减少所需的剂量。
图9示出了a)实施例1、b)实施例14和c)实施例17的电子束结果。获得图案所需的剂量减少了b)17%和c)33%。
实施例20
聚合物1+光酸
用pgmea稀释实施例1中的聚合物以获得具有2%固体含量的溶液。向聚合物溶液中加入一定量的5重量%的为光酸的(4-甲基苯基)[4-(2-甲基丙基)苯基]碘鎓六氟磷酸盐(1-)。使用磁力搅拌器将光酸分散在聚合物溶液中。
与不含光酸的聚合物相比,含光碱的聚合物显示出稍微增强的电子束敏感性。
实施例21
聚合物1+光碱
用pgmea稀释实施例1中的聚合物以获得具有2%固体含量的溶液。向聚合物溶液中加入一定量的5重量%的为光碱的(z)-{[双(二甲基氨基)亚甲基]氨基}-n-环己基(环己基氨基)-甲亚铵四(3-氟苯基)硼酸盐和2重量%的作为敏化剂的2-异丙基噻吨酮。
与不含光碱和敏化剂的聚合物相比,含光碱的聚合物显示出稍微增强的电子束敏感性。
结果评价:实施例20和21证实(图10),这些材料可与常用的光酸和光碱产生剂产生剂一起配制,以获得性能与实施例1相似的树脂组合物。
缩写:
car化学放大光刻胶
ebl电子束光刻
euv极紫外(波长)
euvl极紫外光刻
gpc凝胶渗透色谱
hsq氢倍半硅氧烷
ler线边缘粗糙度
lwr线宽度粗糙度
nil纳米压印光刻
pbl光束光刻
pcd涂覆后延迟
sem扫描电子显微镜
teos四乙氧基硅烷
tmah四甲基氢氧化铵。
参考文献列表
非专利文献
1.okoroanyanwu,u.moleculartheoryoflithography.(spiepress,2015).
2.gangnaik,a.s.,georgiev,y.m.&holmes,j.d.newgenerationelectronbeamresists:areview.chem.mater.29,1898–1917(2017).
3.simone,d.deetal.progressesandchallengesofeuvlithographymaterials.j.photopolym.sci.technol.27,601–610(2014).
4.higgins,c.d.etal.resolution,line-edgeroughness,sensitivitytradeoff,andquantumyieldofhighphotoacidgeneratorresistsforextremeultravioletlithography.jpn.j.appl.phys.50,1–8(2011).
5.desimone,d.,vesters,y.&vandenberghe,g.photoresistsinextremeultravioletlithography(euvl).adv.opt.technol.6,163–172(2017).
6.takeowatanabe.currentstatusandprospectforeuvlithography.20177thint.conf.integr.circuit,des.verif.3–8(2017).
7.grigorescu,a.e.&hagen,c.w.resistsforsub-20-nmelectronbeamlithographywithafocusonhsq:stateoftheart.nanotechnology20,292001(2009).
8.yang,j.k.w.etal.understandingofhydrogensilsesquioxaneelectronresistforsub-5-nm-half-pitchlithography.j.vac.sci.technol.bmicroelectron.nanom.struct.27,2622(2009).
- 还没有人留言评论。精彩留言会获得点赞!