一种基于相似性和双随机游走的微生物‑疾病关系预测方法与流程
本发明属于系统生物学领域,涉及一种基于相似性和双随机游走的微生物-疾病关系预测方法。
背景技术:
越来越多的研究表明微生物在许多人类复杂疾病中起着非常重要的作用。随着目前下一代dna测序技术的快速发展促进了人类身体之间微生物与疾病关联关系的发现,比如微生物群和各种癌症疾病,心血管疾病,代谢综合征(例如肥胖症和糖尿病),中枢神经系统疾病和自身炎症性疾病等。这些研究不但有助于对疾病机制的了解,也有利于对疾病的新治疗和诊断方案的发展。比如,确认粪便微生物群移植是治疗梭菌感染的安全有效的治疗方案,其通过重新引入正常的菌群到供体粪便,校正不平衡并重新建立正常的肠功能。所以对生物与疾病的关系的系统理解变得越来越紧迫。
目前普遍使用的是通过常规的基于实验的方法发现微生物与疾病的关系,其缺点是耗时且昂贵,同时也受到实验环境的限制,比如一些细菌不能在现有的种植实验室环境中进行培养。与此同时,通过计算模型对微生物与疾病之间的关系进行预测方式并没有得到大力的应用发展。到目前为止,很少有通过计算模型对微生物与疾病的关系进行预测的方法出现。katzhmda方法是目前通过已知的微生物与疾病的关系来进行预测新的微生物与疾病的关系的第一个模型,其通过集成疾病表征相似性、高斯核相似性以及微生物高斯核相似性,利用katz度信息来预测新的微生物与疾病的关系。当前微生物在计算方面的研究,大都集中在微生物分类方面,而对其与疾病的关系的关注严重不够。当前对微生物与疾病的关系的计算预测模型的发展程度和预测结果还不足以让生物实验人员认识计算模型预测的有效性,并进一步以此作为后续实验研究的基础。
受制于生物实验验证的效率、通过计算模型预测微生物与疾病的关系的关注和进展不够以及其预测结果的有待进一步提高,当前对微生物与疾病的关系的系统理解还是有限的。迫切需要提出更加有效的预测模型,充分利用已有的生物信息,通过更加科学的方式发现新的微生物与疾病的关系,为后续其关系预测的研究奠定基础,并进一步为生物实验研究提供重要的基本依据。此外,随着当前计算生物学和下一代dna测序技术的发展,了解到微生物对疾病的重要程度越来越高,进而对微生物与疾病的关系预测模型的发展提出了紧迫的需求。因此,为了进一步确认微生物与疾病的关系的重要性,为后续其关系预测模型的发展和生物实验验证提供帮助,有必要设计一种有效的微生物疾病关联关系预测的方法。
技术实现要素:
本发明所要解决的技术问题是,针对现有技术的不足,提供一种基于相似性和双随机游走的微生物-疾病关系预测方法,能够较准确的预测微生物与疾病的关系,为后续其关系预测模型的发展提供基础并进一步有效的避免生物化学实验所消耗的大量人力物力。
发明的技术解决方案如下:
一种基于相似性和双随机游走的微生物-疾病关系预测方法,包括以下步骤:
步骤1:分别构建疾病功能相似性矩阵dfunsim、疾病高斯核相似性矩阵kgip,d和微生物高斯核相似性矩阵kgip,m;
步骤2:集成疾病功能相似性矩阵dfunsim和疾病高斯核相似性矩阵kgip,d得到疾病最终相似性矩阵sd;将微生物高斯核相似性矩阵kgip,m作为微生物最终相似性矩阵sm;
步骤3:根据已知的微生物-疾病关系、微生物最终相似性矩阵sm和疾病最终相似性矩阵sd,构建一个双层的异构网络,利用双随机游走方法对微生物-疾病对进行关联分数预测;关联分数越大,则对应的微生物-疾病对存在关系的可能性越大。
进一步地,所述步骤1中,首先根据已知的疾病-基因关系和基因的功能相似性计算两种疾病之间的功能相似性,然后由所有疾病两两之间的功能相似性构建疾病功能相似性矩阵dfunsim;
对于任意两种疾病a和b,其功能相似性计算公式如下:
其中,ga={ga1,ga2,......,gam}为与疾病a相关联的基因集合,同样,gb={gb1,gb2,......,gbn}为与疾b相关联的基因集合,m和n分别为基因集合ga和gb中的基因数目;
其中f(gai,gbj)为基因gai和gbj之间的语义相似性值,humannet数据库提供了基于对数似然函数的语义相似性值计算值,具体计算方式如下:
f(gai,gbj)=lls(gai,gbj).
其中lls表示对数似然函数(在humannet数据库中,使用对数似然函数计算基因语义相似性值为现有技术)。
进一步地,所述步骤1中,根据已知的微生物-疾病关系,分别构建疾病高斯核相似性矩阵kgip,d和微生物高斯核相似性矩阵kgip,m,过程如下:
首先,定义
然后,计算所有疾病两两之间的高斯核相似性;对于任意两种疾病d1和d2,其高斯核相似性计算方式如下:
kgip,d(d1,d2)=exp(-γd||yd1-yd2||2)
其中,
再计算所有微生物两两之间的高斯核相似性;对于任意两种微生物m1和m2,其高斯核相似性计算方式如下:
kgip,m(m1,m2)=exp(-γm||ym1-ym2||2).
其中,
最后,由所有疾病两两之间的高斯核相似性构建疾病高斯核相似性矩阵kgip,d,由所有微生物两两之间的高斯核相似性构建微生物高斯核相似性矩阵kgip,m。
进一步地,所述步骤2中,集成疾病功能相似性矩阵dfunsim和疾病高斯核相似性矩阵kgip,d得到疾病最终相似性矩阵sd,具体集成方式计算如下:
即疾病最终相似性为功能相似性和高斯核相似性的平均值。
进一步地,所述步骤5中,根据微生物最终相似性sm,疾病最终相似性sd,已知的微生物-疾病数据邻接矩阵y集成一个双层的异构网络,利用双随机游走方法继续预测,其预测流程如下:
首先,对微生物最终相似性矩阵sm数据做列归一化处理,得到随机游走的微生物相似性关系矩阵mm∈nm×nm,其计算方式如下:
同样,对疾病最终相似性矩阵sd数据做列归一化处理,得到随机游走的疾病相似性关系矩阵md∈nd×nd,其计算方式如下:
然后,在这个双层的异构网络中同时游走,过程如下:
在微生物网络中迭代进行左游走:
在疾病网络中迭代进行右游走:
其中,t为当前迭代的次数,pt∈nm×nd表示第t次迭代预测得到的微生物-疾病关联分数矩阵,pt(i,j)表示微生物i和疾病j的关联分数(关联程度);l_pt表示微生物网络上进行第t次迭代预测得到的新的微生物-疾病关联分数矩阵,r_pt表示在疾病网络上进行第t次迭代预测得到的微生物-疾病关联分数矩阵;p0为邻接矩阵y∈nm×nd的归一化矩阵,
当pt收敛(pt+1-pt小于某个很小的阈值时(比如10-10),认为游走达到稳定状态)或者在微生物网络和疾病网络中的迭代游走均达到最大迭代次数时,结束迭代,最终的pt即为预测得到的微生物-疾病关联分数矩阵。
有益效果:
本发明提出了一种基于相似性和双随机游走的微生物-疾病关系预测方法来预测新的微生物与疾病的关系。首先计算通过疾病基因关系和基因功能相似性信息来计算疾病的功能相似性,再根据已知的微生物-疾病关系计算疾病的高斯核相似性,进一步集成得到疾病最终相似性。同样,也根据已知的微生物-疾病关系计算出微生物高斯核相似性并作为最终的微生物相似性。再将微生物最终相似性、疾病最终相似性和已知的微生物-疾病关系集成到一个双层的异构网络中。最后在微生物相似性网络和疾病相似性网络中设置不同随走游走步数,通过迭代到一定的稳定状态预测最终的微生物疾病关系对关联关系分数。通过五倍交叉和留一验证与其他方法的预测结果比较表明,本发明能够对微生物与疾病之间的关系进行更有效的预测。能够为后续预测微生物-疾病关系的计算模型的发展提供重要基础,为生物医学实验提供基本的指导作用,节省其人力物力成本。
本发明在对构建双层异构网络过程中对微生物相似性矩阵和疾病相似性矩阵进行了列归一化处理。在随机游走过程中对微生物和疾病网络中分别设置了随机游走的迭代步数限制。通过结合两个网络中相似的疾病关联相似的微生物和相似的微生物关联相似的疾病来预测最终的关联分数。并采取了和katzhmda方法中同样的五倍交叉验证和留一验证方法进行了预测性能的比较,通过对auc指标的分析表明本发明的预测性能。
本发明针对微生物-疾病关系领域,提供了一种通过计算模型预测其关系的有效的微生物-疾病关联关系预测方法,能够为后续这个领域的计算模型的研究提供重要基础,对疾病机制的整体理解提供帮助,并进一步推动药物的开发和复杂疾病的诊断治疗。
附图说明
图1为本发明总体流程图;
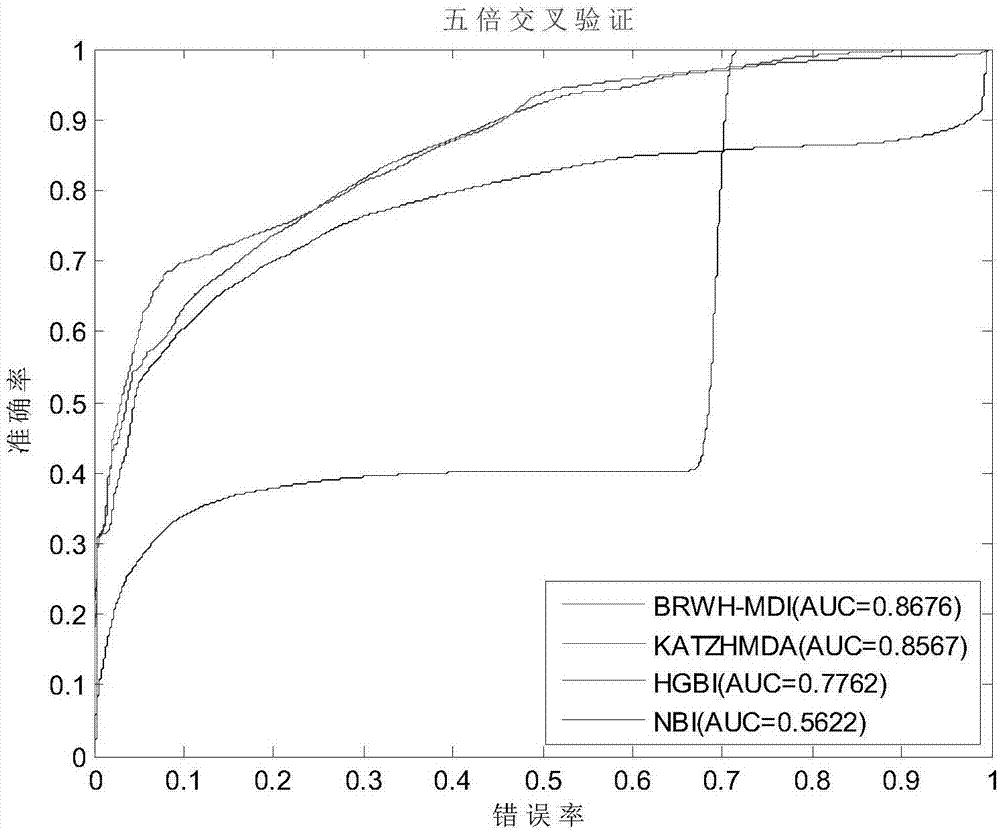
图2为本发明在数据集上五倍交叉验证比较图;
图3为本发明在数据集上留一交叉验证比较图;
具体实施方式
以下将结合附图和具体实施例对本发明做进一步详细说明:
实施例1:
首先利用疾病基因关系和基因相似性信息计算疾病功能相似性;基于已知的微生物-疾病关系计算疾病高斯核相似性和微生物高斯核相似性;利用疾病功能相似性和高斯核相似性集成疾病最终相似性,具体集成方式为取疾病高斯核相似性和疾病功能相似性均值。将微生物高斯核相似性作为微生物最终相似性。再将微生物相似性信息、疾病相似性信息和已知的微生物-疾病关系信息集成到一个双层异构网络中。基于相似的微生物关联相似的疾病和相似的疾病关联相似的微生物的出发点利用双随机游走方法在异构网络中进行微生物-疾病关系预测。对随机游走方法的关键过程在微生物相似性网络和疾病相似性网络中设置不同随走游走步数,迭代到一定的稳定状态获取最终的微生物-疾病关系对的关联分数。
本发明用到的已知微生物-疾病关系来自于hmdad(http://www.cuilab.cn/hmdad)数据库,总共包括39种疾病和292种微生物,其已知的微生物-疾病关系数目为483。通过去重处理,最终的关系数目为450,疾病和微生物数目分别为39和292。疾病基因关系数据来自于disgenet数据库。
基于相似性和双随机游走的微生物-疾病关系预测的整个流程如图1所示,可以划分为以下几个步骤:
(1)计算疾病功能相似性dfunsim的具体过程为:
首先,对于疾病对a和b,定义其功能相似性计算公式如下:
其中,ga={ga1,ga2,......,gam}为与疾病a相关联的基因集合,同样,gb={gb1,gb2,......,gbn}为与疾b相关联的基因集合,m和n分别为基因集合ga和gb中的基因数目;
其中f(gai,gbj)为基因gai和gbj之间的语义相似性值,humannet数据库提供了基于对数似然函数的语义相似性值计算值,具体计算方式如下:
f(gai,gbj)=lls(gai,gbj).
在humannet数据库中,给出的基因6188和6209功能相似性值为0.9697,根据疾病关联的基因,以及基因的功能相似性,疾病gastricandduodenalulcer和gastro-oesophagealreflux的功能相似性值为0.1655。
(2)根据已知的微生物-疾病关系,构建微生物高斯核相似性的过程如下:
首先,定义
kgip,m(m1,m2)=exp(-γm||ym1-ym2||2).
其中,
其中γ'm依照高斯核使用经验设置为1。依据上述计算公式,微生物actinobacillus和微生物actinobacteria的高斯核相似性值为0.0390。
同样,定义疾病d1和d2的高斯核相似性计算方式如下:
kgip,d(d1,d2)=exp(-γd||yd1-yd2||2)
其中,γ'd依照经验也设置为1。
(3)根据计算的疾病功能相似性dfunsim和疾病高斯核相似性kgip,d集成最终的疾病相似性过程,具体集成方式计算如下:
最终疾病相似性为功能相似性和高斯核相似性的平均值。
(4)将微生物高斯核相似性矩阵kgip,m作为微生物最终相似性矩阵sm:
sm=kgip,m
微生物只有一个高斯核相似性,故其最终相似性sm为高斯核相似性。
(5)根据微生物最终相似性sm,疾病最终相似性sd,已知的微生物-疾病数据集成一个双层的异构网络,利用双随机游走方法继续预测,其预测流程为:
首先,对微生物最终相似性矩阵sm数据做列归一化处理得到随机游走的微生物相似性关系矩阵mm∈nm*nm,其计算方式如下:
同样,对疾病最终相似性矩阵sd数据做列归一化处理得到随机游走的疾病相似性关系矩阵md∈nd*nd,其计算方式如下:
我们定义矩阵p∈nm*nd表示预测的微生物-疾病关系,p(i,j)表示微生物i和疾病j的关联分数(关联程度)。随机游走模型的预测过程是在这个双层的异构网络中同时游走,故我们对微生物和疾病网络分别设置了最大迭代次数参数il和ir。异构网络中的游走过程如下:
微生物网络中的左游走:
疾病网络中的右游走:
其中,t为当前迭代的次数,pt∈nm×nd表示第t次迭代预测得到的微生物-疾病关联分数矩阵,pt(i,j)表示微生物i和疾病j的关联分数(关联程度);l_pt表示微生物网络上进行第t次迭代预测得到的新的微生物-疾病关联分数矩阵,r_pt表示在疾病网络上进行第t次迭代预测得到的微生物-疾病关联分数矩阵;p0为邻接矩阵y∈nm×nd的归一化矩阵,
当pt收敛(pt+1-pt小于某个很小的阈值时(比如10-10),认为游走达到稳定状态)或者在微生物网络和疾病网络中的迭代游走均达到最大迭代次数时,结束迭代,最终的pt即为预测得到的微生物-疾病关联分数矩阵。对矩阵中的各关联分数由大到小进行排序,排名越靠前的微生物-疾病对存在关联关系的可能性越大。
为了验证本发明的有效性,我们参考其他算法的验证标准,采用了两种验证方式:(1)5倍交叉验证;(2)留一验证。在五倍交叉验证中,将已知的微生物-疾病关系随机分成5份,依次轮流选择1份为测试集,剩下的4份为训练集,其测试验证次数为100次。在留一验证中,依次从已知的微生物-疾病关系中选择一条已知微生物-疾病关系为测试集,剩下的为训练集。采用的评价指标为auc(theareasunderroccurves)值。
图2显示了集成疾病功能相似性和高斯核相似性在五倍交叉验证中的auc图。从图中可以看出,本发明(predictingmicrobe-diseaseinteractionsbasedonsimilaritiesandbi-randomwalkontheheterogeneousnetwork,简称为brwh-mdi)的auc值为0.8676,优于其他3个基于疾病高斯核相似性、疾病表征相似性和微生物高斯核相似性的方法(katzhmda:0.8567,hgbi:0.7762,nbi:0.5622)。特别是在错误率(fpr值)低的时候,正确率(tpr值)更高,证明了本发明的预测结果中排名在前的微生物-疾病关系越正确。
图3描述了集成疾病功能相似性和高斯核相似性在留一验证中各方法的性能比较图。从图中也可以看出,本发明brwh的auc值为0.8780,也同样由于其他3个方法的表现(katzhmda:0.8644,hgbi:0.7866,nbi:5553)。同样在错误率(fpr值)低的时候,正确率(tpr值)更高,也表明了本发明的预测结果中排名在前的微生物-疾病关系的更高准确性。
通过上述应用案例的表现,本发明能够较准确的预测新的微生物-疾病关系,为后续的生物医学实验提供指导作用,提高疾病诊断和治疗水平。
- 还没有人留言评论。精彩留言会获得点赞!