制备碳酸锂的方法与流程
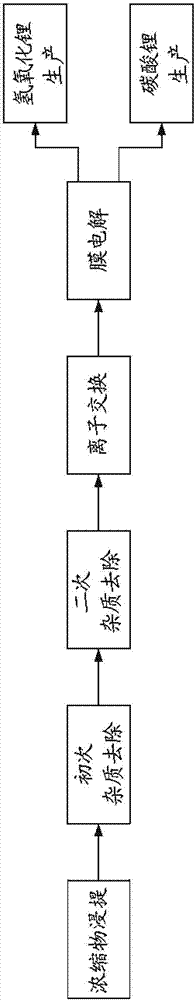
本公开涉及应用于碳酸锂制造的化学领域的改进。例如,此类方法可用于从含锂的原料中制备碳酸锂。例如,该公开还涉及其它锂产物如氢氧化锂和硫酸锂的生产。对于碳酸锂的需求正迅速增长。碳酸锂市场正在扩张,并且当前的全球生产能力可能将不能满足需求的预期增加。例如,碳酸锂被用作铝熔盐电解中的添加物以及珐琅和玻璃中的添加物。碳酸锂还可用于控制躁郁症,用于铌酸锂、钽酸锂和氟化锂电子级晶体的生产,以及用于锂电池的新兴技术。由于锂电池的高的能量密度与重量比及其与其它类型的电池相比的相对长的使用寿命,其已成为在一些现有的和建议的新应用中的电池的选择。锂电池被用于一些应用中,如便携式计算机、手机、医疗器械和植入物(例如心脏起搏器)。锂电池也是新型汽车例如混合动力汽车和电动汽车的发展中的感兴趣的选择,所述新型汽车因为排放降低和对碳氢燃料的依赖降低,因此既是环境友好的也是“绿色的”。使用的碳酸锂可能需要高纯度,例如,对于不同的电池应用而言。碳酸锂生产者的数量有限。作为对锂产物的增加的需求的直接结果,电池制造商正在寻找高质量锂产物(例如碳酸锂)的另外的且可靠的来源。迄今为止提出了少数的用于制备碳酸锂的方法。例如可以通过利用含锂的盐水或者利用海水来制备碳酸锂。一些提出的方法包括所生产的碳酸锂的几个纯化步骤。例如,已提出的方法需要用碳酸钠沉淀,并包括所生产的碳酸锂的几个纯化步骤。因此,需要为用于制备碳酸锂的现有溶液提供另一选择。根据一方面,提供了制备碳酸锂的方法,所述方法包括:在适于将至少一部分硫酸锂转化为氢氧化锂的条件下,将包含硫酸锂的水性组合物进行电解或电渗析,其中在电解或电渗析期间,所述包含硫酸锂的水性组合物被至少基本上维持在具有约1至约4的值的ph;以及将氢氧化锂转化为碳酸锂。根据另一方面,提供了制备碳酸锂的方法,所述方法包括:在适于将至少一部分锂化合物转化为氢氧化锂的条件下,将包含锂化合物的水性组合物进行电解或电渗析;以及将氢氧化锂转化为碳酸锂。根据另一方面,提供了制备碳酸锂的方法,所述方法包括:用水浸提经酸焙烧的含锂的材料,以获得包含li+和至少一种金属离子的水性组合物;将所述包含li+和至少一种金属离子的水性组合物与碱反应,以获得约4.5至约6.5的ph,并由此以至少一种氢氧化物的形式至少部分地沉淀所述至少一种金属离子,以获得包含所述至少一种氢氧化物的沉淀物以及包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;任选地,将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与另一种碱反应,以获得约9.5至约11.5的ph,以及任选地与至少一种金属碳酸盐反应,由此以任选地为至少一种碳酸盐的形式至少部分地沉淀至少一种金属离子,以获得任选地包含所述至少一种碳酸盐的沉淀物以及包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与离子交换树脂接触,以从所述组合物中至少部分地去除至少一种金属离子,由此获得包含锂化合物的水性组合物;将所述包含锂化合物的水性组合物进行电渗析或电解;以及将氢氧化锂转化为碳酸锂。根据另一方面,提供了制备碳酸锂的方法,所述方法包括:用水浸提经酸焙烧的含锂的材料,以获得包含li+和至少一种金属离子的水性组合物;将所述包含li+和至少一种金属离子的水性组合物与碱反应,以获得约4.5至约6.5的ph,并由此以至少一种氢氧化物的形式至少部分地沉淀所述至少一种金属离子,以获得包含所述至少一种氢氧化物的沉淀物以及包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;任选地,将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与另一种碱反应,以获得约9.5至约11.5的ph,以及任选地与至少一种金属碳酸盐反应,由此以至少一种碳酸盐形式至少部分地沉淀至少一种金属离子,以获得任选地包含所述至少一种碳酸盐的沉淀物以及包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与离子交换树脂接触,以从所述组合物中至少部分地去除至少一种金属离子,由此获得包含锂化合物的水性组合物;根据如本公开中所限定的方法将所述包含锂化合物的水性组合物进行电渗析或电解;以及如本公开中所限定的那样将氢氧化锂转化为碳酸锂。因此根据本公开的一方面,提供了制备氢氧化锂的方法,所述方法包括:在适于将锂化合物转化为氢氧化锂的条件下,将包含锂化合物的水性组合物进行第一电膜过程,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流;以及在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行第二电膜过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流。本公开还包括制备氢氧化锂的方法,所述方法包括:在适于将锂化合物转化为氢氧化锂的条件下,将包含锂化合物的水性组合物进行第一电膜过程,以进行至预先确定的程度,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流;以及在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行第二电膜过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流。本公开还包括制备氢氧化锂的方法,所述方法包括:在适于将硫酸锂和/或硫酸氢锂转化为氢氧化锂的条件下,将包含硫酸锂和/或硫酸氢锂的水性组合物进行第一电膜过程,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流;以及在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行第二电膜过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流。本公开还包括制备氢氧化锂的方法,所述方法包括:在适于将硫酸锂和/或硫酸氢锂转化为氢氧化锂的条件下,将包含硫酸锂和/或硫酸氢锂的水性组合物进行第一电膜过程,以进行至预先确定的程度,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流;以及在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行第二电膜过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流。本公开还包括制备氢氧化锂的方法,所述方法包括:在适于将硫酸锂和/或硫酸氢锂转化为氢氧化锂的条件下,将包含硫酸锂和/或硫酸氢锂的水性组合物进行包括双室膜过程的第一电膜过程,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流;以及在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行包括三室膜过程的第二电膜过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流。本公开还包括制备氢氧化锂的方法,所述方法包括:在适于将硫酸锂和/或硫酸氢锂转化为氢氧化锂的条件下,将包含硫酸锂和/或硫酸氢锂的水性组合物进行包括双室膜过程的第一电膜过程,以进行至预先确定的程度,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流;以及在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行包括三室膜过程的第二电膜过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流。本公开还包括制备氢氧化锂的方法,所述方法包括:在适于将锂化合物转化为氢氧化锂的条件下,将包含锂化合物的水性组合物进行双室单极或双极膜电解过程,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流,所述电解过程在包含通过阳离子交换膜而与阴极电解液室隔开的阳极电解液室的第一电化学池中进行;以及在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行三室单极或双极膜电解过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流,所述电解过程在包含通过阴离子交换膜而与中央室隔开的阳极电解液室和通过阳离子交换膜而与中央室隔开的阴极电解液室的第二电化学池中进行。本公开还包括制备氢氧化锂的方法,所述方法包括:在适于将硫酸锂和/或硫酸氢锂转化为氢氧化锂的条件下,将包含硫酸锂和/或硫酸氢锂的水性组合物进行双室单极或双极膜电解过程,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流,所述电解过程在包含通过阳离子交换膜而与阴极电解液室隔开的阳极电解液室的第一电化学池中进行;以及在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行三室单极或双极膜电解过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流,所述电解过程在包含通过阴离子交换膜而与中央室隔开的阳极电解液室和通过阳离子交换膜而与中央室隔开的阴极电解液室的第二电化学池中进行。本公开还包括制备氢氧化锂的方法,所述方法包括:在适于将锂化合物转化为氢氧化锂的条件下,将包含锂化合物的水性组合物进行双室单极或双极膜电解过程,以进行至预先确定的程度,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流,所述电解过程在包含通过阳离子交换膜而与阴极电解液室隔开的阳极电解液室的第一电化学池中进行;以及在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行三室单极或双极膜电解过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流,所述电解过程在包含通过阴离子交换膜而与中央室隔开的阳极电解液室和通过阳离子交换膜而与中央室隔开的阴极电解液室的第二电化学池中进行。本公开还包括制备氢氧化锂的方法,所述方法包括:在适于将硫酸锂和/或硫酸氢锂转化为氢氧化锂的条件下,将包含硫酸锂和/或硫酸氢锂的水性组合物进行双室单极或双极膜电解过程,以进行至预先确定的程度,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流,所述电解过程在包含通过阳离子交换膜而与阴极电解液室隔开的阳极电解液室的第一电化学池中进行;以及在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行三室单极或双极膜电解过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流,所述电解过程在包含通过阴离子交换膜而与中央室隔开的阳极电解液室和通过阳离子交换膜而与中央室隔开的阴极电解液室的第二电化学池中进行。因此根据本公开的一方面,提供了制备碳酸锂的方法,所述方法包括:在适于将锂化合物转化为氢氧化锂的条件下,将包含锂化合物的水性组合物进行第一电膜过程,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流;在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行第二电膜过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流;以及将氢氧化锂转化为碳酸锂。本公开还包括制备碳酸锂的方法,所述方法包括:在适于将锂化合物转化为氢氧化锂的条件下,将包含锂化合物的水性组合物进行第一电膜过程,以进行至预先确定的程度,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流;在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行第二电膜过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流;将氢氧化锂转化为碳酸锂。本公开还包括制备碳酸锂的方法,所述方法包括:在适于将硫酸锂和/或硫酸氢锂转化为氢氧化锂的条件下,将包含硫酸锂和/或硫酸氢锂的水性组合物进行第一电膜过程,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流;在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行第二电膜过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流;将氢氧化锂转化为碳酸锂。本公开还包括制备碳酸锂的方法,所述方法包括:在适于将硫酸锂和/或硫酸氢锂转化为氢氧化锂的条件下,将包含硫酸锂和/或硫酸氢锂的水性组合物进行第一电膜过程,以进行至预先确定的程度,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流;在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行第二电膜过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流;以及将氢氧化锂转化为碳酸锂。本公开还包括制备碳酸锂的方法,所述方法包括:在适于将硫酸锂和/或硫酸氢锂转化为氢氧化锂的条件下,将包含硫酸锂和/或硫酸氢锂的水性组合物进行包括双室膜过程的第一电膜过程,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流;在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行包括三室膜过程的第二电膜过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流;以及将氢氧化锂转化为碳酸锂。本公开还包括制备碳酸锂的方法,所述方法包括:在适于将硫酸锂和/或硫酸氢锂转化为氢氧化锂的条件下,将包含硫酸锂和/或硫酸氢锂的水性组合物进行包括双室膜过程的第一电膜过程,以进行至预先确定的程度,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流;在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行包括三室膜过程的第二电膜过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流;以及将氢氧化锂转化为碳酸锂。本公开还包括制备碳酸锂的方法,所述方法包括:在适于将锂化合物转化为氢氧化锂的条件下,将包含锂化合物的水性组合物进行双室单极或双极膜电解过程,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流,所述电解过程在包含通过阳离子交换膜而与阴极电解液室隔开的阳极电解液室的第一电化学池中进行;在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行三室单极或双极膜电解过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流,所述电解过程在包含通过阴离子交换膜而与中央室隔开的阳极电解液室和通过阳离子交换膜而与中央室隔开的阴极电解液室的第二电化学池中进行;以及将氢氧化锂转化为碳酸锂。本公开还包括制备碳酸锂的方法,所述方法包括:在适于将硫酸锂和/或硫酸氢锂转化为氢氧化锂的条件下,将包含硫酸锂和/或硫酸氢锂的水性组合物进行双室单极或双极膜电解过程,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流,所述电解过程在包含通过阳离子交换膜而与阴极电解液室隔开的阳极电解液室的第一电化学池中进行;在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行三室单极或双极膜电解过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流,所述电解过程在包含通过阴离子交换膜而与中央室隔开的阳极电解液室和通过阳离子交换膜而与中央室隔开的阴极电解液室的第二电化学池中进行;以及将氢氧化锂转化为碳酸锂。本公开还包括制备碳酸锂的方法,所述方法包括:在适于将锂化合物转化为氢氧化锂的条件下,将包含锂化合物的水性组合物进行双室单极或双极膜电解过程,以进行至预先确定的程度,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流,所述电解过程在包含通过阳离子交换膜而与阴极电解液室隔开的阳极电解液室的第一电化学池中进行;在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行三室单极或双极膜电解过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流,所述电解过程在包含通过阴离子交换膜而与中央室隔开的阳极电解液室和通过阳离子交换膜而与中央室隔开的阴极电解液室的第二电化学池中进行;以及将氢氧化锂转化为碳酸锂。本公开还包括制备碳酸锂的方法,所述方法包括:在适于将硫酸锂和/或硫酸氢锂转化为氢氧化锂的条件下,将包含硫酸锂和/或硫酸氢锂的水性组合物进行双室单极或双极膜电解过程,以进行至预先确定的程度,并获得第一锂减少的水性流和第一富含氢氧化锂的水性流,所述电解过程在包含通过阳离子交换膜而与阴极电解液室隔开的阳极电解液室的第一电化学池中进行;在适于制备至少另一部分氢氧化锂的条件下,将所述第一锂减少的水性流进行三室单极或双极膜电解过程,并获得第二锂减少的水性流和第二富含氢氧化锂的水性流,所述电解过程在包含通过阴离子交换膜而与中央室隔开的阳极电解液室和通过阳离子交换膜而与中央室隔开的阴极电解液室的第二电化学池中进行;以及将氢氧化锂转化为碳酸锂。根据另一方面,提供了制备碳酸锂的方法,所述方法包括:通过将co2喷射进包含氢氧化锂的水性组合物而使所述组合物与co2反应,由此获得包含碳酸锂的沉淀物,所述喷射在约10至约12.5的ph下进行;将至少一部分沉淀物加至澄清器中,并获得包含碳酸氢锂的上清液和包含碳酸锂的固体,将所述固体与所述上清液分离;以及在至少约85℃的温度下加热上清液,以将碳酸氢锂至少部分转化为碳酸锂。根据另一方面,提供了制备碳酸锂的方法,所述方法包括:在适于将至少一部分锂化合物转化为氢氧化锂的条件下,将包含锂化合物的水性组合物进行电渗析或电解,其中在电渗析或电解期间,所述包含锂化合物的水性组合物被至少基本上维持在具有约9.5至约12.5的值的ph;以及将氢氧化锂转化为碳酸锂。根据另一方面,提供了制备碳酸锂的方法,所述方法包括:在适于将至少一部分锂化合物转化为氢氧化锂的条件下,将包含锂化合物的水性组合物进行电渗析或电解,其中在电渗析或电解期间,所述水性组合物具有高于7的ph;以及将氢氧化锂转化为碳酸锂。根据另一方面,提供了制备碳酸锂的方法,所述方法包括:用水浸提经酸焙烧的含锂的材料,以获得包含li+和至少一种金属离子的水性组合物;将所述包含li+和至少一种金属离子的水性组合物与碱反应,以获得约4.5至约6.5的ph,并由此以至少一种氢氧化物的形式至少部分地沉淀所述至少一种金属离子,以获得包含所述至少一种氢氧化物的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;任选地将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与另一种碱反应,以获得约9.5至约11.5的ph,以及任选地与至少一种金属碳酸盐反应,由此以任选地为至少一种碳酸盐的形式至少部分地沉淀至少一种金属离子,以获得任选地包含所述至少一种碳酸盐的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与离子交换树脂接触,以从所述组合物中至少部分地去除至少一种金属离子,由此获得包含锂化合物的水性组合物;在适于将至少一部分锂化合物转化为氢氧化锂的条件下,将所述包含锂化合物的水性组合物进行电渗析或电解;以及将氢氧化锂转化为碳酸锂。根据另一方面,提供了制备碳酸锂的方法,所述方法包括:用水浸提经碱焙烧的含锂的材料,以获得包含li+和至少一种金属离子的水性组合物;将所述包含li+和至少一种金属离子的水性组合物与碱反应,以获得约4.5至约6.5的ph,并由此以至少一种氢氧化物的形式至少部分地沉淀所述至少一种金属离子,以获得包含所述至少一种氢氧化物的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;任选地将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与另一种碱反应,以获得约9.5至约11.5的ph,以及任选地与至少一种金属碳酸盐反应,由此以任选地为至少一种碳酸盐的形式至少部分地沉淀至少一种金属离子,以获得任选地包含所述至少一种碳酸盐的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与离子交换树脂接触,以从所述组合物中至少部分去除至少一种金属离子,由此获得包含锂化合物的水性组合物;在适于将至少一部分锂化合物转化为氢氧化锂的条件下,将所述包含锂化合物的水性组合物进行电渗析或电解;以及将氢氧化锂转化为碳酸锂。根据另一方面,提供了制备碳酸锂的方法,所述方法包括:用水浸提经碱焙烧的含锂的材料,以获得包含li+和至少一种金属离子的水性组合物;任选地将所述包含li+和至少一种金属离子的水性组合物与碱反应,以获得约4.5至约6.5的ph;以至少一种氢氧化物的形式至少部分地沉淀所述至少一种金属离子,以获得包含所述至少一种氢氧化物的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;任选地将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与另一种碱反应,以获得约9.5至约11.5的ph,以及任选地与至少一种金属碳酸盐反应,由此以任选地为至少一种碳酸盐的形式至少部分地沉淀至少一种金属离子,以获得任选地包含所述至少一种碳酸盐的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与离子交换树脂接触,以从所述组合物中至少部分地去除至少一种金属离子,由此获得包含锂化合物的水性组合物;在适于将至少一部分锂化合物转化为氢氧化锂的条件下,将所述包含锂化合物的水性组合物进行电渗析或电解;以及将氢氧化锂转化为碳酸锂。根据另一方面,提供了制备碳酸锂的方法,所述方法包括:在适于将至少一部分硫酸锂转化为氢氧化锂的条件下,将包含硫酸锂的水性组合物进行电渗析或电解,其中在电渗析或电解期间,所述包含硫酸锂的水性组合物被至少基本上维持在具有约9.5至约12.5的值的ph;以及将氢氧化锂转化为碳酸锂。根据另一方面,提供了制备碳酸锂的方法,所述方法包括:在适于将至少一部分硫酸锂转化为氢氧化锂的条件下,将包含硫酸锂的水性组合物进行电渗析或电解,其中在电渗析或电解期间,所述包含硫酸锂的水性组合物具有高于7的ph;以及将氢氧化锂转化为碳酸锂。根据另一方面,提供了制备氢氧化锂的方法,所述方法包括:在适于将至少一部分锂化合物转化为氢氧化锂的条件下,将包含锂化合物的水性组合物进行电渗析或电解。根据另一方面,提供了制备氢氧化锂的方法,所述方法包括:在适于将至少一部分锂化合物转化为氢氧化锂的条件下,将包含锂化合物的水性组合物进行电渗析或电解,其中在电渗析或电解期间,所述包含锂化合物的水性组合物被至少基本上维持在具有约9.5至约12.5的值的ph。根据另一方面,提供了制备氢氧化锂的方法,所述方法包括:在适于将至少一部分硫酸锂转化为氢氧化锂的条件下,将包含硫酸锂的水性组合物进行电渗析或电解,其中在电渗析或电解期间,所述包含硫酸锂的水性组合物被至少基本上维持在具有约9.5至约12.5的值的ph。根据另一方面,提供了制备氢氧化锂的方法,所述方法包括:在适于将至少一部分锂化合物转化为氢氧化锂的条件下,将包含锂化合物的水性组合物进行电渗析或电解,其中在电渗析或电解期间,所述包含锂化合物的水性组合物具有高于7的ph。根据另一方面,提供了制备氢氧化锂的方法,所述方法包括:在适于将至少一部分硫酸锂转化为氢氧化锂的条件下,将包含硫酸锂的水性组合物进行电渗析或电解,其中在电渗析或电解期间,所述包含硫酸锂的水性组合物具有高于7的ph。根据另一方面,提供了制备氢氧化锂的方法,所述方法包括:用水浸提经酸焙烧的含锂的材料,以获得包含li+和至少一种金属离子的水性组合物;将所述包含li+和至少一种金属离子的水性组合物与碱反应以获得约4.5至约6.5的ph,并由此以至少一种氢氧化物的形式至少部分地沉淀所述至少一种金属离子,以获得包含所述至少一种氢氧化物的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与离子交换树脂接触,以从所述组合物中至少部分地去除至少一种金属离子,由此获得包含锂化合物的水性组合物;以及在适于将至少一部分锂化合物转化为氢氧化锂的条件下,将所述包含锂化合物的水性组合物进行电渗析或电解。根据另一方面,提供了制备氢氧化锂的方法,所述方法包括:用水浸提经碱焙烧的含锂的材料,以获得包含li+和至少一种金属离子的水性组合物;将所述包含li+和至少一种金属离子的水性组合物与碱反应,以获得约4.5至约6.5的ph,并由此以至少一种氢氧化物的形式至少部分地沉淀所述至少一种金属离子,以获得包含所述至少一种氢氧化物的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;任选地将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与另一种碱反应,以获得约9.5至约11.5的ph,以及任选地与至少一种金属碳酸盐反应,由此以任选地为至少一种碳酸盐的形式至少部分地沉淀至少一种金属离子,以获得任选地包含所述至少一种碳酸盐的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与离子交换树脂接触,以从所述组合物中至少部分地去除至少一种金属离子,由此获得包含锂化合物的水性组合物;以及在适于将至少一部分锂化合物转化为氢氧化锂的条件下,将所述包含锂化合物的水性组合物进行电渗析或电解。根据另一方面,提供了制备氢氧化锂的方法,所述方法包括:用水浸提经碱焙烧的含锂的材料,以获得包含li+和至少一种金属离子的水性组合物;任选地将所述包含li+和至少一种金属离子的水性组合物与碱反应,以获得约4.5至约6.5的ph;以至少一种氢氧化物的形式至少部分地沉淀所述至少一种金属离子,以获得包含所述至少一种氢氧化物的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;任选地将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与另一种碱反应,以获得约9.5至约11.5的ph,以及任选地与至少一种金属碳酸盐反应,由此以任选地为至少一种碳酸盐的形式至少部分地沉淀至少一种金属离子,以获得任选地包含所述至少一种碳酸盐的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与离子交换树脂接触,以从所述组合物中至少部分地去除至少一种金属离子,由此获得包含锂化合物的水性组合物;以及在适于将至少一部分锂化合物转化为氢氧化锂的条件下,将所述包含锂化合物的水性组合物进行电渗析或电解。根据另一方面,提供了制备氢氧化锂的方法,所述方法包括:用水浸提经酸焙烧的含锂的材料,以获得包含li+和至少一种金属离子的水性组合物;将所述包含li+和至少一种金属离子的水性组合物与碱反应,以获得约4.5至约6.5的ph,并由此以至少一种氢氧化物的形式至少部分地沉淀所述至少一种金属离子,以获得包含所述至少一种氢氧化物的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;任选地将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与另一种碱反应,以获得约9.5至约11.5的ph,以及任选地与至少一种金属碳酸盐反应,由此以任选地为至少一种碳酸盐的形式至少部分地沉淀至少一种金属离子,以获得任选地包含所述至少一种碳酸盐的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与离子交换树脂接触,以从所述组合物中至少部分地去除至少一种金属离子,由此获得包含锂化合物的水性组合物;以及在适于将至少一部分锂化合物转化为氢氧化锂的条件下,将所述包含锂化合物的水性组合物进行电渗析或电解。根据另一方面,提供了制备硫酸锂的方法,所述方法包括:用水浸提经酸焙烧的含锂的材料,以获得包含li+和至少一种金属离子的水性组合物,其中所述含锂的材料是之前已经与h2so4反应的材料;将所述包含li+和至少一种金属离子的水性组合物与碱反应,以获得约4.5至约6.5的ph,并由此以至少一种氢氧化物的形式至少部分地沉淀所述至少一种金属离子,以获得包含所述至少一种氢氧化物的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;以及将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与离子交换树脂接触,以从所述组合物中至少部分地去除至少一种金属离子,由此获得包含硫酸锂的水性组合物。根据另一方面,提供了制备硫酸锂的方法,所述方法包括:用水浸提经酸焙烧的含锂的材料,以获得包含li+和至少一种金属离子的水性组合物,其中所述含锂的材料是之前已经与h2so4反应的材料;将所述包含li+和至少一种金属离子的水性组合物与碱反应,以获得约4.5至约6.5的ph,并由此以至少一种氢氧化物的形式至少部分地沉淀所述至少一种金属离子,以获得包含所述至少一种氢氧化物的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;任选地将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与另一种碱反应,以获得约9.5至约11.5的ph,以及与至少一种金属碳酸盐反应,由此以至少一种碳酸盐形式至少部分地沉淀至少一种金属离子,以获得包含所述至少一种碳酸盐的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;以及将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与离子交换树脂接触,以从所述组合物中至少部分地去除至少一种金属离子,由此获得包含硫酸锂的水性组合物。根据另一方面,提供了制备碳酸锂的方法,所述方法包括:用水浸提经碱焙烧的含锂的材料,以获得包含li+和至少一种金属离子的水性组合物;将所述包含li+和至少一种金属离子的水性组合物与碱反应,以获得约4.5至约6.5的ph,并由此以至少一种氢氧化物的形式至少部分地沉淀所述至少一种金属离子,以获得包含所述至少一种氢氧化物的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;任选地将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与另一种碱反应,以获得约9.5至约11.5的ph,以及任选地与至少一种金属碳酸盐反应,由此以任选地为至少一种碳酸盐的形式至少部分地沉淀至少一种金属离子,以获得任选地包含所述至少一种碳酸盐的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与离子交换树脂接触,以从所述组合物中至少部分地去除至少一种金属离子,由此获得包含锂化合物的水性组合物;在适于将至少一部分锂化合物转化为氢氧化锂的条件下,将所述包含锂化合物的水性组合物进行电渗析或电解;以及将氢氧化锂转化为碳酸锂;或者用水浸提经碱焙烧的含锂的材料,以获得包含li+和至少一种金属离子的水性组合物;任选地将所述包含li+和至少一种金属离子的水性组合物与碱反应,以获得约4.5至约6.5的ph;以至少一种氢氧化物的形式至少部分地沉淀所述至少一种金属离子,以获得包含所述至少一种氢氧化物的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;任选地将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与另一种碱反应,以获得约9.5至约11.5的ph,以及任选地与至少一种金属碳酸盐反应,由此任选地以至少一种碳酸盐形式至少部分地沉淀至少一种金属离子,以获得任选地包含所述至少一种碳酸盐的沉淀物和包含li+且含有降低含量的所述至少一种金属离子的水性组合物,并将所述水性组合物与所述沉淀物分离;将所述包含li+且含有降低含量的所述至少一种金属离子的水性组合物与离子交换树脂接触,以从所述组合物中至少部分地去除至少一种金属离子,由此获得包含锂化合物的水性组合物;在适于将至少一部分锂化合物转化为氢氧化锂的条件下,将所述包含锂化合物的水性组合物进行电渗析或电解;以及将氢氧化锂转化为碳酸锂。根据另一方面,提供了制备氢氧化锂的方法,所述方法包括:在适于将至少一部分硫酸锂转化为氢氧化锂的条件下,将包含硫酸锂的水性组合物进行电解或电渗析,其中在电解期间,所述包含硫酸锂的水性组合物具有高于7的ph。根据另一方面,提供了制备氢氧化锂的方法,所述方法包括:在适于将至少一部分锂化合物转化为氢氧化锂的条件下,将包含锂化合物的水性组合物进行电解或电渗析,其中在电解或电渗析期间,所述包含硫酸锂的水性组合物具有高于7的ph。在仅以示例的方式呈现本公开的不同实施方案的下述图中:图1为关于根据本公开的方法的实例的框图;图2为关于根据本公开的方法的另一实例的流程图;图3为显示根据本公开的方法的另一实例中锂含量(tenor)随时间的变化的图;图4为显示根据本公开的方法的另一实例中铁含量随时间的变化的图;图5为显示根据本公开的方法的另一实例中铝含量随时间的变化的图;图6为显示根据本公开的方法的另一实例中不同金属的含量随时间的变化的图;图7为显示根据本公开的方法的另一实例中不同金属的含量随时间的变化的图;图8为显示根据本公开的方法的另一实例中钙含量随摩尔过量的碳酸钠的变化的图;图9为显示根据本公开的方法的另一实例中镁含量随摩尔过量的碳酸钠的变化的图;图10为根据本公开的方法的另一实例的原理图示。图10描述了如何使用离子交换树脂以从所述组合物中至少部分地去除至少一种金属离子;图11为显示根据本公开的方法的另一实例中钙含量随离子交换过程中的柱床体积的变化的图;图12为显示根据本公开的方法的另一实例中镁含量随离子交换中的柱床体积的变化的图;图13为显示根据本公开的方法的另一实例中钙含量随离子交换中的柱床体积的变化的图;图14为显示根据本公开的方法的另一实例中镁含量随离子交换中的柱床体积的变化的图;图15为显示根据本公开的方法的另一实例中锂含量随离子交换中的柱床体积的变化的图;图16为为显示根据本公开的方法的另一实例中不同金属的含量随离子交换中的柱床体积的变化的图;图17为可用于实施根据本公开的方法的另一实例的单极膜电解池的实例的原理图示;图18为显示根据本公开的方法的另一实例中,于40℃下,在单极膜电解期间,电流效率以及阳极电解液中产生的h2so4的浓度、阴极电解液室中产生的lioh的浓度随通过的电荷的变化的图;图19为显示根据本公开的方法的另一实例中,在40℃下,电流效率和浓度随通过的电荷的变化的图;图20为显示根据本公开的方法的另一实例中电流效率和浓度随通过的电荷的变化的图;图21为显示根据本公开的方法的另一实例中电流密度、ph和电导率特性随通过的电荷的变化的图;图22为显示根据本公开的方法的另一实例中电流密度、ph和电导率特性随通过的电荷的变化的图;图23为显示根据本公开的方法的另一实例中电流效率和浓度随通过的电荷的变化的图;图24为显示根据本公开的方法的另一实例中电流效率和浓度随通过的电荷的变化的图;图25为显示根据本公开的方法的另一实例中电流密度、ph和电导率特性随通过的电荷的变化的图;图26为显示根据本公开的方法的另一实例中电流密度、ph和电导率特性随通过的电荷的变化的图;图27为显示根据本公开的方法的另一实例中电流效率和浓度随通过的电荷的变化的图;图28为显示根据本公开的方法的另一实例中电流密度、ph和电导率特性随通过的电荷的变化的图;图29为显示根据本公开的方法的另一实例中浓度随通过的电荷的变化的图;图30为显示根据本公开的方法的另一实例中电流密度、ph和电导率特性随通过的电荷的变化的图;图31显示根据本公开的方法的另一实例中电流效率和浓度随通过的电荷的变化的图;图32为关于根据本公开的方法的另一实例的流程图;图33为关于根据本公开的方法的另一实例的流程图;图34为显示根据本公开的方法的另一实例中锂含量随时间的变化的图;图35为显示根据本公开的方法的另一实例中锂含量随时间的变化的图;图36为可被用于实施根据本公开的方法的另一实例的膜电解池的实例的原理图示;图37显示了关于在约60℃下利用n324/aha膜的根据本公开的方法的图:图37a为显示电流和电压随通过的电荷的变化的图,图37b为显示给料电导率、电流密度和酸ph随通过的电荷的变化的图,图37c为显示“酸性”室、给料以及碱中不同离子的浓度随通过的电荷的变化的图,以及图37d为显示硫酸根的电流效率随通过的电荷的变化的图;图38显示了关于在约60℃下利用n324/aha膜的根据本公开的方法的图:图38a为显示电流和电压随通过的电荷的变化的图,图38b为显示给料电导率、电压、给料ph和酸ph随通过的电荷的变化的图,图38c为显示电流/电压斜线上升(ramp)的图,图38d为显示给料中不同离子的浓度随通过的电荷的变化的图,图38e为显示酸性室(或阳极电解液室)中的铵和硫酸盐的浓度随通过的电荷的变化的图,图38f为显示碱中不同离子的浓度随通过的电荷的变化的图,以及图38g为显示硫酸根的电流效率随通过的电荷的变化的图;图39显示了关于在约60℃下利用n324/aha膜的根据本公开的方法的图:图39a为显示电流和电压随通过的电荷的变化的图;图39b为显示给料电导率、电压、给料ph和酸ph随通过的电荷的变化的图,图39c为显示给料中不同离子的浓度随通过的电荷的变化的图,图39d为显示碱中不同离子的浓度随通过的电荷的变化的图,图39e为显示“酸性”室中铵和硫酸盐的浓度随通过的电荷的变化的图,图39f为显示硫酸根的电流效率随通过的电荷的变化的图,以及图39g为显示给料中不同离子的浓度随通过的电荷的变化的图;图40显示了关于在约60℃和约200ma/cm2下利用n324/aha膜的根据本公开的方法的图:图40a为显示电流和电压随通过的电荷的变化的图,图40b为显示给料电导率、电压、给料ph和酸ph随通过的电荷的变化的图,图40c为显示给料中不同离子的浓度随通过的电荷的变化,图40d为显示“酸性”室中铵和硫酸盐的浓度随通过的电荷的变化的图,图40e为显示碱中不同离子的浓度随通过的电荷的变化的图,以及图40f为显示硫酸根的电流效率随通过的电荷的变化的图;图41显示了关于在约80℃和约200ma/cm2下利用n324/aha膜的根据本公开的方法的图:图41a为显示电流和电压随通过的电荷的变化的图,图41b为显示给料电导率、电压、给料ph和酸ph随通过的电荷的变化的图,图41c为显示电流/电压斜线上升的图,图41d为显示给料中不同离子的浓度随通过的电荷的变化的图,图41e为显示“酸性”室中铵和硫酸盐的浓度随通过的电荷的变化的图,图41f为显示碱中不同离子的浓度随通过的电荷的变化的图,以及图41g为显示硫酸根的电流效率随通过的电荷的变化的图;图42显示了关于在约60℃和约200ma/cm2下利用n324/aha膜的根据本公开的方法的图:图42a为显示电流和电压随通过的电荷的变化的图;图42b为显示给料中不同离子的浓度随通过的电荷的变化的图,图42c为显示给料电导率、电压、给料ph和酸ph随通过的电荷的变化的图,图42d为显示给料中不同离子的浓度随通过的电荷的变化的图,图42e为显示“酸性”室中铵和硫酸盐的浓度随通过的电荷的变化的图,图42f为显示碱中不同离子的浓度随通过的电荷的变化的图,以及图42g为显示硫酸根的电流效率随通过的电荷的变化的图;图43为显示在约60℃和约200ma/cm2下利用n324/aha膜的根据本公开的方法的实例中电流密度、ph和电导率随通过的电荷的变化的图;图44为根据本公开的实施方案的方法和系统的原理图;图45为根据本公开的实施方案的双室膜池的原理图示,所述双室膜池可用于包括含有锂化合物(如硫酸锂和/或硫酸氢锂)的水溶液的电解的方法中;图46显示了关于在约80℃的温度下和约3ka/m2的电流密度下在双室膜电解池中利用nafion324阳离子交换膜的用于制备氢氧化锂的方法的实例的图:图46a为显示不同离子的给料浓度和转化率随通过的电荷的变化的图,图46b为显示电流效率、转化率、比率和给料ph随通过的电荷的变化的图,图46c为显示电压和电流密度随通过的电荷的变化的图,以及图46d为显示氢氧根浓度随通过的电荷的变化的图;图47显示了关于在约80℃的温度下和约4ka/m2的电流密度下在双室膜电解池中利用nafion324阳离子交换膜的用于制备氢氧化锂的方法的实例的图:图47a为显示电压和电流密度随通过的电荷的变化的图,图47b为显示不同离子的给料浓度随通过的电荷的变化的图,图47c为显示电流效率、转化率和比率随通过的电荷的变化的图,以及图47d为显示氢氧根浓度随通过的电荷的变化的图;图48显示了关于在约80℃的温度下和约5ka/m2的电流密度下在双室膜电解池中利用nafion324阳离子交换膜的用于制备氢氧化锂的方法的实例的图:图48a为显示电压和电流密度随通过的电荷的变化的图,图48b为显示不同离子的给料浓度和比率随通过的电荷的变化的图,图48c为显示电流效率、转化率和比率随通过的电荷的变化的图,以及图48d为显示氢氧根浓度随通过的电荷的变化的图;图49显示了关于在约80℃的温度下和约200ma/cm2的电流密度下在三室膜电解池中利用nafion324阳离子交换膜和astomaha阴离子交换膜的用于制备氢氧化锂同时产生硫酸铵的方法的实例的图:图49a为显示三室膜电解池的不同室中不同离子的浓度随通过的电荷的变化的图,图49b为显示电流密度、池的电压以及给料ph和酸ph随通过的电荷的变化的图,图49c为显示三室膜电解池的不同室的电流效率和比率随通过的电荷的变化的图,以及图49d为显示电压和电流密度随通过的电荷的变化的图;以及图50显示了关于在约60℃的温度下和约100ma/cm2的电流密度下在三室膜电解池中利用nafion324阳离子交换膜和fumatechfab阴离子交换膜的用于制备氢氧化锂同时产生硫酸的方法的实例的图:图50a为显示三室膜电解池的不同室中的浓度随通过的电荷的变化的图,图50b为显示三室膜电解池的不同室的电流效率和比率随通过的电荷的变化的图,图50c为显示电流密度、通过的电荷以及给料ph随通过的电荷的变化的图,以及图50d为显示电压和电流密度随通过的电荷的变化的图。根据通过示例的方式阐明的下述不同实施方案的描述,其它特征和优势将更加显而易见。本文使用的术语“适合的”意为特定条件的选择将取决于待实施的具体处理或操作,但是所述选择完全在本领域技术人员的技能范围内。本文描述的所有方法均将在足以提供期望的产物的条件下进行。在理解本公开的范围时,本文使用的术语“包括”及其衍生语意图为开放性术语,其指定存在所规定的特征、元素、组分、组、整数和/或步骤,但是不排除存在其它未规定的特征、元素、组分、组、整数和/或步骤。前述也适用于具有相似含义的词,如术语“包含”、“具有”极其衍生语。本文使用的术语“由…组成”及其衍生语意图为封闭性术语,其指定存在所规定的特征、元素、组分、组、整数和/或步骤,同时排除了存在其它未规定的特征、元素、组分、组、整数和/或步骤。本文使用的术语“基本上由…组成”意图指定存在所规定的特征、元素、组分、组、整数和/或步骤以及在实质上不影响特征、元素、组分、组、整数和/或步骤的基本特征和新特征的那些特征、元素、组分、组、整数和/或步骤。本文使用的程度术语如“约(about)”和“大约(approximately)”意为修改项的合理量的偏差,以使最终结果不明显改变。这些程度术语应被理解为包括修改项的至少±5%或至少±10%的偏差,条件是该偏差不会否定其所修改的词的含义。本文使用的表述“至少一种金属离子”指例如至少一种金属的至少一种类型的离子。例如,至少一种金属离子可以是mx+。在该实例中,mx+是金属m的离子,其中x+是金属m的特殊形式或氧化态。因此,mx+是至少一种金属(m)的至少一种类型的离子(氧化态x+)。例如,my+可以是金属m的另一类型的离子,其中x和y是不同的整数。本文使用的表述“至少基本上维持”,当其指在本公开的方法或其部分(例如喷射、加热、电渗析、电解等)期间维持的ph值或ph范围时,指的是在所述方法或其部分期间至少75%、80%、85%、90%、95%、96%、97%、98%或99%的时间中维持所述ph值或ph范围。本文使用的表述“至少基本上维持”,当指在本公开的方法或其部分(例如喷射、加热、电渗析、电解等)期间维持的浓度值或浓度范围时,指的是在所述方法或其部分期间至少75%、80%、85%、90%、95%、96%、97%、98%或99%的时间中维持所述浓度值或浓度范围。本文使用的表述“至少基本上维持”,当指在本公开的方法或其部分(例如喷射、加热、电渗析、电解等)期间维持的温度值或温度范围时,指的是在所述方法或其部分期间至少75%、80%、85%、90%、95%、96%、97%、98%或99%的时间中维持所述温度值或温度范围。本文使用的表述“至少基本上维持”,当指在本公开的方法或其部分(例如喷射、加热、电渗析、电解等)期间维持的氧化电位值或氧化电位范围时,指的是在所述方法或其部分期间至少75%、80%、85%、90%、95%、96%、97%、98%或99%的时间中维持所述氧化电位值或氧化电位范围。本文使用的表述“至少基本上维持”,当指在本公开的方法或其部分(例如电渗析、电解等)期间维持的电流值或电流范围时,指的是在所述方法或其部分期间至少75%、80%、85%、90%、95%、96%、97%、98%或99%的时间中维持所述电流值或电流范围。本文使用的表述“至少基本上维持”,当指在本公开的方法或其部分(例如电渗析、电解等)期间维持的电压值或电压范围时,指的是在所述方法或其部分期间至少75%、80%、85%、90%、95%、96%、97%、98%或99%的时间中维持所述电压值或电压范围。本文使用的术语“电膜过程”指例如利用离子交换膜,并将电位差作为离子物质的驱动力的过程。电膜过程可以是例如(膜)电渗析或(膜)电解。例如,电膜过程可以是(膜)电解。下文提供的实例是非限制性的,并且被用于更好地示例说明本公开的方法。本公开的方法可以有效地处理不同的含锂的材料。含锂的材料可以是含锂的矿石、锂化合物或者回收的含锂的工业实体。例如,含锂的矿石可以是,例如α-锂辉石、β-锂辉石、锂云母、伟晶岩、透锂长石、锂霞石、锂磷铝石、水辉石、贾达尔石、蒙脱石、粘土或其混合物。锂化合物可以是,例如licl、li2so4、lihco3、li2co3、lino3、lic2h3o2(醋酸锂)、lif、硬脂酸锂或柠檬酸锂。含锂的材料还可以是回收的含锂的工业实体,如锂电池、其它锂产物或其衍生物。本领域技术人员会理解各种反应参数将会根据多种因素而改变,如原料的性质、它们的纯度水平、反应的规模以及所有参数(因为它们可以是相互依赖的),并且可以相应地调整反应条件以将收率最优化。例如,在可用于制备碳酸锂的本公开的方法中,所述方法可以包括在至少约85℃的温度下加热上清液,以将碳酸氢锂至少部分地转化为碳酸锂,并沉淀其中包含的任何溶解的碳酸锂。例如,在可用于制备碳酸锂的本公开的方法中,原料可以是,例如氢氧化锂。例如,可以是通过如本公开中所述的方法产生的氢氧化锂。例如,可以通过下述来实施氢氧化锂至碳酸锂的转化:通过将co2喷射进包含氢氧化锂的水性组合物使所述组合物与co2反应,由此获得包含碳酸锂的沉淀物,所述喷射在约10至约12.5的ph下实施;将至少一部分沉淀物加至澄清器中,并获得包含碳酸氢锂的上清液和包含碳酸锂的固体,将所述固体与所述上清液分离;以及在至少约85℃的温度下加热上清液,以将碳酸氢锂至少部分地转化为碳酸锂。本公开的方法可以有效地处理不同的含锂的材料。含锂的材料可以是含锂的矿石、锂化合物或回收的含锂的工业实体。例如,含锂的矿石可以是,例如α-锂辉石、β-锂辉石、锂云母、伟晶岩、透锂长石、锂霞石、锂磷铝石、水辉石、蒙脱石、粘土或其混合物。锂化合物可以是,例如licl、li2so4、lihco3、li2co3、lino3、lic2h3o2(醋酸锂)、硬脂酸锂、柠檬酸锂或lif。含锂的材料还可以是回收的含锂的工业实体,如锂电池、其它锂产物或其衍生物。本领域技术人员会理解各种反应参数将会根据多种因素而改变,所述反应参数如例如反应时间、反应温度、反应压力、反应物比例、流动速率、反应物纯度、电流密度、电压、保留时间、ph、氧化/还原电位、柱床体积、使用的树脂的类型和/或再循环速率,所述多种因素如原料的性质、它们的纯度水平、反应的规模以及之前提及的所有参数(因为它们可以是相互依赖的),并且可以相应地调整反应条件以将收率最优化。例如,当所述方法包括在至少约85℃的温度下加热上清液以将碳酸氢锂至少部分地转化为碳酸锂时,其可以进一步包括沉淀其中包含的任何溶解的碳酸锂。例如,当喷射时,ph可以被至少基本维持在约10至约12.5,约10.5至约12.0,约10.5至约11.5,约10.7至约11.3,约10.8至约11.2,约10.9至约11.1,或者约11的值。例如,可以在至少约87℃,至少约89℃,至少约91℃,至少约93℃,至少约95℃,至少约97℃,约85℃至约105℃,约90℃至约100℃,约92℃至约98℃,约93℃至约97℃,约94℃至约96℃或者约95℃的温度下加热上清液。例如,在所述过程期间,包含氢氧化锂的水性组合物可以被至少基本上维持在约30至约70g/l,约40至约60g/l或者约48至约55g/l的氢氧化锂浓度。例如,可以在约10至约40℃,约15至约30℃或者约20至约30℃的温度下实施喷射。例如,当加热上清液时,后者可以维持在约1至约10g/l,约2至约6g/l或者约3至约5g/l的li浓度。例如,在电渗析或电解期间,ph可以被至少基本上维持在约1至约4,约1至约2,约1至约3,约2至约3,或者约2至约4的值。例如,在电解期间,ph可以被至少基本上维持在约1至约4,约2至约4或者约2的值。例如,在电渗析期间,ph可以被至少基本上维持在约1至约4或者约1至约2的值。例如,可以在三室膜电解池中实施电渗析或电解。例如,可以在双室膜电解池中实施电渗析或电解。例如,可以在三室膜池中实施电渗析或电解。例如,可以在双室膜池中实施电渗析或电解。例如,可以在单极或双极电解池中实施电解。例如,可以在单极或双极三室电解池中实施电解。例如,可以在双极电解池中实施电解。例如,可以在双极三室电解池中实施电解。例如,可以在双极电渗析池中实施电渗析。例如,可以在双极三室电渗析池中实施电渗析。例如,可将包含硫酸锂或锂化合物的水性组合物进行单极或双极膜电解过程。例如,可将包含硫酸锂或锂化合物的水性组合物进行单极或双极三室膜电解过程。例如,可将包含硫酸锂或锂化合物的水性组合物进行双极膜电渗析过程。例如,可将包含硫酸锂或锂化合物的水性组合物进行双极三室电渗析过程。例如,可在电解池中实施电渗析或电解,其中通过阴极膜将阴极室与中央室或阳极室隔开。例如,可以在双极膜中实施电渗析。例如此类膜为分解水分子(h+和oh-)的膜,其中产生酸性和碱性溶液,例如低浓度的酸性或碱性溶液。例如,可以通过使用单极或双极膜来实施电解。例如,可以通过利用包含配置有单极或双极膜和双极电极的三室池的电解堆(electrolysisstack)来实施电解。例如,此类电极有效地在阴极电极释放氢气(h2),并在阳极电极释放氧气(o2)或氯气(cl2)。例如,此类电极有效地分解水分子。例如,膜可以是全氟膜或苯乙烯/二乙烯基苯膜。例如,膜可以是阳离子交换膜、peek增强膜。例如,可以通过下述来实施电渗析或电解:将包含锂化合物(例如licl、lif、li2so4、lihco3、li2co3、lino3、lic2h3o2(醋酸锂)、硬脂酸锂或柠檬酸锂)的水性组合物引入中央室,将包含氢氧化锂的水性组合物引入阴极室,并在阳极室(或酸性室)产生包含酸(例如hcl、h2so4、hno3或醋酸)的水性组合物。本领域技术人员将会理解,例如当向中央室中引入licl时,在阳极室中产生hcl,例如单极或双极膜电解池。例如,当在中央室中使用lif时,在阳极室中产生hf。例如,当在中央室中使用li2so4时,在阳极室中产生h2so4。例如,当在中央室中使用lihco3时,在阳极室中产生h2co3。例如,当在中央室中使用lino3时,在阳极室中产生hno3。例如,当在中央室中使用lic2h3o2时,在阳极室中产生醋酸。例如,当在中央室中使用硬脂酸锂时,在阳极室中产生硬脂酸。例如,当在中央室中使用柠檬酸锂时,在阳极室中产生柠檬酸。例如,锂化合物可以包含氯化锂(licl)、氟化锂(lif)、硫酸锂(li2so4)、硫酸氢锂(lihso4)、碳酸氢锂(lihco3)、碳酸锂(li2co3)、硝酸锂(lino3)、醋酸锂(lic2h3o2)、硬脂酸锂和/或柠檬酸锂,基本上由氯化锂(licl)、氟化锂(lif)、硫酸锂(li2so4)、硫酸氢锂(lihso4)、碳酸氢锂(lihco3)、碳酸锂(li2co3)、硝酸锂(lino3)、醋酸锂(lic2h3o2)、硬脂酸锂和/或柠檬酸锂组成,或者由氯化锂(licl)、氟化锂(lif)、硫酸锂(li2so4)、硫酸氢锂(lihso4)、碳酸氢锂(lihco3)、碳酸锂(li2co3)、硝酸锂(lino3)、醋酸锂(lic2h3o2)、硬脂酸锂和/或柠檬酸锂组成。例如,锂化合物可以包含硫酸锂和/或硫酸氢锂,基本上由硫酸锂和/或硫酸氢锂组成,或者由硫酸锂和/或硫酸氢锂组成。例如,包含硫酸锂和/或硫酸氢锂的组合物还可包含h2so4。例如,可以通过下述来实施电渗析或电解:将硫酸锂引入中央室,将包含氢氧化锂的水性组合物引入阴极室,并在阳极室中产生包含硫酸的水性组合物。例如,在所述过程期间使用的阳极电解液可以包含氨、硫酸氢铵、硫酸铵和/或nh4oh。例如,在所述过程期间使用的阳极电解液可以包含氨、硫酸氢铵、硫酸铵和/或nh4oh,由此产生铵盐。例如,所述方法还可以包括在阳极电解液室中,在酸性室中,在阳极电解液中,在阳极或其周围添加氨和/或nh4oh,例如气态氨或液态氨,例如nh3和/或nh4oh,其中在所述方法中使用阳极。例如,所述方法还可以包括在阳极电解液室,在阳极电解液,在阳极或其周围添加氨和/或nh4oh,由此产生铵盐,其中在所述方法中使用阳极。例如,所述方法还可包括在所述方法中使用的阳极电解液室或阳极电解液中添加氨和/或nh4oh。例如,所述方法还可包括在方法中使用的阳极电解液中添加氨和/或nh4oh,由此产生铵盐。例如,铵盐可以是(nh4)2so4。例如,产生的铵盐的浓度可以为约1至约4m,约1至约3m,或者约1.5m至约2.5m。例如,阳极电解液中存在的硫酸氢铵的浓度可以为约1至约4m,约1至约3m,或者约1.5m至约3.5m的浓度。例如,阳极电解液中存在的硫酸铵的浓度可以为约1至约4m,约1至约3m,或者约1.5m至约3.5m的浓度。例如,阳极电解液的ph被维持在约-0.5至约4.0,约-0.5至约3.5,约-0.25至约1.5,或者约-0.25至约1.0的值。例如,与产生的硫酸相比,可以以亚化学计量的量添加氨。例如,可以以0.5:1至2:1或者1:1至1.9:1的氨:硫酸的摩尔比添加氨。例如,可以通过下述来实施电渗析或电解:将包含锂化合物(例如licl、lif、li2so4、lihco3、li2co3、lino3、lic2h3o2(醋酸锂)、硬脂酸锂或柠檬酸锂)的水性组合物引入中央室,将包含氢氧化锂的水性组合物引入阴极室,以及将包含nh3的水性组合物引入阳极室。例如,当将包含nh3的水性组合物引入阳极室时,可能不需要质子阻挡膜(proton-blockingmembrane),并且可以使用能够例如在约80℃的温度下运行且可以例如具有较低电阻的膜。例如,包含锂化合物的水性组合物还可包含na+。例如,在电渗析或电解期间,包含氢氧化锂的水性组合物可以被至少基本上维持在约30至约90g/l,约40至约90g/l,约35至约70g/l,约40至约66g/l,约45至约65g/l,约48至约62g/l,或者约50至约60g/l的氢氧化锂浓度。例如,在电渗析或电解期间,包含氢氧化锂的水性组合物可以被至少基本上维持在约1至约5m,约2至约4m,约2.5至约3.5m,约2.7至约3.3m,约2.9至约3.1m,或者约3m的氢氧化锂浓度。例如,在电渗析或电解期间,包含硫酸的水性组合物可以被至少基本上维持在约30至约100g/l,约40至约100g/l,约60至约90g/l,约20至约40g/l,约20至约50g/l,约25至约35g/l,或者约28至约32g/l的硫酸浓度。例如,在电渗析或电解期间,包含硫酸的水性组合物可以被至少基本上维持在约0.1至约5m,约0.2至约3m,约0.3至约2m,约0.3至约1.5m,约0.4至约1.2m,约0.5至约1m,或者约0.75m的硫酸浓度。例如,在电渗析或电解期间,包含硫酸的水性组合物可以被至少基本上维持在约0.5m至约1.4m,约0.6m至约1.3m,约0.65至约0.85m,约0.7m至约1.2m,约0.8m至约1.1m,约8.5m至约1.05m,或者约0.9m至约1.0m,约20至约50g/l,或者35至约70g/l的硫酸浓度。例如,在电渗析或电解期间,包含硫酸锂的水性组合物可以被至少基本上维持在约5至约30g/l,约5至约25g/l,约10至约20g/l,或者约13至约17g/l的硫酸锂浓度。例如,在电渗析或电解期间,包含硫酸锂的水性组合物可以被至少基本上维持在约0.2至约3m,约0.4至约2.5m,约0.5至约2m,或者约0.6至约1.8m的硫酸锂浓度。例如,在电渗析或电解期间,包含硫酸锂的水性组合物可以被至少基本上维持在约0.2至约3m,约0.4至约2.5m,约0.5至约2m,或者约0.6至约1.8m的硫酸根(so42-)浓度。例如,在电渗析或电解期间,包含硫酸锂的水性组合物可以包含基于组合物中的钠和锂的总重量的约1至约30重量%,约1至约25重量%,约5至约25重量%,约10至约25重量%的钠。例如,在电渗析或电解期间,包含硫酸锂的水性组合物可以包含钠。li:na(g/g)的比例可以是约2:1至约10:1,或者约3:1至约5:1。例如,在电渗析或电解期间,包含硫酸锂的水性组合物的温度可以为约20至约80℃,约20至约60℃,约30至约40℃,约35至约65℃,约40至约60℃,约35至约45℃,约55至约65℃,约50至约60℃,约50至约70℃,或者约46至约54℃。例如,在电渗析或电解期间,包含硫酸锂的水性组合物的温度可以被至少基本上维持在约20至约60℃,约30至约40℃,约50至约60℃,或者约46至约54℃的值。本领域技术人员将会理解此温度可以随电解池中选择的膜的变化而改变。例如,在电渗析或电解期间,包含硫酸锂的水性组合物的温度可以被至少基本上维持在约20至约80℃,约20至约60℃,约30至约40℃,约35至约65℃,约40至约60℃,约35至约45℃,约55至约65℃,约50至约70℃,约50至约60℃,或者约46至约54℃的值。例如,当在电渗析或电解期间使用asahiaav或类似的阴离子交换膜时,包含硫酸锂的水性组合物的温度可以被至少基本上维持在约40℃的值。例如,当在电渗析或电解期间使用fumatechfab或类似的阴离子交换膜时,包含硫酸锂的水性组合物的温度可以被至少基本上维持在约60℃的值。例如,在电渗析或电解期间可以使用nafion324或类似的阳离子交换树脂或膜。可将其它膜如nafion902、fumatechfkb或neoseptacmb用于氢氧化物的浓缩。例如,当在电渗析或电解期间将包含nh3的水性组合物引入阳极室时,包含硫酸锂的水性组合物的温度可以被至少基本上维持在约20至约100℃,约20至约95℃,约20至约90℃,约45至约95℃,约65至约95℃,约20至约80℃,约20至约80℃,约75至约85℃,约20至约60℃,约30至约40℃,约35至约65℃,约40至约60℃,约35至约45℃,约55至约65℃,约50至约60℃,约50至约70℃,或者约46至约54℃的值。例如,在电渗析或电解期间,电流可以被至少基本上维持在约300至约6000a/m2,约2000至约6000a/m2,约3500至约5500a/m2,约4000至约5000a/m2,约400至约3000a/m2,约500至约2500a/m2,约1000至约2000a/m2,约400至约1200a/m2,约400至约1000a/m2,约300至约700a/m2,约400至约600a/m2,约425至约575a/m2,约450至约550a/m2,或者约475至约525a/m2的密度。例如,在电渗析或电解期间,电流可以被至少基本上维持在约30至约250ma/cm2,50至约250ma/cm2,约75至约200ma/cm2,或者约100至约175ma/cm2的密度。例如,在电渗析或电解期间,电流可以被至少基本上维持在约50至约150a/m2,约60至约140a/m2,约70至约130a/m2,约80至约120a/m2,,或者约90至约110a/m2的密度。例如,在电渗析或电解期间,电流可以被至少基本上维持在约400至约3000a/m2,约400至约1200a/m2,约400至约1000a/m2,约400至约600a/m2,约425至约575a/m2,或者约450至约550a/m2的密度。例如,在电解期间,电流可以被至少基本上维持在约700至约1200a/m2的密度。例如,在电解期间,池的电压可以被至少基本上维持在约2至约10v,约3.0v至约8.5v,约6.5v至约8v,约5.5v至约6.5v,或者约6v的值。例如,在电渗析或电解期间,电压可以被至少基本上维持在约4.5v至约8.5v,约6.5v至约8v,约5.5v至约6.5v,或者约6v的值。例如,在电渗析或电解期间,电流可以被至少基本上维持在恒定值。例如,在电渗析或电解期间,电压可以被至少基本上维持在恒定值。例如,在所述过程期间,电压可以被至少基本上维持在约3至约10v或者约4至约7v的恒定值。例如,池的电压可以被至少基本上维持在约1.0v至约8.5v,约1.0v至约3.0v,约2.0v至约3.0v,约3.0v至约8.5v,约6.5v至约8v,约5.5v至约6.5v,或者约6v的值。例如,在电渗析或电解期间,总电流效率可以为约50%至约90%,约60%至约90%,约60%至约85%,约60%至约70%,约60%至约80%,约65%至约85%,约65%至约80%,约65%至约75%,约70%至约85%,或者约70%至约80%。例如,在电渗析或电解期间,总的lioh电流效率可以为约50%至约90%,约60%至约90%,约60%至约70%,约60%至约80%,约65%至约85%,约65%至约80%,约65%至约75%,约70%至约85%,或者约70%至约80%。例如,在电渗析或电解期间,总的h2so4电流效率可以为约55%至约95%,55%至约90%,约60%至约85%,约65%至约80%,约85%至约95%,或者约70%至约80%。例如,在电渗析或电解期间,总的h2so4电流效率可以为约55%至约90%,约60%至约85%,约65%至约80%,或者约70%至约80%。例如,用电解或电渗析产生lioh之后,可以获得包含li2so4和/或lihso4以及h2so4的混合物。例如,可以通过实施电渗析从所述混合物中至少部分地回收li2so4。例如,可将包含li+和至少一种金属离子的水性组合物与碱反应,以获得约4.8至约6.5,约5.0至约6.2,约5.2至约6.0,约5.4至约5.8,或者约5.6的ph。例如,可将包含li+和至少一种金属离子的水性组合物与石灰反应。例如,与碱反应以获得约4.5至约6.5的ph的水性组合物中包含的至少一种金属离子可选自fe2+、fe3+和al3+。例如,与碱反应以获得约4.5至约6.5的ph的水性组合物中包含的至少一种金属离子可以包括fe3+。例如,与碱反应以获得约4.5至约6.5的ph的水性组合物中包含的至少一种金属离子可以包括al3+。例如,与碱反应以获得约4.5至约6.5的ph的水性组合物中包含的至少一种金属离子可以包括fe3+和al3+。例如,沉淀物中包含的至少一种氢氧化物可以选自al(oh)3和fe(oh)3。例如,沉淀物可以包含为al(oh)3和fe(oh)3的至少两种氢氧化物。例如,经使用以获得约4.5至约6.5的ph的碱可以是石灰。例如,石灰可以以具有约15重量%至以重量计约25重量%的浓度的水性组合物形式提供。例如,所述方法还可包括将经与碱反应以获得约4.5至约6.5的ph的包含li+和至少一种金属离子的水性组合物维持在至少约350mv的氧化电位。例如,通过在水性组合物中喷射包含o2的气体使所述水性组合物可以被至少基本上维持在至少约350mv的氧化电位。例如,气体可以是空气。可选地,气体可以是o2。例如,所述方法可以包括将包含li+且含有降低含量的至少一种金属离子的水性组合物与另一种碱反应,以获得约9.5至约11.5,约10至约11,约10至约10.5,约9.8至约10.2,或者约10的ph。例如,经使用以获得约9.5至约11.5的ph的碱可以是naoh、koh或lioh。例如,经使用以获得约9.5至约11.5的ph的碱可以是naoh。例如,至少一种金属碳酸盐可选自na2co3、nahco3和(nh4)2co3。例如,碱和金属碳酸盐可以是水性naoh、nahco3、lioh和lihco3的混合物。例如,至少一种金属碳酸盐可以是na2co3。例如,包含li+且含有降低含量的至少一种金属离子的水性组合物可与另一种碱反应一段时间,所述时间足以将水性组合物中的所述至少一种金属离子的含量降低至低于预先确定的值。例如,至少一种金属离子可选自mg2+、ca2+和mn2+。例如,反应可以实施一段时间,所述时间足以将ca2+的含量降低至低于约250mg/l,约200mg/l,约150mg/l,或者约100mg/l。例如,反应可以实施一段时间,所述时间足以将mg2+的含量降低至低于约100mg/l,约50mg/l,约25mg/l,约20mg/l,约15mg/l或者约10mg/l。例如,离子交换树脂可以是阳离子树脂。例如,离子交换树脂可以是基本上对二价和/或三价金属离子具有选择性的阳离子树脂。例如,与离子交换树脂接触可允许将组合物的ca2+的含量降低至低于约10mg/l,约5mg/l,约1mg/l,或者约0.5mg/l。例如,与离子交换树脂接触可允许将组合物的mg2+的含量降低至低于约10mg/l,约5mg/l,约1mg/l,或者约0.5mg/l。例如,与离子交换树脂接触可允许将组合物的总二价离子的含量(如ca2+、mg2+和mn2+)降低至低于约10mg/l,约5mg/l,约1mg/l,或者约0.5mg/l。例如,可以用水浸提经酸焙烧的含锂的材料,以获得包含li+和选自下述金属的至少三种金属离子的水性组合物:铁、铝、锰以及镁。例如,可以用水浸提经酸焙烧的含锂的材料,以获得包含li+和选自下述中的至少三种金属离子的水性组合物:al3+、fe2+、fe3+、mg2+、ca2+、cr2+、cr3+、cr6+、zn2+以及mn2+。例如,可以用水浸提经酸焙烧的含锂的材料,以获得包含li+和选自下述中的至少四种金属离子的水性组合物:al3+、fe2+、fe3+、mg2+、ca2+、cr2+、cr3+、cr6+、zn2+以及mn2+。例如,在电渗析或电解期间,ph可以被至少基本上维持在约10至约12,约10.5至约12.5,或者约11至约12的值。例如,在第一电膜过程期间,消耗硫酸锂和/或硫酸氢锂以制备氢氧化锂可进行至预先确定的程度。例如,在本公开的过程中,在适于将锂化合物如硫酸锂和/或硫酸氢锂转化为氢氧化锂的条件下,将包含锂化合物如硫酸锂和/或硫酸氢锂的水性组合物进行第一电膜过程,以进行至预先确定的程度。对于本公开的具体方法而言合适的预先确定的程度的选择可由本领域技术人员做出。例如,在适于消耗锂化合物如硫酸锂和/或硫酸氢锂以制备氢氧化锂直至一种或多种竞争性副反应进行至预先确定的程度、例如进行至氢氧化锂的制备不再有效的程度的条件下,将包含锂化合物如硫酸锂和/或硫酸氢锂的水性组合物进行第一电膜过程。例如,其中所述第一电膜过程为在包括通过阳离子交换膜而与阴极电解液室隔开的阳极电解液室的第一电化学池中实施的双室单极或双极膜电解过程,锂化合物如硫酸锂和/或硫酸氢锂至氢氧化锂的转化可以继续进行直至氢氧根的电流效率不再有效,例如不再至少基本上维持氢氧根的电流效率,因而其减少。例如,其中第一电膜过程为在包括通过阳离子交换膜而与阴极电解液室隔开的阳极电解液室的第一电化学池中实施的双室单极或双极膜电解过程,锂化合物如硫酸锂和/或硫酸氢锂至氢氧化锂的转化可以继续进行直至阳极电解液室中的ph为约0.4至约1.2,约0.5至约0.8,约0.5至约0.7,或者约0.6的值。例如,其中第一电膜过程为在包括通过阳离子交换膜而与阴极电解液室隔开的阳极电解液室的第一电化学池中实施的双室单极或双极膜电解过程,锂化合物如硫酸锂和/或硫酸氢锂至氢氧化锂的转化可以继续进行,直至水性组合物中包含的硫酸锂和/或硫酸氢锂消耗特定的量。例如,预先确定的程度可以包括基于水性组合物中包含的硫酸锂和/或硫酸氢锂的总量,消耗水性组合物中包含的硫酸锂和/或硫酸氢锂的约30至约60重量%或者约30至约50重量%。例如,预先确定的程度可以包括消耗水性组合物中包含的硫酸锂和/或硫酸氢锂的约35至约45重量%。例如,预先确定的程度可以包括消耗水性组合物中包含的硫酸锂和/或硫酸氢锂的约38至约42%。例如,水性组合物可以包含硫酸锂,以及预先确定的程度可以包括消耗水性组合物中包含的硫酸锂的约30至约50%。例如,水性组合物可以包含硫酸锂,以及预先确定的程度可以包括消耗水性组合物中包含的硫酸锂的约35至约45%。例如,水性组合物可以包含硫酸锂,以及预先确定的程度可以包括消耗水性组合物中包含的硫酸锂的约38至约42%。例如,第一电膜过程可以包括三室膜电解过程,例如三室单极或双极膜电解过程;基本上由三室膜电解过程,例如三室单极或双极膜电解过程组成;或者由三室膜电解过程,例如三室单极或双极膜电解过程组成。例如,第一电膜过程可以包括双室膜电解过程,例如双室单极或双极膜电解过程;基本上由双室膜电解过程,例如双室单极或双极膜电解过程组成;或者由双室膜电解过程,例如双隔室单极或双极膜电解过程组成。例如,第一电膜过程可以包括三室膜电解过程,例如三室双极膜电解过程;基本上由三室膜电解过程,例如三室双极膜电解过程组成;或者由三室膜电解过程,例如三室双极膜电解过程组成。例如,第一电膜过程可以包括双室膜电解过程,例如双室双极膜电解过程;基本上由双室膜电解过程,例如双室双极膜电解过程组成;或者由双室膜电解过程,例如双室双极膜电解过程组成。例如,可以在包括通过阳离子交换膜而与阴极电解液室隔开的阳极电解液室的第一电化学池中实施双室膜电解过程如双室单极或双极膜电解过程。例如,阳离子交换膜可以包括磺化的聚四氟乙烯如nafiontm324阳离子交换膜或用于苛性碱浓缩的其它膜如fuma-techfkb或astomcmb阳离子交换膜;基本上由磺化的聚四氟乙烯如nafiontm324阳离子交换膜或用于苛性碱浓缩的其它膜如fuma-techfkb或astomcmb阳离子交换膜组成;或者由磺化的聚四氟乙烯如nafiontm324阳离子交换膜或用于苛性碱浓缩的其它膜如fuma-techfkb或astomcmb阳离子交换膜组成。对于本公开的具体方法而言合适的阳离子交换膜的选择可由本领域技术人员做出。例如,在双室膜电解过程如双室单极或双极膜电解过程期间,可将包含锂化合物如硫酸锂和/或硫酸氢锂的水性流引入阳极电解液室,可从阳极电解液室中移出第一锂减少的水性流,以及可从阴极电解液室中移出第一富含氢氧化锂的水性流。例如,在双室单极或双极膜电解过程的阴极电解液室中,氢氧化锂可以被至少基本上维持在约2m至约4m,约2.5至约3.5m,约2.8至约3.2m,或者约3m的浓度。例如,在双室单极或双极膜电解过程期间,可在约10℃至约100℃,约10℃至约100℃,约10℃至约90℃,约20℃至约85℃,或者约80℃的温度下,将包含锂化合物如硫酸锂和/或硫酸氢锂的水性流引入阳极电解液室。例如,在双室单极或双极膜电解过程期间,可在约20℃至约100℃,约20℃至约85℃,约20℃至约85℃,约60℃至约85℃,约70℃至约85℃,或者约80℃的温度下,从阳极电解液室中移出第一锂减少的水性流。例如,在双室单极或双极膜电解过程期间,第一电化学池中的温度可以被至少基本上维持在约60℃至约110℃,约60℃至约100℃,约60℃至约90℃,约60℃至约85℃,约75℃至约85℃,约50至约70℃,约55至约65℃,或者约80℃的值。例如,在双室单极或双极膜电解期间,电流密度可以被至少基本上维持在从约0.1ka/m2至约8000ka/m2,0.5ka/m2至约6ka/m2,约1ka/m2至约6ka/m2,约2ka/m2至约6ka/m2,或者约3ka/m2至约5ka/m2的值。例如,电流密度可以被至少基本上维持在选自约3ka/m2,约4ka/m2以及约5ka/m2的值。例如,电流密度可以被至少基本上维持在约4ka/m2的值。例如,在双室单极或双极膜电解过程中,电压可以被至少基本上维持在约3v至约8v,约5v至约10v,约4v至约6v,约4至约5,或者约4.5的值。例如,第一电化学池可具有约100m2至约2000m2,约100m2至约1000m2,约400m2至约500m2,或者约430m2的表面积。例如,第二电膜过程可以包括双室膜电解过程,例如双室单极或双极膜电解过程;基本上由双室膜电解过程,例如双室单极或双极膜电解过程组成;或者由双室膜电解过程,例如双室单极或双极膜电解过程组成。例如,第二电膜过程可以包括三室膜电解过程,例如三室单极或双极膜电解过程;基本上由三室膜电解过程,例如三室单极或双极膜电解过程组成;或者由三室膜电解过程,例如三室单极或双极膜电解过程组成。例如,可在包括通过阴离子交换膜而与中央室隔开的阳极电解液室和通过阳离子交换膜而与中央室隔开的阴极电解液室的第二电化学池中实施三室膜电解过程,如三室单极或双极膜电解过程。例如,阳离子交换膜可以包括磺化的聚四氟乙烯如nafiontm324阳离子交换膜或者用于苛性碱浓缩的其它膜如fuma-techfkb或astomcmb阳离子交换膜;基本上由磺化的聚四氟乙烯如nafiontm324阳离子交换膜或者用于苛性碱浓缩的其它膜如fuma-techfkb或astomcmb阳离子交换膜组成;或者由磺化的聚四氟乙烯如nafiontm324阳离子交换膜或者用于苛性碱浓缩的其它膜如fuma-techfkb或astomcmb阳离子交换膜组成。对于本公开的具体方法而言合适的阳离子交换膜的选择可由本领域技术人员做出。例如,在三室膜电解过程如三室单极或双极膜电解过程期间,可将第一锂减少的水性流引入中央室,可从中央室中移出第二锂减少的水性流,以及可从阴极电解液室中移出第二富含氢氧化锂的水性流。例如,三室膜电解过程如三室单极或双极膜电解过程还可包括在阳极电解液室中产生酸如硫酸,以及从阳极电解液室中移出含酸的水性流如含硫酸的水性流。对于本公开的具体方法而言合适的阴离子交换膜的选择可由本领域技术人员做出。例如,本领域技术人员将会理解,质子阻挡膜可以,例如用于同时产生酸如硫酸的方法中。例如,在三室单极或双极膜电解过程中,阴离子交换膜可以是质子阻挡膜。例如,质子阻挡膜可以是如fumatechfab、astomacm或asahiaav阴离子交换膜。例如,在三室单极或双极膜电解过程的阳极电解液室中,酸(如硫酸)可以被至少基本上维持在约0.1m至约2m的酸(如硫酸)浓度。例如,在三室单极或双极膜电解过程的阳极电解液室中,硫酸可以被至少基本上维持在约0.5m至约1.5m,约0.7m至约1.2m,或者约0.8m的硫酸浓度。例如,在三室膜电解过程的阴极电解液室中,氢氧化锂可以被至少基本上维持在约1m至约5.0m,约1m至约4.0m,约1.5m至约2.5m,约1.8m至约2.2m,或者约2m的浓度。例如,在三室单极或双极膜电解期间,可在约20℃至约85℃,约40℃至约85℃,约40℃至约75℃,约50℃至约70℃,约50℃至约65℃,或者约60℃的温度下,将第一锂减少的水性流引入中央室。例如,在三室单极或双极膜电解过程期间,可在约20℃至约80℃,约30℃至约70℃,约40℃至约80℃,或者约60℃的温度下,从阳极电解液室中移出第二锂减少的水性流。例如,在三室单极或双极膜电解过程中,第二电化学池中的温度可以被至少基本上维持在约30℃至约90℃,约40℃至约85℃,约50℃至约80℃,约50℃至约70℃,约50℃至约65℃,或者约60℃的值。例如,在三室单极或双极膜电解过程中,电流密度可以被至少基本上维持在约0.5ka/m2至约5ka/m2,约1ka/m2至约2ka/m2,约3ka/m2至约5ka/m2,约4ka/m2,或者约1.5ka/m2的值。例如,在三室单极或双极膜电解过程中,电压可以被至少基本上维持在约5v至约9v,约6v至约8v,约6.5v至约7.5v,或者约7v的值。例如,第二电化学池可以具有约的1000m2至约4000m2,约2000m2至约3000m2,或者约2700m2的池面积。可选地,例如,在本公开的方法中,三室单极或双极膜电解过程还可以包括将氨引入阳极电解液室中,在阳极电解液室中产生铵化合物如硫酸铵,以及从阳极电解液室中移出含铵化合物的水性流如含硫酸铵的水性流。对于本公开的具体方法而言合适的阴离子交换膜的选择可由本领域技术人员做出。例如,本领域技术人员将会理解,在不同时产生酸如硫酸的方法中,可使用不是质子阻挡膜的阴离子交换膜,因为其可能例如与质子阻挡膜相比能够经受更高的温度和/或具有更低的电阻。例如,在三室单极或双极膜电解过程中,阴离子交换膜可以不是质子阻挡膜。例如,阴离子交换膜可以是诸如astomaha阴离子交换膜或fuma-techfap的膜。例如,在三室单极或双极膜电解过程的阳极电解液室中,铵化合物(如硫酸铵)可以被至少基本上维持在约0.5m至约5m,约1m至约4m,或者约3m的铵化合物(如硫酸铵)浓度。例如,在三室单极或双极膜电解过程的阴极电解液室中,氢氧化锂可以被至少基本上维持在约1m至约4.0m,约1.5m至约2.5m,或者约2m的浓度。例如,本公开的方法还可包括使至少一部分的第二锂减少的水性流再循环至第一电膜过程。例如,再次使用双室单极或双极膜电解池以获得更高浓度的氢氧化锂是可能的。本领域技术人员还将理解,用于制备氢氧化锂的连续过程也可能是有用的。例如,当在第二电膜过程中,第二电化学池的中央室的ph达到约2至约12,约3至约10,约4至约9,约5至约8,或者约8的值时,第二锂减少的水性流可以再循环至第一电膜过程,以控制第一锂减少的水性流的ph的值高于0.4至约1.2,约0.5至约0.8,约0.5至约0.7,或者约0.6。例如,所述方法还可包括将再循环的第二锂减少的水性流呈送至第一电膜过程,直至阳极电解液室中的ph为约0.4至约1.2,约0.5至约0.8,约0.5至约0.7,或者约0.6的值,然后将第一锂减少的水性流再呈送至第二电膜过程。例如,可以至少基本上维持双室单极或双极膜电解过程的阳极电解液室和/或三室单极或双极膜电解过程的中央室中的ph。例如,可以通过调整双室单极或双极膜电解过程的电流密度、三室单极或双极膜电解过程的电流密度、第一锂减少的水性流的流动速率和第二锂减少的水性流的流动速率中的至少一种来至少基本上维持ph。例如,在双室单极或双极膜电解过程期间,硫酸锂和/或硫酸氢锂至氢氧化锂的转化可以进行至预先确定的程度。例如,在双室单极或双极膜电解过程期间,可将包含锂化合物如硫酸锂和/或硫酸氢锂的水性流引入阳极电解液室,可从阳极电解液室中移出第一锂减少的水性流,以及可从阴极电解液室中移出第一富含氢氧化锂的水性流;以及在三室单极或双极膜电解过程期间,可将第一锂减少的水性流引入中央室,可从中央室中移出第二锂减少的水性流,以及可从阴极电解液室中移出第二富含氢氧化锂的水性流。例如,所述方法还可包括将至少一部分的第二锂减少的水性流再循环至双室单极或双极膜电解过程。本领域技术人员将会理解,视情况,利用本文公开的实例,所述方法也可以发生改变。例如,可将至少一部分的本公开的方法作为分批过程进行操作。可选地,例如可将所述方法作为连续过程或半连续过程进行操作。例如,本领域技术人员将会理解,例如如本文所述的,可以通过调整双室单极或双极膜电解过程的电流密度和/或三室单极或双极膜电解过程的电流密度和/或在所述过程之间流动的流的流动速率,从而至少基本上维持双室单极或双极膜电解过程的阳极电解液室和/或三室单极或双极膜电解池的中央室中的ph。例如,可以至少基本上维持双室单极或双极膜电解过程的阳极电解液室和/或三室单极或双极膜电解过程的中央室中的ph。例如,可以通过调整双室单极或双极膜电解过程的电流密度、三室单极膜电解过程的电流密度、第一锂减少的水性流的流动速率和第二锂减少的水性流的流动速率中的至少一种来至少基本上维持ph。用于测量和/或监测ph的合适的手段的选择可由本领域技术人员做出。合适的电流密度和/或合适的流动速率的选择可由本领域技术人员做出。例如,本公开的方法还可进一步包括将至少一部分的第二富含氢氧化锂的水性流再循环至第一电膜过程。例如,方法还可包括从第一电化学池的阴极电解液室中移出第一含氢的流。例如,所述方法还可包括从第二电化学池的阴极电解液室中移出第二含氢的流。例如,所述方法还可包括从第一电化学池的阳极电解液室中移出第一含氧的流。例如,所述方法还可包括从第二电化学池的阳极电解液室中移出第二含氧的流。例如,经酸焙烧的含锂的材料可以是之前已与h2so4反应的β-锂辉石。例如,可以通过利用如ca504,477中所述的方法获得经酸焙烧的含锂的材料,在此通过引用将其整体并入。例如,经酸焙烧的含锂的材料可以是之前已与h2so4反应的α-锂辉石、β-锂辉石、锂云母、伟晶岩、透锂长石、锂磷铝石、水辉石、贾达尔石、蒙脱石、粘土或其混合物。例如,经碱焙烧的含锂的材料可以是之前已与na2co3以及与co2反应并最终经加热的β-锂辉石。例如,当实施经碱焙烧的锂材料的浸提时,在焙烧的矿石中可形成碳酸锂(在水中的溶解度很低)。然后可使其成浆,并喷射co2(例如在高压釜中),以将碳酸锂转化为可溶于水的碳酸氢锂,以及在约85至约95℃的温度下加热以驱除co2并再次沉淀更纯的碳酸锂。可以重复碳酸氢盐步骤,以获得更高的纯度等级。用氢氧化钠焙烧β-锂辉石并浸出可能需要纯化的氢氧化锂是可能的。在本公开的方法中,因此可以通过进一步添加一些碱、一些酸或者通过稀释来控制ph。可以如之前对通过喷射空气所指出的那样来控制orp。例如,当将所述包含li+和至少一种金属离子的水性组合物与碱反应,以获得约4.5至约6.5的ph并由此以至少一种氢氧化物的形式至少部分地沉淀至少一种金属离子以获得沉淀物时,至少一种金属离子的金属可以是fe、al、cr、zn或其混合物。例如,当将包含li+且含有降低含量的至少一种金属离子的水性组合物与另一种碱反应,以获得约9.5至约11.5的ph,以及任选地与至少一种金属碳酸盐反应,由此至少部分地沉淀至少一种金属离子时,至少一种金属离子的金属可以是mn、mg、ca或其混合物。例如,当将包含li+且含有降低含量的至少一种金属离子的水性组合物与离子交换树脂接触,以至少部分地去除至少一种金属离子时,至少一种金属离子可以是mg2+、ca2+或其混合物。实施例1如图1所示,可以通过例如利用这样的方法以及通过利用预浸提的含锂的材料作为原料,从而获得氢氧化锂。例如,可以使用不同的浸提的矿石,如经酸焙烧的β-锂辉石。图1中所示的方法还可用于生产碳酸锂。根据另一实施方案,原料可以是锂化合物,如硫酸锂、氯化锂或氟化锂。在此种情况下,所述过程将变得更短,并将在名为“膜电解”的框处开始。经酸焙烧的β-锂辉石(arβ-锂辉石)对两种不同的arβ-锂辉石的混合物进行测试。样品由不同比例的浮选和重介选(dms)浓缩物组成。样品被鉴定为75/25和50/50。在前的样品含有约75重量%的浮选浓缩物和约25重量%的dms浓缩物。在后的样品含有以质量计的基本上等份的两种浓缩物。给料样品的分析数据总结于表1中。两种样品具有十分相似的分析特征。75/25样品比50/50样品具有更高水平的fe、mn、mg、ca和k。两种样品都含有arβ-锂辉石的典型组合物。表1.arβ-锂辉石样品的分析数据浓缩物浸提(cl)和初次杂质去除(pir)浓缩物浸提(cl)和初次杂质去除(pir)的目的是1)溶解arβ-锂辉石中包含的硫酸锂,和2)从由给料固体中与锂共同浸提的过程溶液中去除主要杂质。将四槽级联用于组合的cl和pir过程回路(参见图2)。利用配置有振动给料器的给料漏斗添加arβ-锂辉石。各反应器均配置有下述:连接有4叶片斜叶轮(4-bladepitchimpeller)的顶置式混合器电机(0.5hp)、ph和orp(氧化还原电位)探针。pir反应器还具有直接位于叶轮下方的空气喷布器。过程泥浆经溢流口,通过重力从一个反应器流动至下一反应器。设置cl反应器的溢流口以使槽的有效容积为约32l。各pir反应器具有约14l的有效容积。来自pir槽3(槽队列的最后一个反应器)的溢出物被泵送至过滤站。在约85小时的操作中,浸提了约1,200kg的75/25arβ-锂辉石样品和约1,400kg的50/50arβ-锂辉石样品。从一种给料至另一给料的变化发生在操作的第37小时。操作的0时为浆状物开始从cl反应器溢出时。在cl步骤中,在搅拌槽中将水和固体以50:50的重量比合并,并在常温常压条件下混合约30至约45分钟。锂与非期望的脉石金属一同被提取,所述非期望的脉石金属例如:铁、铝、硅、锰以及镁。因此获得的泥浆(cl泥浆)包含固体组合物和含有溶解的li+(锂离子)以及溶解的上文提及的金属的离子的水性组合物(液体)。监测但不控制cl泥浆的ph和orp。可选地,最终可以通过进一步添加一些碱、一些酸或者通过稀释来控制ph。同时可以如之前对通过喷射空气所指示的那样来控制orp。cl泥浆通过重力流动至pir槽1。可选地,在引入pir槽1之前,可将水性组合物与固体组合物分离。在此种情况下,水性组合物(在本实施例的情况下而非全部cl泥浆)将被加进槽1中。操作9小时之后,存在足以再循环回cl的容积的清洗物1部分(清洗合并的cl和pir固体残余物时产生的第一置换清洗物部分)。清洗物1的初始再循环速率被设置为cl的水添加需求的约50%。在操作37小时之后,增加该量以组成添加至过程中的水的60%。该清洗流含有平均约12g/l的li(约95g/l的li2so4)。实施初次杂质去除(pir),以例如从水性组合物中基本上去除fe、al和si,同时基本上不沉淀任何锂。在该过程中,通过向三个pir槽中添加石灰泥浆,浓缩物浸提泥浆(包含水性组合物和固体组合物)的ph升高至约5.6。将石灰以具有约20wt%的浓度的泥浆形式添加。因此,cl泥浆转化为沉淀物和水性组合物。诸如fe、al和si的杂质被至少基本上沉淀为不可溶的金属氢氧化物,并存在于沉淀物中,而锂离子基本上存在于水性组合物中。pir回路的保留时间为约45至约60分钟。将空气喷射至pir槽中,以将过程泥浆的氧化电位维持在约350mv或高于350mv。在该水平下,以亚铁(fe2+)形式存在的铁将可能被氧化为三价铁(fe3+),在此ph下三价铁形式适于沉淀。因此,获得包含例如fe、al和si的金属氢氧化物的沉淀物,并且其最终与包含锂离子的水性组合物分离。因此在pir中,可以通过进一步添加一些碱、一些酸或者通过稀释来控制ph。可以如之前对通过喷射空气所指示的那样来控制orp。将产生的泥浆(包含水性组合物和固体组合物(包含沉淀物))在盘式过滤器(panfilter)上过滤。将滤液(包含锂离子且具有降低含量的上文提及的金属(如fe、al和si)的水性组合物)进行二次杂质去除(sir)。pir滤饼经历三次置换清洗。分别从后两个清洗物中收集第一清洗物部分。将第一清洗流作为水给料流的一部分而再循环至cl过程,以回收含有的锂。将清洗物部分2和3合并,并作为溶液存储。该溶液可被用于石灰泥浆补充物,以回收锂单元(lithiumunits)。图3中显示了cl和pir中的锂含量。在9小时,来自pir的第一清洗物部分被再循环回cl槽,以组成添加至浸提物的水的一半。因此整个回路中的锂含量增加至约18g/l(约142.6g/l的li2so4)。在37.5小时,增加再循环速率以组成添加至浸提物的水的60%,并且锂含量增加至约25g/l(约198g/l的li2so4)。pir第一清洗物的锂含量的范围为约12至约15g/l(约95g/l至约118.8g/l的li2so4)。一旦生产量降低,在整个操作中ph基本稳定。在操作期间,pir槽3中的泥浆的orp基本上稳定,并且高于约350mv。图4中显示了cl和pir的铁含量。在10小时和54小时,pir3的ph接近约5.6的值,但是pir3液中的铁含量增加。图4和图5中显示了铁和铝的分布。在整个运行中,cl槽中的铁和铝均显示出水平增加。尽管在cl中观察到了增加,但在大部分运行中,pir3中的铁水平维持低于约5mg/l。在前40小时,pir3中的铝低于约10mg/l,然后在剩余的操作时间中范围为约20至约65mg/l。表2显示了cl和pir回路的物料平衡。基于固体分析计算了锂提取物和杂质沉淀。物料平衡显示arβ-锂辉石给料中存在的总共约82%的锂进行至二次杂质去除(sir)。具体而言,对于75/25混合物,实现了约79%的锂提取,以及对于50/50混合物,实现了约86%的锂提取。铝和铁的未浸提的或者被沉淀的部分分别共计约96%以及约99%。其它测试已经显示可以从arβ-锂辉石获得约95%的提取收率。表2.cl与pir回路的物料平衡二次杂质去除对pir滤液(包含锂离子且含有降低含量的上文提及的金属(如fe、al和si)的水性组合物)进行二次杂质去除(sir),以基本上沉淀并从中去除ca、mg和mn杂质。向sir回路的给料添加在操作小时6(从cl槽溢出后6小时)时开始。在一个级联中存在四个处理槽(参见图2)。在运行期间,可以通过改变槽的溢流口而从约11.8至约17.5l调节槽容积。所有槽均带有挡板,并通过置顶式混合器进行搅拌。监测所有槽的ph、orp和温度。在前两个搅拌槽中,利用约2m的氢氧化钠(naoh)(另一种碱),将ph增加至约10。在该ph调节之后,向第三个槽中添加基于给料中的目标杂质水平的过量的碳酸钠(na2co3),以将剩余的二价杂质转化为不可溶的碳酸盐。将来自第三个槽的泥浆泵送至澄清器。将底流固体移出,并通过过滤回收,同时在1000l的箱(tote)中收集溢出的溶液。表3和图6中显示了从浓缩物浸提阶段直至二次杂质去除的最后的槽的溶液的平均杂质含量。表3.所选择的杂质的分布浸提阶段引入的杂质包括铁、铝、铬、锌、镁、锰以及钙。基本上所有的铬以及超过约98%的铁和铝被基本上沉淀于第一pir槽(pir1)中。在后两个pir(pir2和pir3)槽中发生最少的沉淀。通过sir的第一个槽(sir1),溶液中基本上仅剩余的杂质为镁和钙。所有其它元素均少于约1mg/l。尽管大部分沉淀发生于sir1中,但sir2的额外的保留时间使镁含量从约40mg/l降低至约20mg/l。从sir2至sir4,随着更多的保留时间,镁和钙的含量显示出稳定的下降。在实验装置运行期间,sir4的杂质水平平均为约1mg/lmn,约14mg/lmg,以及约241mg/lca。然而,通过将关键参数的优化,可以获得低至约200mg/lca以及约2mg/lmg的水平。在整个操作中监测ph和orp。仅在前两个槽中控制ph。最初,对sir2所选的ph为约10。在操作小时30处,sir2中的ph增加至约10.5。除sir2中的ph降低至约10的小时50处的两小时时期之外,其余运行时间的ph保持在约10.5。在两个阶段中达到的平均ph值为约10.1和约10.5,以及产生的氢氧化钠消耗分别为每小时约0.022kg氢氧化钠和约0.024kg氢氧化钠。总氢氧化钠消耗是每大约1000kg碳酸锂当量(lce)为约10千克氢氧化钠溶液。在图7中随时间绘出了sir2溶液的杂质含量。这些溶液的ph已经通过氢氧化钠调节至10以上,但是还未由碳酸钠计量。调节之后镁含量降低,但是其水平显示出似乎在设定值改变之前开始的逐渐向下的趋势。应当注意,在实验装置的后期,所有sir槽的保留时间均增加,这可能也导致了改善的沉淀能力。离开sir4的溶液中的钙和镁含量标绘于图8和图9中。这些图涉及关于在取样时使用的碳酸钠用量的杂质含量(仅mg和ca)。此外,基于在各样品的时间处的整个sir回路的保留时间标绘数据。在测试的范围内,随着碳酸钠的增加,金属含量降低。应当注意,最低的杂质含量也与较高的回路保留时间对应。将碳酸钠用量表示为在添加碳酸钠之前存在的钙杂质的摩尔过量(利用来自sir2的分析)。该数据指示ca的溶液含量可降低至低于约200mg/l。当来自sip回路的产物离开最后一个槽(sir4)时,每4小时对其进行分析(参见图2)。将sir4产物泵送至100l澄清器,并通过0.5μm的螺旋缠绕式筒式过滤器(spiralwoundcartridgefilter)过滤来自澄清器的溢出物,然后在1000l塑料箱中收集。对这些箱再次进行分析,以确认用于离子交换(ix)的大部分的钙给料含量(bulkcalciumfeedtenors)。当对所述箱进行取样时,在各个箱的底部观察到了浅棕色固体。分析显示从离开回路最后一个槽(sir4)的液体至箱中静置未混合的溶液中的钙含量的显著降低。对两种流的平均分析的比较显示于下文表4中。表4.时间对sir产物的影响sir回路的物料平衡显示于表5中。物料平衡显示总共约92%的镁和全部的锰存在于固体中。对于约99.1%的总sir锂回收,分配至固体中的锂为约0.9%。表5.sir回路的物料平衡离子交换通过离子交换(ix)回路处理sir产物,以在锂产物产生之前进一步降低ca和mg含量。ix回路包含三个填充有purolitetms950的柱,purolitetms950是对二价和三价金属离子具有选择性的可以钠的形式使用的阳离子树脂。purolitetms950包含负载于大孔径交联聚合物上的氨基膦酸树脂。可将其用于去除重金属阳离子。在高ph下,它可具有去除第2族金属阳离子(mg、ca和ba)以及cd、ni和co的活性。在高ph下,二价金属阳离子与单价金属阳离子(例如li、na、k)相比被优先吸收。将适于基本上选择性去除二价金属阳离子如ca2+和mg2+和/或三价金属阳离子的任何离子交换树脂均可被可选地用于本公开。可选地,可以使用多于一种类型的树脂以选择性地去除不同的金属阳离子。因此,对于不同的金属阳离子,可以使用不同的离子交换树脂。用于ix回路的操作原理为超前-滞后再生(lead-lagregeneration)过程(参见图2和图10)。回路的ix柱中的两个参与ca和mg的去除,而树脂再生循环在第三个柱上进行。图10中提供了阐明通过ix回路的溶液流动和超前-滞后再生过程的原理图。ca和mg的负载将发生于表示为超前和滞后的两个柱上,并且将产生具有低于约10mg/l的ca和mg溶液含量的流出物。作为用于下一负载循环的滞后柱,在再次引入之前,负载柱经历剥离和再生阶段。柱由干净的pvc管制备。各个柱具有约15cm的直径和约76cm的高度。各个柱的柱床体积为约10l。ix操作的参数总结于表6中。这些参数基于实验室测试结果,并且设计超前-滞后柱配置,以在滞后流出物中的ca和mg含量超过确定的上限之前处理75个柱床体积(bv)的给料溶液,确定的各阳离子的上限为约10mg/l。处理75个bv的给料溶液之后,超前和滞后柱中的树脂的合并的吸收能力将不足以产生ca和mg含量均低于约10mg/l的最终流出物。在此时,负载循环完成。超前柱进入再生阶段。滞后柱取代超前柱的位置。再生柱成为滞后柱。再生阶段包括用反渗透(ro)水清洗滞后柱,以排出柱中富含li的溶液。将该溶液传递至滞后柱。给料清洗阶段之后利用约2mhcl进行脱酸。这从树脂上去除了吸收的ca、mg、li以及其它金属阳离子。树脂当前为酸性形式。然后进行酸清洗阶段,以从柱上冲洗剩余的hcl(aq)。然后通过将约2mnaoh穿过柱而将树脂转化为na形式(再生阶段)。最终步骤包括利用反渗透(ro)水从柱上清洗过量的naoh。现在树脂被再生,并准备好进入用于下一负载周期的滞后位置。分别收集来自脱酸循环的流出物。将来自酸清洗、再生和再生清洗循环的流出物全部捕获在同一桶中。脱酸阶段产生了含有li、ca和mg的溶液。数据指示,首先从柱中洗脱出li,然后是ca和mg。分别捕获li部分并因而产生氯化锂溶液可以是可能的。表6.ix实验操作参数ix阶段溶液柱床体积(bv)速率,bv/h负载ix给料755给料清洗ro水1.55脱酸2mhcl35酸清洗ro水55再生2mnaoh35再生清洗ro水351bv=10l通过ix回路,在四个循环中处理了总共约2154lsir产物。各循环的给料溶液中的平均li、ca和mg含量总结于表7中。表7.ix-给料溶液中的平均li、ca和mg含量最初设计了循环以操作75个bv的负载阶段。平均负载流动速率为约832ml/分钟(约49.9l/h)。循环1是75个bv的给料溶液经过超前-滞后柱的仅有循环。图11显示了循环1的ca负载曲线,其中绘制了针对累积的经处理的柱床体积的来自超前和滞后柱的流出物的ca含量。在该图上还绘制了给料溶液中的平均ca含量以及对于本实施例所选择的滞后流出物中的ca含量的限度(约10mg/l)。超前柱的ca突破点发生在7.5个bv。75个bv之后,超前流出物中的ca含量为约82.3mg/l,指示对于ca未到达超前柱的负载能力。滞后柱的ca突破点发生在约35个bv。在第60个bv和第65个bv,滞后流出物中的ca含量增加至高于约10mg/l。决定继续循环1的负载阶段直至第75个bv的点,尽管滞后流出物中的ca高于约10mg/l。将来自第65个bv至第75个bv的点的流出物转移至200l桶中,并保持与循环1的主要产物溶液分离。当确定产生的合并的溶液中的ca含量将不会超过约10mg/l时,稍后将转移的溶液与循环1的主要产物合并。图12中显示了循环1的mg的类似负载特征。在该图中还包括给料溶液中的平均mg含量以及例如滞后流出物中的mg含量的上限(约10mg/l)。超前柱的mg的突破点发生在7.5个bv。在75个bv之后,超前流出物中的mg含量为约9.5mg/l。滞后柱的mg的突破点发生在52.5个bv。根据该实施例,在75个bv之后,滞后流出物的mg含量为约0.8mg/l,完全低于所选择的ix产物溶液中mg的限制水平。必须在75个bv的给料溶液可被经柱处理之前停止循环2和3。图13中绘制了针对累积的bv,各ix循环中滞后流出物的ca含量。在循环2的情况下,超前和滞后柱的ca突破点分别发生在<约7.5个bv以及约23个bv。在约68个bv之后停止循环2。在约60个bv之后,滞后流出物中的ca已达到约13mg/l。循环3的滞后柱的ca突破点发生在前5个bv内。在约30个bv之后停止循环3。在30个bv的点处的滞后流出物中的ca含量为约7.7mg/l。循环3给料溶液的平衡的处理超过循环4中的约36.4个bv。循环的超前柱和滞后柱的ca突破点分别发生在<约7.5个bv以及约7.5个bv。循环4滞后流出物ca含量数据的外推指示,在60个bv之后,产物溶液将具有>约10mg/l的ca含量。图14中绘制了针对累积的bv,各ix循环的滞后流出物的mg含量。明确的是,滞后流出物中的mg含量从未接近于临近约10mg/l水平的水平。图15中绘制了针对累积的bv,各ix循环的超前流出物的平均li含量。在该图中还包括给料溶液的平均li含量。数据指示,基本上无li负载于树脂上。图16中绘制了针对累积的bv,循环1和2的脱酸流出物中的li、ca和mg含量。数据指示,li首先从树脂中脱出,并达到例如在约0.5个bv和约1.5个bv范围内的上限含量。在大约1个bv开始从树脂上洗脱ca和mg,并且在约2个bv时两者都达到例如上限含量。在3个bv之后,从树脂上洗脱三种金属。循环3和4的ca和mg分布类似。基于kg/约1000kg,记录相对于的产生的lce的试剂消耗。从离子交换产生的硫酸锂流含有约39.1kg的li(这包含未经历sir和ix的pirpls样品中100%的锂单元)。假定下游过程中无损失,能够产生的该碳酸锂的等效质量将等于约187.7kg。ix回路产生了约2006l产物溶液。ix产物溶液的分析数据总结于表8中。li含量的范围为约15.7至约21.9g/l。ca和mg含量的范围分别为约2.4至约5.7mg/l以及<约0.07至约0.2mg/l。其它显要的组成分别为平均约3.5g/l以及约0.1g/l的na和k。表8中也列出了在分析技术的检测限度以下分析的元素。表8.ix产物溶液分析表9中提供了ix回路的物料平衡。获得了li的良好可计量性。在剥离/再生过程溶液中损失了约2.7%的li。该过程移除了给料溶液中包含的约97.6%的ca以及约99.0%的mg。通过将产物溶液中的ca和mg含量降低至低于约10mg/l的各金属阳离子,ix回路满足了所述方法的目的。此外,产生了高质量的硫酸锂溶液。表9.ix物料平衡对cl/pir残余物的混合样品的半定量x-射线衍射(sq-xrd)数据的检查显示,各样品既含有α-锂辉石又含有β-锂辉石。由两种给料样品(75/25和50/50)分别产生的cl/pir残余物的sq-xrd数据总结于表10中。α-锂辉石的存在指示,由第三方供应商进行的相变步骤(α-锂辉石的酸焙烧)不是100%有效的。因此以该形式存在的任何li将不能在化学上用于湿法冶金方法。应当注意,相变步骤(从α-锂辉石转化为β-锂辉石)的效率不是100%,因此湿法冶金方法的给料中所包含的li的一部分是α-锂辉石。表10.两种cl/pir残余物类型的sq-xrd数据cl/pir残余物中作为β-锂辉石的li单元决不可用于所述方法中,并且因此提供了错误的低li回收值。计算经调整的li回收率,所述回收率不考虑cl/pir残余物中作为β-锂辉石的li单元。该计算的数据总结于表11中。所有输出过程流中的全部li为约63.2kg。这包含cl/pir残余物中约11.7kg的以β-锂辉石存在的li。因此,经调整的总li输出值变为约51.6kg。通过整个过程总共可回收的li为约46.9kg。然后计算经调整的总li回收率为约95.8%。表11.经调整的总li回收率li质量,g基于分析的总li输出60615回收的总li46884cl/pir残余物中作为β-锂辉石的总li11655总li输出减去作为β-锂辉石的li48960经调整的总li回收率,%95.8因而产生了高等级的硫酸锂溶液。根据图1,该溶液可被用作,例如,产生高品质氢氧化锂和/或高品质碳酸锂溶液的锂源。该高等级的硫酸锂溶液还可被用作产生其它高等级的锂产物的给料。实施例2电解:将li2so4转化为liohi.简介使用nafiontm324阳离子交换膜。该膜是增强的全氟化的双层膜,其中设计有磺酸交换基团,以例如降低氢氧根的反迁移(导致较高的电流效率)。这可以通过面向阴极放置较高等效重量的聚合物层来实现。其也可在升高的温度下使用。一些替代物,例如较为便宜的阳离子交换膜可能也适用于本公开的方法,如nafion902、fumatechfkb和neoseptacmb。本文对两种不同的阴离子交换膜进行了测试。asahitmaav阴离子交换膜是例如在酸浓缩应用中使用的弱碱性的质子阻挡膜。在约40℃下对该膜进行测试。本文测试的第二阴离子交换膜是fumatechfab膜。该膜是具有极好的机械稳定性的酸稳定的质子阻挡膜,并且能够经受更高温度。在约60℃下对其进行测试。更高的操作温度可以,例如在过程给料溶液进入电解过程之前需要更少的对所述溶液的冷却,以及通过增加溶液和膜的电导率降低总的能量消耗。其还可以例如降低结晶回路中的氢氧化锂流所需的热量的量以及给料返回至溶解步骤所需的热量的量。ii.实验在配置有dsa-o2阳极、不锈钢阴极以及一对阴离子/阳离子交换膜的electrocellmp池中实施本实验。给料循环由绝缘的约5升的玻璃储存器组成,其中在该玻璃储存器周围缠绕有600瓦特的带状加热器。用iwakitmwmd-30lfx离心式循环泵循环该溶液。溶液ph、流动速率、温度和入口压力(进入池)均被监测和控制。还监测溶液的电导率。当需要时,利用蠕动泵以及作为储存器的量筒,向给料溶液中添加酸(或碱)用于ph控制。阳极电解液循环包括绝缘的约2升的玻璃储存器,其中在该玻璃存储器周围缠绕有300瓦特的加热带。用类似的泵将溶液循环至上文所述的器件。还检测并控制溶液的流动速率、温度和入口压力。利用流动速率可调节的蠕动泵将稀释水(为了控制浓度)直接添加至储存器。允许该储存器溢出至较大的聚丙烯收集储存器,然后溶液经蠕动泵从该较大的聚丙烯收集储存器循环回玻璃储存器。阴极液循环基本与阳极电解液循环类似。电极反应如下:阴极:h2o+e-→1/2h2+oh-阳极:h2o→1/2o2+2h++2e-图17中显示了池配置的图解。整个电解装备包含于通风橱内,以促进在电极处产生的氢气和氧气的适当排放。在实验期间取样,并利用简单的酸/碱滴定分析样品的酸度和碱度。还通过离子色谱法分析选择的样品的阴离子(硫酸根)和阳离子(锂和钠)。iii.结果与讨论在约40℃下用nafion324/asahiaav膜进行的实验.在该配置中进行两次实验(856-04和856-11)。表12总结了该实验中使用的参数。对于两种实验,应用约6.8伏特的恒定量。该电压最初是基于关于这些膜的操作条件的先前的经验而选择的。表12:使用aav的结果的总结.*在ic分析之前,通过用于样品中和的koh校正添加的na.实验#856-0485611膜naf324/aavnaf324/aav温度℃4040模式恒定6.8v恒定6.8v通过的电荷(摩尔e/%理论li)5.73/58.35.01/100.7时间(hr)14.2512.78avgcd(ma/cm2)107.7105初始[h2s04](摩尔)0.240.49最终[h2s04](摩尔)0.970.53酸ce62.465.1酸的水运送(mol/molso4)1.6-2.7初始酸中的[li]和[na](毫摩尔)0/0*0/2.4*最终酸中的[li]和[na](毫摩尔)0/0*0/2.1*初始碱中的[li]/[na]/[oh](摩尔)0.49/0/0.463.1/0.18/2.85最终碱中的[li]/[na]/[oh](摩尔)2.97/0.18/3.133.55/0.23/3.63碱ce82.473.3碱的水运送(mol/molli+na)7.47.0初始碱/最终碱中的[so4](毫摩尔)0.4/1.91.9/1.8初始给料中的[li]/[na]/[so4](摩尔)3.27/0.18/1.683.18/0.18/1.65最终给料中的[li]/[na]/[so4](摩尔)2.39/0.08/1.251.95/0.05/0.90移出的li%33.462.3ph控制在4.0的lioh(电荷%)18.25.7li物料平衡%10399so4物料平衡%101.597在第一个实验(#856-04)中,酸和碱的浓度都起始于约0.5n(约0.25m硫酸),并且经由电解而允许增加。允许通过添加稀释水而使酸强度达到约1m,然后在此处维持恒定,而允许碱浓度持续增加。图18中显示了浓度和产生的电流效率的图表。在整体电流效率为约82%时,达到了约3.13m的最终碱浓度。整体酸电流效率为约62%,其中最终酸强度为约0.97m。在实验期间,通过添加酸,给料ph最初降低至大约4,然后在此处得以维持。这需要在ph控制下对氢氧化锂进行计量,其也指示阳离子交换膜比阴离子交换膜更有效地发挥作用。维持该ph所需的氢氧化锂的量占电荷的约18%,并且如所期望的,其接近碱与酸的电流效率之间的差异。对于约33%的理论锂移出,整体电流密度为约108ma/cm2。作为与穿过膜的离子一同运送的水的量的度量的水运送被测量为:穿过nafion324膜进入碱性室的水运送为约7.4摩尔/摩尔li+na,以及穿过asahiaav膜进入酸性室的水运送为约1.6摩尔/摩尔硫酸根。在具有该膜配置的第二个实验(#856-11)中,酸强度在约0.5m的降低的浓度下维持恒定,以及最初使用更高的碱浓度(约2.85m),并且允许碱浓度升高至约3.63m。此外,使用较少的起始给料以便达到更高的消耗。在这些条件下,将给料ph维持在约4.0需要较少的氢氧化锂(当前对应于约6%),指示尽管两种膜的效率在一起更接近,但是nafion324膜的效率仍高于aav膜的效率。图19中显示了浓度和产生的电流效率的图表。整体的碱电流效率为约73%,以及酸电流效率为约65%。效率之间的差异再次与维持给料ph所需的氢氧化锂的量(约6%)充分对应。对于约62%的理论锂移出而言,该实验的整体电流密度与之前运行的约105ma/cm2十分相似。穿过nafion324的水运送率大约为约7.0摩尔/摩尔li+na。穿过asahiaav的水运送测量为约-2.7摩尔/摩尔硫酸根(即由于使用的酸浓度较低,因而水运送为从酸至给料)。在约60℃下用nafion324/fumatechfab膜进行的实验.初始基础测试在该配置中总共进行6次实验(#856-22至#856-63)。表13总结了前三次实验的结果,其被用于确定操纵过程变量时的不同的影响。表13:使用fab的结果的总结.*在ic分析之前,通过用于样品中和的koh校正添加的na.在第一个实验中(#856-22),酸强度最初为约0.46m,并通过添加稀释水而允许酸强度在维持恒定之前升高至约1m。初始氢氧化锂强度为约3.08m,并也通过添加稀释水允许其在维持恒定之前升高至约3.5m。图20中显示了浓度和产生的电流效率的图表。将给料ph预先调节至约4.0,然后在此处得以维持。这最初需要添加酸(fab膜比nafion324更有效),但稍后随着酸强度增加约两倍,需要添加氢氧化锂(nafion324变得更有效),并且反迁移至给料室的质子增加。在与用asahiaav膜进行的实验相同的恒定电压(池中约为6.8v)下运行池。测量的整体的酸电流效率为约65%,以及测量的碱电流效率为约70%。达到的平均电流密度为约102ma/cm2。图21中显示了电流密度、ph和电导率的分布。在第一部分实验期间观察到电流密度高至约123ma/cm2的突然增加,其随后在实验剩余时间逐渐下降。在不希望被理论所限制的情况下,该增加被认为与有助于降低fab膜的电阻的在此期间的硫酸强度的增加有关。fab膜的电导率可取决于它的ph(例如,在约为中性的硫酸钠溶液中fab膜可以具有约50ωcm2的电阻,但是在约为0.5m的硫酸溶液中电阻可降低至约16ωcm2(两个测量均在约25℃下),其为所划分的两种溶液的函数,即它是给料ph和酸的浓度这两项的函数。在该实验中途出现的电流密度和电导率的峰值是由于在稳定下来之前,在两天实验的第二天开始时溶液温度超过了约60℃的设定值。由于处理最小体积的给料所需的时长,在该次运行中移出的锂的量低至约56%。改进设备以使其可连续运行过夜,这将允许完成处理更大的体积。以此种方式运行下一实验,并作出其它改进,例如尽量增加电流密度和电流效率。酸和碱的浓度起始于较低浓度,其目的是在大部分时间在较低的浓度下运行并具有较高的效率,然后通过停止添加水允许两者的浓度增加至期望的值。作出的其它改变是为了在较低的ph(ph约为3或3以下)下运行给料,从而尽量降低fab膜的电阻。如图22中所示,观察到了显著不同且较低的电流密度分布。较低的酸和碱浓度将具有较低的电导率,并将导致较低的电流密度,但是其不足以解释观察到的所有降低。在不希望被理论所限制的情况下,对在稍后的运行之后的池的拆卸的观察结果提示,主要原因可能是nafionn324膜表面的污染。该污染似乎是膜表面(给料侧)的碳酸盐形成物,并且可能是在系统未运行期间形成的。在工作中稍后移出的膜含有少量的白色沉淀,其容易用酸去除(形成气体)。尚不清楚该沉淀是否是在较高ph下运行给料时形成的或者是否是在池被排干并允许来自空气的二氧化碳在膜表面(具有高ph)反应时形成的。在上述任一情况下,当系统在较低ph运行时,低电流密度看上去并不是问题。一旦给料ph达到约2(设定ph计量不允许记录低于约2的ph),电流密度显著增加。在夜间,实验被设定为在估计量的电荷处关闭。然而,由于该方法的效率略好于估计的效率,因而池继续运行,并且给料几乎全部被消耗(移出约99.7%的li)。尽管大致完全消耗是可能的,但是电流密度急剧降低。完全消耗还可能对膜不利,因为系统中的任何杂质均被迫使通过膜来运送。实验结束时的ph也显著增加,因为锂/钠浓度变得与质子运送相当。此时硫酸根的浓度为约18mm,并且主要以硫酸氢根形式存在。最终的酸和碱浓度分别低于之前运行的约0.8m以及约2.6m。对于酸产生而言,较低浓度产生了约为77%的较高整体电流效率,以及对于碱产生而言,约为73%。图23中显示了在运行期间计算的浓度以及电流效率。对于氢氧化锂产生而言的电流效率主要取决于它的浓度,同时也取决于给料溶液的ph。较高浓度的氢氧化锂导致穿过阳离子膜的羟基物质的较高反迁移,从而降低电流效率。同样地,给料溶液的ph越低,与锂离子竞争运送至阴极电解液室的可用的质子越多,同时导致较低的电流效率。氢氧化锂浓度也受运行给料至完成的影响。在低电流期间,将发生较低的电流效率,以及大量的渗透水从低浓度的给料移动至碱中。在测量的约8.3mol水/mol运送的锂/钠的相对高的水运送速率中反映了这种影响。此外,给料室的ph也十分依赖于正在产生的酸的浓度。酸产物的浓度越高,越多的质子穿过阴离子膜迁移至给料室,导致较低的酸电流效率以及较低的给料ph(如上文所讨论的,这影响苛性碱的电流效率)。用新的膜重建该池,并且除了使用较高的起始酸和碱浓度之外,重复实施之前的实验。图24显示在整个实验中,酸浓度被保持在从约0.9至约1.0m。碱以约2.4m起始,并且在整个运行中被允许增加至几乎约3m。对于酸和碱产生而言,电流效率分别为约77%和约75%。图25显示与第一次运行(856-22)相比,该运行的电流密度仍相对较低。其更类似于第二次运行(856-34),但是由于这次运行比856-34更早地结束(在约91%的锂移出时而非约99.7%时),平均电流密度明显较高,约为83ma/cm2。由于质子反向迁移的量,溶液的最终ph为约1.8。在该ph下,约60%的硫酸根以硫酸氢根形式存在于溶液中,其中溶液中仅含有约0.015m的质子。具有较低给料ph的n324/fab运行(生产运行)将三次实验的最终设置用于产生用于结晶研究的产物。表14中显示了测试的总结。使用较大体积,并试图通过在恒定的酸浓度和较低的给料ph下运行来增加先前运行的电流密度。通过在较低给料ph下运行,在运行之间不存在任何在较高ph(>约3)下运行给料时所见到的膜污染相关的问题。然而,酸和碱的电流效率均受损。这些运行中的其它差异为池使用其它电压:约7.8v而非约6.8v。在856-49期间,在早期做出这一改变,导致电流密度从约55ma/cm2至约95ma/cm2的增加。在确定功耗细节中,将使用较高的电压数。表14:用fab进行的生产运行的总结.*在ic分析之前,通过用于样品中和的koh校正添加的na.图26至图31显示了浓度和电流效率的图表。在较低ph下启动系统并允许给料ph降低对于该过程的电流效率是不利的。在商业化设备的情况下,可比在这些实验室实验中更好地控制给料ph。在较长期的运行中,向给料中添加硫酸,以在实验开始之前将其ph从约10降低至约3。这样做是为了完成给料体积,然后在操作中给料ph持续降低。然而,在设备中,可将较少的(asmallerhealof)给料溶液酸化,并且随着实验的继续,在约为10的ph下可以添加更多的给料。如果该过程持续运行而非以批处理模式运行,则产生类似的益处。根据这些实验估计实验结束时给料中超过一半的酸是因酸预处理而产生的。通过持续添加给料,质子浓度可从约0.15m下降至约0.075m,这将增加测量的电流效率。尽管在后三次运行中作出了小的改变以增加可实现的电流密度,但是获得的结果非常一致且是可重复的。碱电流效率和水运送的轻微改变是因为给料ph的变化。在测试期间,产生了约25l氢氧化锂以及约45l硫酸。iii.结论已显示利用使用nafion324阳离子交换膜以及asahiaav或fumatechfab阴离子交换膜进行的电解,在约40℃或约60℃的温度下,可高效率地从硫酸锂处理流中成功回收氢氧化锂。这两种阴离子膜在酸产生中都是有效的,但是在相似的电流效率下,fab膜允许更高的酸浓度。fab膜还可在更高温度(约60℃)下运行,因此其可以例如降低所需冷却的量。基于这些考虑,利用n324和fab的组合来阐述下述方法。利用n324/fab膜的方法基于进行的测试,预期该方法将具有下述特征:·产生的硫酸的浓度为约0.75m·产生的氢氧化锂的浓度为约3.2m·平均电流密度为约100ma/cm2·电流效率为约75%·池电压为约6v(对于计算,参见下文)·从给料至碱的水运送为约8mol水/mol阳离子·从给料至酸的水运送为<约1mol水/mol阳离子mp池中该过程的池电压为约7.8v。然而,实验室的池在电极和膜之间具有很大的流动缝隙(约10mm),这在较大设备的池中将大幅减少。缝隙通常可降低至约2mm,这将从总的池电压去除约1.8v(分别基于约275ms/cm,约400ms/cm和约70ms/cm的酸、碱和给料的电导率)。利用该降低的池电压和预测的电流效率,该过程将需要约8.9kwh/kg的lioh功耗(在约3.2m溶液中)。对于产生约3吨/小时lioh的设备,该设备将包含约4500m2的池面积,这将是与中等尺寸的氯-碱设备相当的大型电化学设备。除在较高的ph下运行时,对于膜或电极未发现稳定性问题。总结在本公开的研究中已经显示,利用使用nafion324阳离子交换膜以及asahiaav或fumatechfab阴离子交换膜进行的电解,在约40℃或约60℃的温度下,可高效率地从硫酸锂处理流中成功回收氢氧化锂。在这两种情况下,均产生作为副产物的硫酸。在测试的两种电解配置中均使用了nafion324膜。阳离子膜具有很好的锂产生效率,在超过约70%的电流效率下产生高至约3.6m的氢氧化物。在较低浓度下的较高效率显示是可能的,但是阴离子膜的低效限制了该需求。在不希望被理论所限制的情况下,较低的酸效率有效降低了给料溶液的ph,导致使用产生的一些氢氧化锂以维持ph或者导致质子与锂/钠竞争穿过阳离子膜。这有效地使该过程的效率与两种膜的最低效率相等。硫酸锂给料含有大浓度的钠离子。阳离子膜不是选择性的,因此产生的碱含有与给料中存在的钠离子比例粗略相同的钠离子。碱还包含约2mm(约200ppm)硫酸盐。并入asahiaav(在约40℃下)和fumatechfab(在约60℃下)这两种膜,获得约100ma/cm2的电流密度是可能的。然而,当酸浓度高于约0.5m时,aav膜给出低于约65%的电流效率。fab的酸效率更依赖于酸浓度,在约0.9m的酸浓度下给出约75%的电流效率。在高于该数值时,酸效率显著降低。当使用fab膜时,达到的电流密度十分依赖于给料溶液的ph(由于其在较高ph下的电阻)。为了达到与用aav膜的那些情况相似的电流密度,维持较低的给料ph是必要的。这通过下述中的一种来完成:增加产生的酸的强度,因此也增加穿过阴离子膜进入给料室的质子的反迁移;或者在较低给料ph下运行。这两种条件均被发现通过增加给料中的质子/li比例,因此也增加竞争进入阴极电解液室的质子,从而导致对酸产生以及氢氧化锂产生而言较低的电流效率。基于在本公开的研究中进行的测试,预期该方法将具有下述特征:·产生的硫酸的浓度为约0.75m·产生的氢氧化锂的浓度为约3.2m·平均电流密度为约100ma/cm2·电流效率为约75%·池电压为约6v(对于该方法,在工程化的池中)·从给料至碱的水运送为约8mol水/mol阳离子·从给料至酸的水运送为<约1mol水/mol阳离子尽管上述方法显示出前景,也可以采用产生硫酸铵而非硫酸的替代方法,并且下文给出所述方法的细节及其至少一些益处。实施例3lioh转化为li2co3碳酸锂产生小试设备包括两个回路——氢氧化锂碳化回路(lc)和碳酸氢锂分解回路(dc)。与过程溶液接触的所有装置均由玻璃、塑料或中的一种制成。由于液体的高腐蚀性和品质敏感性,未向过程中引入金属。将由实施例2产生的氢氧化锂溶液用作用于碳酸锂产生的给料。表15中列出了给料中选择的金属的含量。因此li的含量范围为约14g/l至约15.5g/l(或者lioh的含量范围为约48.3g/l至约53.5g/l)。表15.氢氧化锂溶液的选择分析数据图32中提供了lc回路方案。氢氧化锂碳化(lc)过程在封闭的4l反应器中进行。反应器配置有顶置式叶轮、喷射器、水平控制器、ph探针以及热电偶。例如,可将打嗝式喷射器(burp-typesparger)用于co2的添加。喷射器位于叶轮下方。例如,下方配置的喷射器可确保气体完全分散。利用电磁阀,通过反应泥浆的ph来控制co2的流动。将蠕动泵用于转移溶液和泥浆。来自lc的过程泥浆被持续泵送至lc澄清器,其中允许固体沉降,并且溶液相可以持续溢流回至lc反应器。从每一移动基底上的澄清器底流获得澄清器固体,并通过#3滤纸过滤。用热的反渗透水将滤饼浸没清洗三次,然后在设定至约105℃至约110℃的灶中的盘上干燥。回收的滤液被返回至lc回路。通过控制至dc回路的排放泵的水平传感器,将lc反应器水平维持在约3l的恒定容积。lc回路渗料线路(bleedline)使lc澄清器溢出物行进至dc反应器。图33中提供了dc回路方案。在封闭的4l反应器中进行dc过程。将反应器置于电热炉中,并配置有顶置式叶轮、ph探针和热电偶。将dc反应器中的溶液加热至约95℃,以分解碳酸氢锂,并从溶液中驱出剩余的碳酸锂。将产生的泥浆泵送至热的澄清器。从澄清器顶部采取渗料(bleed),并收集于dc过滤桶中。将澄清器中的dc渗料管入口置于固定的水平,并将排放泵设定成比至dc反应器的给料的流动速率更高的流动速率,从而维持dc反应器中的泥浆水平。在每一移动基底上收集增稠的浆。以与lc反应器固体相同方式处理滤饼。产生的固体代表次要的碳酸锂产物。该dc固体流与主要的碳酸盐流保持分离,并独立地进行表征。试验设备操作碳酸锂生产试验设备持续运行3天,每天24小时,其中每8小时换班,换三次。采取每小时的读数以监测lc和dc反应器中的温度和ph以及给料、co2和用过的溶液的投入速率和排放速率。每4小时收集来自lc回路渗料和dc回路渗料的随机采样的样品(grabsample),并使之进行用于锂分析的原子吸收光谱(被称为li-aas)。这些分析提供了该过程的性能的快速反馈。每4小时从lc和dc渗料流中收集混合样品,并合并成12小时混合样品。对混合样品进行li-aas分析以及利用电感耦合等离子体(icp扫描)的一系列其它元素的分析。每天采取给料的随机采样的样品,并使之进行li-aas和icp扫描分析。在试验设备操作期间,流向lc反应器的给料从约30ml/分钟增加至约60ml/分钟,以观察保留时间对lioh碳化效率的影响。试验设备的操作条件列于表16中。表16:试验设备操作的条件在3天的实验设备运行期间,产生了约12.5kg碳酸锂;从lc反应器获得了约9.9kg产物,以及从dc反应器获得了约2.6kg产物。在试验设备运行期间,产生的li2co3的质量总结于表17和18中。表17.从lc回路获得的碳酸锂固体表18.从dc回路获得的碳酸锂固体处理了约184升含有约14.7g/l的锂(或者约50.8g/l的氢氧化锂)的氢氧化锂溶液,并产生了约161升含有约1.39g/l的锂(或者约7.39g/l的碳酸锂)的用过的li2co3溶液。每天使用的材料的质量和体积总结于表9中。表19.用于试验设备操作的材料结果与讨论在测试的开始,将lc反应器填充氢氧化锂溶液并搅动。起始二氧化碳流,并且在一个半小时内,反应泥浆的ph从约12.6降低至约ph11.0的设定值。当目标ph接近时,开始试验设备操作的连续模式。开始向lc反应器中添加新鲜的氢氧化锂溶液,并且通过控制co2(g)的添加将反应泥浆的ph维持在约ph11.0的值。在操作约2.5小时之后,来自lc澄清器的溢流开始,并且来自lc回路的渗料被推进至dc反应器。预期来自lc反应器的渗料溶液将含有约3.5至约4g/l作为碳酸锂的li。lc回路溢出物中的li含量在4g/l附近波动,并且在图34中绘制了针对运行时间的含量数值。表20中总结了来自lc回路的混合溶液中的金属的分析数据,其中所述金属的浓度超过了分析检测限度。lc渗料含量与lc给料溶液的渗料含量的比较(表15)指示na和k含量仅最低限度地受lc过程的影响。表20:来自lc回路的渗料的混合样品中选择的金属的含量在试验设备运行期间,dc渗料中的锂含量为约1240mg/l至约1490mg/l。在dc过程中,观察到了碳酸锂溶液中锂含量的相当大的消耗(与lc渗料中的约2800mg/l至约4760mg/l的li相比)。来自dc回路的渗料中的选择的金属的分析结果总结于表21中。与lc过程类似,观察到了经过dc过程的na和k含量的最小改变(与表20和表21中的lc渗料以及dc渗料相比)。表21:来自dc回路的渗料的混合样品中选择的金属的含量图35中绘制了针对操作时间,来自的dc回路的渗料中的锂含量。表22总结了关于每12小时的试验设备操作时期的lioh给料溶液和二氧化碳气体用量的数据。表22中还包括关于用于分批或连续模式以及用于用增加的给料流动速率进行的测试的材料的数据。对于整体试验设备而言,以约90.2%的效率利用二氧化碳。将至lc反应器的给料流动速率从约30ml/分钟增加至约60ml/分钟对co2利用效率影响不大。表22:关于二氧化碳利用的数据试验设备期间产生的碳酸锂固体的分析数据总结于表23和表24中。来自除“lc固体批次13r”之外的所有批次的碳酸锂样品(表23)均符合所需的约99.9%纯度的碳酸锂规格。将来自批次“lc固体批次12”和“lc固体批次13r”的li2co3固体重新制浆以及重新清洗,以试图减少固体的na和k含量。将干燥的产物进行分析。重新制浆的碳酸锂含有显著较低量的na和k。由清洗测试推断,可通过额外的清洗从碳酸锂固体中去除na和k。表23.从lc回路获得的li2co3固体的分析结果表24:从dc回路获得的li2co3固体的分析结果表25:合并的li2co3产物的分析数据注意:li2co3等级由差异确定此外,dc回路产物具有比来自lc回路的固体更小的粒度:与lc产物中约80%的颗粒在约104μm以下相比,dc产物中约80%的颗粒在约57μm以下。整体试验设备的物料平衡总结于表26中。根据表中提供的数据,明显的是,约88%的锂被转化为碳酸锂固体。钠和钾不与碳酸锂一起沉淀。表26.物料平衡总结:因此证明用二氧化碳气体喷射氢氧化锂溶液是将氢氧化锂转化为高纯度和高品质的碳酸锂的有效方法。事实上,该过程的平均二氧化碳利用效率为约90%。还证明来自氢氧化锂的碳酸锂的产生可以以连续的方式操作。包括i)氢氧化锂碳化以及ii)碳酸氢锂分解和沉淀的碳酸锂产生方法显示是有效的。(i)和(ii)均产生了高等级的碳酸锂产物。试验设备产生约12.5kg的li2co3等级>99.9%的碳酸锂固体。实现的从lioh至li2co3的li转化为约88%。钠和钾不与li2co3共沉淀。实施例4利用氨中和酸的替代方法申请人之前已经在us61/788292(在此通过引用整体并入)中表明,利用使用nafion324阳离子交换膜以及asahiaav或fumatechfab阴离子交换膜进行的电解,在约40℃或约60℃的温度下,可高效率地从硫酸锂处理流中成功回收氢氧化锂。在这两种情况下,均产生作为副产物的硫酸。可以使用产生硫酸铵而非硫酸的替代方法,并且本公开的细节研究证实了其可行性。利用与us61/788292中相似的电解池实施测试,除了用neoseptatmaha膜替换高电阻的质子阻挡fumatechtmfab膜。aha膜是由astomtm(日本)制造的阴离子膜,其具有较高温度的稳定性(约80℃),对硫酸根运送具有良好的电阻。对于氢氧化物产生而言的电流效率(在约3m为约80%)与在当将给料维持在约中性ph时的之前的研究中获得的最高电流效率相匹配。以非常高的效率产生盐在最初是可能的。然而,随着批处理的进行,氢氧根的低效(约20%)引起给料ph的增加以及给料中与穿过aha膜运送的硫酸根竞争的氢氧根的增加。基于本研究中进行的测试,预期在约60℃下,利用nafion324膜和aha膜的连续方法将具有下述特征,并且在下文表27中将其与已知的硫酸方法的结果进行比较。表27.硫酸方法和硫酸铵方法的比较硫酸方法硫酸铵方法推荐的方法批处理连续处理膜n324/fabn324/aha硫酸/硫酸铵0.75m3m氢氧化锂3-3.2m3-3.2m平均电流密度100ma/cm2150ma/cm2氢氧根的电流效率75%80%自制池中的池电压6v4.6v水运送:给料至碱8mol水/mol阳离子8mol水/mol阳离子水运送:给料至酸<1mol水/mol阳离子12mol水/mol阳离子之前的研究(us61/788292)已经显示利用使用nafion324阳离子交换膜以及asahiaav或fumatechfab阴离子交换膜进行的电解,在约40℃或约60℃的温度下,可高效率地从硫酸锂处理流中成功回收氢氧化锂。在这两种情况下,均产生作为副产物的硫酸。硫酸的产生可以限制,例如系统中阴离子膜的选择、能够达到的酸浓度以及操作温度。某些阴离子交换膜如具有高电阻的质子阻挡膜,特别是对于硫酸根的运送具有高电阻的质子阻挡膜(如fumatechfab膜或类似的膜)可以,例如限制用于制备氢氧化锂的过程中所达到的电流密度。然而,这些膜可被限制于约60℃的温度。在类似的电解池可以产生高度浓缩的硫酸铵(>约2m),并且由于例如硫酸氢盐的缓冲能力和在溶液中溶解氨的能力,如图36中所示的使得阳极电解质溶液为非酸性的是可能的。这样,例如可能不需要质子阻挡的阴离子交换膜,并且可以使用替代的膜,例如能够在约80℃的温度下运行并应具有较低电阻的neoseptaaha膜。此种方法可能,例如移除较高电阻的fab膜,可能允许在较高电流密度(由此减少膜面积)、较低电压(由此减少功耗)或者这两者的组合下进行操作。其还可能,例如产生替代的商业材料。硫酸铵可作为化肥的组分出售,并且应具有比硫酸更高的价值。其还例如被预期在电解期间从给料中移出更多的水,由此允许在更宽范围的给料转化上的更有效的操作。其还可能,例如允许在需要溶液的较少冷却的较高温度下操作过程。在这些较高的温度下,溶液和膜也具有较低的电阻,降低了功耗。下文总结了在该系统中实施的测试,其中在之前的方法中使用的阴离子膜(fumatechfab)被neosepataaha(astomcorp.)膜替代,并且使用氨以控制池的“酸性”室的ph。在与之前的研究(us61/788292)中所使用的类似配置的electrocellmp池中实施实验,但是其中用neoseptaaha(astomcorp.)膜替代所述阴离子膜。不同的电解回路与之前的研究(us61/788292)中所使用的类似,除了向阳极电解液(酸/盐)回路中增添ph控制。ph控制器驱动电磁阀,这允许直接向阳极电解液储存器中添加氨。注意不允许阳极电解液的ph增加至高于约5,因为在高ph下dsa-o2涂层可被去除。除了之前实施的那些分析之外,通过阳离子离子色谱对铵离子进行分析。实验装置的所有其它方面均与之前描述的相同。在本研究期间,进行不同持续时间的实验。这些实验评估了温度、电流密度、给料转化、酸/盐浓度、碱浓度以及ph控制策略对电流效率、电压和水运送的影响。浓度范围和电流效率总结于表28中。在前两个实验中,允许碱浓度和酸/盐浓度从其起始值增加。第二个实验运行超过两天,以提供更大量的硫酸盐去除。在这种情况下,由于装置的容积限制,必须向给料中添加水以获得多于约90%的去除。在剩余的实验中,仅向酸性室和碱性室中添加水,以试图维持大致恒定的盐和碱浓度(模拟连续生产)。在大致恒定的酸(约2.5-3m硫酸根)和碱(约2.8-3.1m氢氧根)浓度下运行实验856-81至856-86,以调查不同温度和电流密度的影响。最后两个实验的酸性室的控制ph不同,以试图调节与产生的给料ph相关的问题。表28:硫酸铵产生的结果的总结.记录的各产物流的硫酸根的电流效率(ce).通常给料中硫酸根的电流效率应当与酸中硫酸根的电流效率相等。如表28中所示,在一些实验中存在高至约8%的差别。在不希望被理论所限制的情况下,主要误差可能是因为由容纳在设备中而导致的体积测量误差,例如当处理高浓度的溶液时。图37-43是关于表28中总结的实验的图:图37a-d涉及实验856-71;图38a-g涉及实验856-78;图39a-g涉及实验856-81;图40a-f涉及实验856-84;图41a-g涉及实验856-86;图42a-g涉及实验856-88;以及图43涉及实验856-90。下述部分进一步讨论了本研究的结果和所述方法的方面。氢氧化锂生产所述方法产生的氢氧化锂的氢氧根浓度为约3m。在整个测试中效率相当一致,在约150ma/cm2下给出的数值略低于约80%,在更高的电流密度下其增加至超过约80%。在最后的实验中,允许氢氧化锂浓度增加至约3.5m,并且电流效率减少约7%。在这些实验中,效率主要是氢氧根的反迁移,因为与之前的研究不同,给料的ph总是高于约7,这消除了任何质子运送。然而,还可能存在一些与铵运送相关的低效。如图39d中所示,氢氧化物的组合物主要是氢氧化锂/氢氧化钠,其中锂与钠的比例与给料中存在的类似。硫酸铵生产在大部分实验中,如图39e所示的将硫酸铵的浓度保持在约2.5至约3m的硫酸根,这提供了约90%的电流效率。效率的损失不可能是由于铵的反迁移。在硫酸铵处于低浓度的第一个实验中,在给料中存在极少的铵(<约20mm),其占电荷的少于约1%。当铵浓度增加时,铵浓度增加至约100mm,其仍少于电荷的约2%。进一步分析提示,剩余的电荷主要是由于氢氧根从给料至酸的运送。穿过n324膜的氢氧根反迁移引起给料ph增加。由于实验856-78运行至较高百分比的去除,因此该实验在较高的氢氧根浓度下运行持续较长时期,从而降低了穿过aha膜的硫酸根的电流效率。在下一部分中讨论该影响的其它细节及其后果。硫酸锂给料消耗在大部分实验中(除了856-78),未向给料中添加水。由于设备(以及更大批次所需的时间)的限制,大部分实验在约80%的转化之后结束。如图39g所示,由于水运送的量,在测试结束时硫酸锂的浓度仍然是高的。如果不发生水运送,则结束时硫酸根的浓度将为约0.35m。图39g也显示了给料中的氢氧根浓度随着通过的电荷的变化。如所示,即使在实验结束时,由于氢氧根从碱穿过n324膜反迁移,氢氧根的浓度仍然增加。在实验结束时,氢氧根的浓度与硫酸根的浓度类似,这降低了该方法的效率。最后,离开给料至酸性室的氢氧根的量将等于从碱进入的量,并且氢氧根的浓度将达到稳定状态。该浓度可能接近约1m氢氧根浓度。在较低的酸ph(阳极电解液ph)下的实验性试验例如,在本研究的一些实验中,由于给料中的氢氧根的反迁移,使给料ph增加。能够用于规避该问题的一种控制方法是向给料中添加硫酸,以将其ph维持在约7和10之间。由于氢氧化物的生产效率为约80%,因此将需要酸等于电荷的约20%。可选地,可以改变关于酸/盐的ph设定值,以允许一些质子反迁移。在这种情况下,如果给料ph高于某一测量的设定值(例如约9.5,约9.7,或者约10),则停止向酸中添加氨。酸的ph下降,这允许质子反迁移,直至给料ph降低至低于所需的设定值。然后向酸中添加氨,以增加ph,并重复该过程。上文提及的方法允许该过程的自校正,并且不需要任何外部的硫酸。将会理解,高浓度盐的溶液中的ph测量可能不准确,因为钠(和锂)离子,例如干扰测量的ph。通常测量的ph可以是与实际ph不同的一对ph单元;通常碱盐溶液中较低,酸中较高。将会理解,必须注意校正以及抵消该影响,例如当将ph用作控制算法时。本公开中显示的图表如所测量的。最后两个实验利用该类型的控制。856-88在约3.5的ph下,从约2.5m的硫酸铵开始,并允许在未添加任何其它氨的情况下运行。如图42b中所示,给料中的氢氧根的浓度持续增加直至经过该运行的约一半,然后浓度开始略微降低。这与测量的约10的给料ph一同发生,并且如图42c中所示测量的酸ph为约0.5。然而,仍然还未有足够的质子运送以消除给料ph的增加。已发生一些转化的点也与给料中所有硫酸根已经转化为硫酸氢根从而产生一些游离酸的点相对应。如图42e中所示,铵浓度等于约1.9mol电荷时的硫酸根的浓度(约2.5m(nh4)hso4)。最后的实验856-90是在前的实验的延续,除了使用新的给料溶液之外。如图43中所示,给料ph略微增加,然后在下降至ph约为7之前稳定,而酸ph继续降低。在约-0.25的记录的酸ph下,给料ph开始迅速降低,并且重新开始添加氨。酸ph再次增加至质子的反迁移被限制的点,并且给料ph开始增加。就在重新开始添加氨之前以及添加结束之后采取酸的样品。分析添加前的样品为约3.4m硫酸根,含有约0.6m质子(指示约3.1mnh4hso4加约0.3mh2so4)。添加氨之后,溶液再次为约3.4m硫酸根,但是含有约3.3m硫酸氢根和约0.1m硫酸根,指示游离的质子已经被中和。本测试证实,以此种方式运行该方法是可能的。氢氧化物生产、给料中硫酸根的去除以及酸中硫酸根生产的电流效率(如表28中所示)更加接近平衡。然而,该次运行的苛性碱强度略高,使得整体电流效率更接近约73%。在约为0的测量的ph下运行的盐中的铵浓度约为在约为3.5(即nh4hso4而非(nh4)2so4)的ph下运行的同样的硫酸盐浓溶液的浓度的一半,这将会减少铵反迁移的量,因此减少运送至碱的铵的量。池电压和水运送硫酸铵系统与硫酸系统相比的优势在于,当从所述方法中移除高电阻的fumatechfab膜时,可以获得可能较高的电流密度和较低的池电压。表29显示了当前工作中获得的池电压的范围,在约150ma/cm2下需要6v以及在约200ma/cm2下需要6.5v。在之前的工作中,使用约7.8v的恒定池电压,以获得约100ma/cm2的平均电流密度。因此在较低电压下获得了较高的电流密度。在约60℃下,具有约2mm溶液缝隙的池以低至约4.6v运行。将会理解,从prodcell至商业化的池存在较小的改变,因为可在较高的电导率下运行给料。当在约200ma/cm2下运行时,在约80℃下运行池使池电压降低了约0.6v。然而,在商业化的池中该影响可能更小,因为主要的改善是在溶液电导率方面,并且商业化的池具有更小的溶液缝隙。表29:池电压范围及水运送值。该系统中的水运送相当高,平均约为10mol水运送/mol电荷(约22mol水/mol硫酸锂运送)。这大约是维持恒定的给料浓度从而允许系统以完全连续过程而运行所需的水的约一半。在给料流中并入反渗透单元以去除剩余的水,由此允许给料完全转化是可能的。在较低的酸ph下运行的实验具有较低的相关水运送。在不希望被理论所限制的情况下,该影响可能是由于与质子反迁移相关的一些水运送以及进入酸中的较低渗透阻力。尽管两种溶液中硫酸根浓度相同,但是在后两个实验中存在更少的铵。水运送值引用每摩尔电荷。按照碱中阳离子的摩尔数,这些数值需要除以电流效率。按照进入酸中的硫酸根的摩尔数,这些数值需要乘以2并除以电流效率。基于本研究中实施的测试,所述方法可以,例如如果使用较低的ph控制,则产生约3m或更高浓度的硫酸铵;例如如果使用较低的酸的ph,则产生浓度约3m的氢氧化锂,具有约150ma/cm2的平均电流密度,对于氢氧化物生产具有约80%的电流效率,对于自设计的池具有约4.6v的池电压,具有约8mol水/mol阳离子的从给料至碱的水运送,以及具有约12mol水/硫酸根或更低的从给料至酸/盐的水运送。当与之前的硫酸方法相比时,这些条件可以,例如将产生约3吨/小时lioh的设备所需的池面积减少超过约35%。还可以,例如将工业化设计的池的功耗从约8.9kwh/kglioh降低至约6.4kwh/kglioh(在约3m溶液中)。还可以,例如根据给料ph控制方案,产生约8-10吨/小时的硫酸铵(以干重计)。氢氧根穿过n324膜的反迁移增加了给料的ph。该运送可能影响整个过程,以及可以使用不同的控制策略以提供稳定操作。例如可以使用三种不同的控制策略:例如可以使用硫酸将给料ph控制在中性附近至轻微的碱性ph(约7-9)。该方法,例如需要额外的控制回路,并且可能,例如需要购买硫酸。购买的额外的硫酸被转化为硫酸铵。氢氧化锂生产可能仍处于约80%的电流效率下,以及硫酸铵可能处于约90%-100%。低效可能是穿过aha的铵的反迁移。如果例如存在合适的硫酸来源以及产生的硫酸铵的出口,则该选择可能是有用的。例如,可能未实施校正,并且给料ph可能增加直至穿过aha的氢氧根的低效与穿过n324的氢氧根的低效相匹配。这可能,例如使得氢氧化锂和硫酸铵的效率相同。尽管可能是最容易实施的,但是可能例如需要考虑阴离子交换膜在高ph溶液和温度下的稳定性。例如,可以使用碱稳定的阴离子交换膜。例如,可以允许硫酸铵的ph的变化,以允许一些质子反迁移。如果给料ph增加,则停止添加至酸/盐的氨的量,在阳极产生质子直至足够的质子穿过aha迁移以使给料ph更低,然后再次发生氨添加。该方法再次与氢氧化锂和硫酸铵的生产相匹配,但是可能保持aha处的ph是低的。其还例如具有用较低的铵浓度运行酸/盐的益处。例如,约3m硫酸根的溶液在约0的ph下可能包含约0.5m硫酸以及约2.5m硫酸氢铵,但是在约4的ph下可能包含几乎约6m硫酸铵。这可能,例如降低在aha膜上的铵的反迁移的量。然后酸/盐溶液可以,例如用氨进行后中和,以产生约3m的(nh4)2so4溶液。例如还可以使用更高的硫酸根浓度。实施例5进一步考虑li2so4转化为lioh实施例图44中显示了本公开的方法的示例性流程图。其中举例说明的方法10用于制备氢氧化锂。参考图44,在其中举例说明的方法中,在适于消耗锂化合物如硫酸锂和/或硫酸氢锂以制备氢氧化锂的条件下,将包含锂化合物如硫酸锂和/或硫酸氢锂的水性组合物进行第一电膜过程,例如包含双室膜过程(如双室单极膜电解过程)的第一电膜过程,任选地其中用于制备氢氧化锂的锂化合物如硫酸锂和/或硫酸氢锂的消耗进行至预先确定的程度。参考图44,可在第一电化学池12中实施双室膜过程如双室单极膜电解过程,第一电化学池12包含通过膜如阳离子交换膜18而与阴极电解液室16隔开的阳极电解液室14。将会理解,关于锂化合物如硫酸锂和/或硫酸氢锂,本文使用的术语“消耗”指水性组合物中存在的锂化合物如硫酸锂和/或硫酸氢锂的量的降低。例如本领域技术人员将会容易地理解,在双室单极膜电解过程如图44中所示的过程期间,在阳极20处水(h2o)可以转化为质子(h+)和氧气(o2),在阴极22处水可以转化为氢氧根离子(oh–)和氢气(h2),以及最初存在于包含锂化合物如硫酸锂和/或硫酸氢锂的水性组合物中的锂离子(li+)可由电位差驱动从阳极电解液室14穿过膜如阳离子交换膜18而进入阴极电解液室16。由此获得第一锂减少的水性流24和第一富含氢氧化锂的水性流26,如图44所示,可分别将其从第一电化学池12的阳极电解液室14和阴极电解液室16移出。考虑到电流,li+离子通过膜18迁移,由此将li2so4转化为lioh。还可获得第一含氧的流27和第一含氢的流28,如图44所示,可分别将其从第一电化学池12的阳极电解液室14和阴极电解液室16移出。可选地,作为电解反应的产物而产生的氧气和/或氢气,还可以例如保留在水溶液中,并分别作为第一锂减少的水性流24和第一富含氢氧化锂的水性流26的组分被分别从第一电化学池12的阳极电解液室14和阴极电解液室16移出。如图44所示,可以使用包含锂化合物如硫酸锂和/或硫酸氢锂的水性流29,以将锂化合物如硫酸锂和/或硫酸氢锂引入第一电化学池12的阳极电解液室14。如图44所示,然后可在适于制备至少另一部分氢氧化锂的条件下,将第一锂减少的水性流24进行第二电膜过程,例如包括三室膜过程如三室膜电解过程的第二电膜过程。如图44所示,可在第二电化学池30中实施三室膜过程如三室膜电解过程,第二电化学池30包含通过膜如阴离子交换膜36而与中央室34隔开的阳极电解液室32以及通过膜如阳离子交换膜40而与中央室34隔开的阴极电解液室38。例如,本领域技术人员将会容易地理解,在三室单极膜电解过程如图44中所示的过程期间,在阳极42处水(h2o)可以转化为质子(h+)和氧气(o2),在阴极44处水可以转化为氢氧根离子(oh–)和氢气(h2),最初存在于第一锂减少的水性流24中的锂离子(li+)可由电位差驱动从中央室34穿过膜如阳离子交换膜40而进入阴极电解液室38,以及最初存在于第一锂减少的水性流24中的硫酸根离子(so42-)可由电位差驱动从中央室34穿过膜如阴离子交换膜36而进入阳极电解液室32。由此可获得第二锂减少的水性流46和第二富含氢氧化锂的水性流48,如图44中所示,可分别将其从第二电化学池30的中央室34和阴极电解液室38移出。事实上,第二锂减少的水性流46可被运送至阳极电解液室14中,而第二富含氢氧化锂的水性流48可被运送至阴极电解液室16中。如图44中所示,在三室单极膜电解过程期间,可将第一锂减少的水性流引入第二电化学池30的中央室34,可将第二锂减少的水性流46从第二电化学池30的中央室34移出,以及可将富含氢氧化锂的水性流48从第二电化学池30的阴极电解液室38移出。在本公开的方法中,三室单极膜电解过程可进一步包括在阳极电解液室32中产生硫酸。如图44中所示,因此可将作为含硫酸的水性流的流50从第二电化学池30的阳极电解液室32移出。可选地,三室单极膜电解过程可进一步包括将氨引入第二电化学池30的阳极电解液室32(例如经流52),并在第二电化学池30的阳极电解液室32中产生硫酸铵。如图44中所示,因此可将作为含硫酸铵的水性流的流50从第二电化学池30的阳极电解液室32移出。还可获得第二含氧的流54和第二含氢的流56,如图44中所示,可分别将其从第二电化学池30的阳极电解液室32和阴极电解液室38移出。可选地,作为电解反应的产物而产生的氧气和/或氢气,还可以例如保留在水溶液中,并分别作为流50和第二富含氢氧化锂的水性流48的组分被分别从第二电化学池30的阳极电解液室32和阴极电解液室38移出。本领域技术人员将会理解,可以使用其它流如流58、流60以及流62,例如以将其它试剂和/或溶剂引入第一电化学池12的阴极电解液室16,第二电化学池30的阴极电解液室38和/或第二电化学池30的阳极电解液室62。例如,可使用此类流以添加酸(例如h2so4)和/或碱(例如lioh),从而例如维持或改变ph;和/或添加水,从而例如维持或改变方法10的电化学池12、30的室中的浓度。本领域技术人员还将理解,也可将此类试剂和/或溶剂作为图44中显示或未显示的其它流的组分而引入图44中所示的电化学池12、30的不同的室,以维持或改变电化学池12、30的室中的参数如ph和/或反应物浓度(如li2so4、lihso4、lioh、nh3、nh4hso4、(nh4)2so4)。如图44中所示,本公开的方法可进一步包括将至少一部分的第二锂减少的水性流46再循环至第一电膜过程。例如,如图44中所示,可将第二锂减少的水性流46引入第一电化学池12的阳极电解液室14。例如,可使用泵,经合适的导管,将至少一部分的第二锂减少的水性流46从第二电化学池30送至第一电化学池12。如图44中所示,本公开的方法还可进一步包括将至少一部分的第二富含氢氧化锂的水性流48再循环至第一电膜过程。例如,如图44中所示,可将至少一部分的第二富含氢氧化锂的水性流48作为流58的组分而引入第一电化学池12的阴极电解液室16。本领域技术人员将会理解,将至少一部分的第二富含氢氧化锂的水性流48引入第一电化学池12的阴极电解液室16的可选方式是可能的。例如,可将至少一部分的第二富含氢氧化锂的水性流48作为单独的流而引入阴极电解液室16。例如,可使用泵,经合适的导管,将至少一部分的第二富含氢氧化锂的水性流48从第二电化学池30运送至第一电化学池12。例如,当根据li2so4和/或lihso4的消耗(例如通过电流效率降低所观察到的),池12中的li2so4和/或lihso4的电解已达到某预先确定的程度时,或者当阳极电解液室14中的阳极电解液的ph(例如使用ph计测量的ph)低于预先确定的值时,可将阳极电解液室14中的内容物(流24)运送至池30的中央室34。观察到在池12中,阳极电解液室14的ph可具有降低的趋势,并因此当反应效率降低或者不再有效时,将流24转移至室34,其中ph可具有增加的趋势直至达到电解效率降低或者不再有效的点。在此种情况下,可将流46运送至室14,其中ph将降低。li2so4和/或lihso4在室14和34之间的转移可通过相同的运送手段或不同的运送手段进行。此类手段可以是与泵结合的导管。本领域技术人员将会理解,在本公开的方法中,根据起始溶液(或给料溶液)(例如li2so4和/或lihso4的水溶液)的ph,可将起始溶液首先在双室单极膜电解过程池中进行处理(例如,如果ph为中性或碱性),然后在三室单极膜电解过程中进行处理。可选地,可将起始溶液首先在三室单极膜电解过程池中进行处理(例如,如果ph为中性或碱性),然后在双室单极膜电解过程池中进行处理。当在室38中达到某一lioh浓度时,可将流48运送至室16,其中可将lioh进一步浓缩。可将本公开的方法操作为例如批处理过程。可选地,可将本公开的方法操作为半连续过程或连续过程。本领域技术人员将会理解,可以例如通过本领域已知的手段监测本公开的方法的一个或多个参数,例如但不限于ph、温度、电流密度、电压、电流效率和浓度。用于监测本公开的方法中的具体参数的合适的手段的选择可由本领域技术人员做出。例如鉴于本领域技术人员的公知常识并参考本公开,本领域技术人员还可以维持和/或改变此类参数。某些已知的方法已经,例如并入了三室池的使用,因为在图45中所示的双室配置中,阳极反应产生氧气和质子,这导致阳极电解质溶液的ph降低。当使用双室池时,阳离子的完全移出可能变得低效,因为对于穿过阳离子膜的电荷转移而言,质子与锂离子运送相竞争。然而,利用双室膜电解池,锂化合物如硫酸锂部分转化为硫酸氢锂应当是可能的。硫酸氢根具有1.9的pka,因此硫酸根将缓冲硫酸锂水溶液的ph,以使在高至一半的硫酸根转化为硫酸氢根(即25%转化)时,质子浓度将为约0.01m。在该浓度下,由于nafion324(n324)膜处的质子,低效将是可忽略的。之前的工作已经显示,完全转化为硫酸氢根(即50%转化)的溶液的ph为约0.9,或者质子浓度稍高于0.1m。在这种情况下,由于质子比锂离子更可移动,因此穿过n324膜的质子运送将可能是显著的,这可以例如降低氢氧化锂生产的电流效率。因此,硫酸锂的完全转化将是不可能的,并且本公开中总结的测试研究集中于确定效率随着转化的变化。在本公开的方法中,利用双室膜电解过程将水溶液中的硫酸锂部分地转化之后(以将更多的锂转化为氢氧化锂),然后可将溶液送至三室膜电解过程。本文也记录了测试,其中通过两种方法处理双室工作中产生的溶液,以研究当给料溶液具有较低ph时的方法的操作。一般实验细节在配置有dsa-o2阳极、不锈钢(ss316)阴极和nafion324膜的icifm-01实验室电解池(64cm2,icichemicals,uk)中实施双室实验。在与之前研究中使用的三室膜电解池相似配置的electrocellmp池(100cm2)中实施三室研究,并且实验设备的其它方面与之前在其它申请(us61/636,869;us61/755,151;us61/788,292;pct/ca2013/000398)中描述的那些相同。实施例5a:双室膜电解池试验利用双室配置,用包含硫酸锂的水溶液作为给料溶液实施测试。由于这些运行的主要目的是评估电流效率随转化(硫酸氢根/硫酸根)的变化,因此用约2mlioh在阴极电解液室中实施测试。这低于在之前的研究中产生的约3m的浓度。然而,在约3m的浓度下,氢氧根浓度的微小变化能够显著地降低氢氧化锂的电流效率。相对而言,约2m浓度附近的氢氧根浓度的微小变化不明显影响氢氧化锂电流效率,因此效率的任何变化通常可归因于来自给料的质子运送。利用双室池,在不同的电流密度下实施不同的运行。图46-48为关于表30中总结的实验的图:图46a-46d涉及实验号856-96;图47a-47d涉及实验号856-99;以及图48a-48d涉及实验号879-1。下文讨论了利用双室池的实验结果以及这些运行的过程的方面。如例如图46a中所示,随着各运行的进行,锂和钠离子被从给料中移出。随着水被从给料中移出,硫酸根离子的浓度从约1.7m浓缩高至至约2.3m,其同锂离子运送出给料一起将给料中锂离子与硫酸根离子的比例从在电解开始时的超过约2变为结束时的低于约1。在该运行中,进行略多于约50%的转化,以使最终的阳极电解液溶液仅含有亚硫酸氢锂和少量的硫酸。在运行期间定期从两个室取样,并评估电流效率。图46b显示了阴极电解液中氢氧化物生产的累积的电流效率和来自给料的阳离子损失。如所显示的,在约35%的转化和约45%的转化时采集的样品之间的电流效率开始降低。尽管累积的电流效率的变化看上去是小的,但是增加的电流效率的变化(未显示)是相当大的。该变化似乎在测量的给料ph达到约0.6时发生。在较高电流密度下的运行具有相似的趋势。表30提供了在约3ka/m2(实验号856-96),约4ka/m2(实验号856-99)以及约5ka/m2(实验号879-1)的电流密度下实施的三次运行的结果。对于运行的开始部分,运行的氢氧根的电流效率接近约80%。对于利用较高电流密度的运行,电流效率开始降低的点似乎在稍后发生(即在较高转化下)。表30:用硫酸锂给料进行的双室运行的特征.图47a中显示了利用4ka/m2的电流密度的运行的电压分布。在大部分运行中,电压在开始时高,并随着运行的进行而降低。在图47a中,在运行期间,氢氧根浓度从约1.9m增加至约2.4m,这降低了阴极电解液室中的电压下降。所构建的icifm-01池具有约7mm的电极/膜缝隙。在缝隙可降低至约2mm的较大的商业化的池中,估计当利用作为约3m的包含氢氧化锂的水溶液的阳极电解质液时,整体电压将在约4.5-5v之间。因此,在约4ka/m2的电流密度下运行的双室膜电解过程的功耗将为约7kwh/kg(含lioh的3m溶液)。这与观察到的同时产生硫酸铵的三室池所需的功耗相当,除了过程仅在约1.5ka/m2的电流密度下运行之外。如果在约3吨/小时lioh的设备中利用双室池以转化约40%的硫酸锂时,在约400ma/cm2的电流密度下运行的池面积将为约430m2。如本文所讨论的,然后可用三室池处理剩余的约60%的硫酸锂。在讨论三室工作之后,下文将进一步讨论池面积估算。实施例5b:用转化的硫酸锂/硫酸氢锂进行的三室膜电解池试验双室工作可用于由硫酸锂溶液产生氢氧化锂,达到约40%的转化。由于过程溶液的量是小的,因此用合成制备的硫酸氢锂/硫酸锂溶液实施开始的两次运行,以适当地确定测试的条件。将来自双室工作的终溶液重新混合,并通过添加一些氢氧化锂,将其调整为约42%转化的溶液。为了去除可能的氢氧根浓度的影响,氢氧化锂的浓度下降至约2m。a.用于产生硫酸铵的n324/aha三室池将之前研究(us61/636,869;us61/755,151;us61/788,292;pct/ca2013/000398)中使用的三室池再次用于本研究的测试工作,并且其包含nafionn324阳离子交换膜和astomaha阴离子交换膜。图49a-d为关于该实验的图。该部分讨论了利用同时产生硫酸铵的三室池的实验的结果以及该方法的方面。以约200ma/cm2的电流密度,在池中运行含有约1.64mlihso4和约0.2mli2so4(即约85%的硫酸氢根)的起始溶液,其中移出阴极液中产生氢氧化锂/氢氧化钠的硫酸锂以及阳极电解液中的硫酸铵(在ph控制下,向给料中添加氨)。水由给料运送,但是向阳极电解液和阴极液中添加额外的水以基本上维持如图49a中所示的浓度。运行实验,其中从给料中移出约93%的硫酸根。如图49b中所示,在运行期间,由于硫酸根的移出比锂更高效,因此给料ph(其开始于约0.6)增加,在实验结束时达到稍高于约2。同样地,在整个运行中,给料中硫酸氢根的百分比降低直至大部分溶液作为硫酸根存在。池电压非常恒定在约7v,直至接近运行结束,其中给料开始消耗。测量的不同室的电流效率显示于图49c中,其证实了更有效的硫酸根移出。氢氧化物生产效率为约72%,而硫酸根的移出为约114%。高于100%的硫酸根移出是归因于这样的计算,即,假定“硫酸根”作为硫酸根(so42-)通过膜进行运送,而在这些ph下,一些运送必须是作为亚硫酸根(hso4-)。b.用于产生硫酸的n324/fab三室池重新构建三室电化学池,用一片新的fumatechfab膜替代astomaha膜,并实施类似的测试,在阳极电解液中产生硫酸。图50a-d为关于该实验的图。在该部分讨论了利用同时产生硫酸的三室池的实验的结果以及该方法的方面。在该实验中,如图50a中所示,向阳极电解液添加更多的水以将硫酸强度保持低于约0.8m。观察到了电流效率(图50b)和给料ph(图50c)的相似趋势。在这种情况下,由于使用了较低的电流密度(约100ma/cm2),仅约73%的硫酸根被移出,并且在实验运行中发生比实施例5b部分a中所讨论的实验更少的转化。尽管该测试的电流密度为之前的测试的一半,但是获得了相似的池电压。在不希望被理论所限制的情况下,这主要是由于fab膜的高电阻。与之前的研究(us61/636,869;us61/755,151;us61/788,292;pct/ca2013/000398)相比,这些测试中的氢氧根的电流效率低了约10%-15%。将池拆开,并在n324膜中看到了裂缝。裂缝位于衬垫区域,并且不应该造成问题。在不希望被理论所限制的情况下,该裂缝可能是通过因多次重建造成的塑料框的轻微变形(在较高温度下)而形成的。用一片新的n324膜实施运行,并且电流效率略微提高。用较高ph的硫酸锂溶液替代硫酸氢锂/硫酸锂溶液来实施最后的运行,并且电流效率提高至接近正常。在不希望被理论所限制的情况下,较低的给料ph似乎影响三室的生产。随着给料ph增加,电流效率未显著增加,这已经被预期。在不希望被理论所限制的情况下,据了解给料中的钙还可引起效率损失,例如在氯-碱工业中。因此显示,并入双室和三室膜电解池之组合的方法可用于将硫酸锂转化为氢氧化锂。双室池在产生氢氧根方面是有效的,直至约40%的转化。本测试还显示,当将产生的溶液在三室池中处理时,发生对于氢氧化物生产而言电流效率约10-15%之间的降低。对于氢氧化物的形成,观察到同时产生硫酸铵或硫酸的方法表现得相似。在双室池中处理约40%的锂值,显著降低了产生3吨/小时lioh所需的总的池面积。由于在约400ma/cm2的较高的电流密度下操作双室池,对于该方法而言,功率成本将是相似的。本领域技术人员将会理解,利用较低的电流密度将会降低功率,但会增加所需的池面积。表31:不同方法的池面积和功率[1]kwh/kglioh,在3m溶液中。例如由于双室池中使用的高电流密度,获得了对本系统的益处。然而,本领域技术人员将会理解,在这些电流密度下dsa-o2阳极的寿命减少。在本研究的方法中获得的对氢氧化物生产而言较低的电流效率将略微增加三室过程的池面积。然而,该低效假定了溶液的连续处理,其中将溶液从双室系统给送至运行三室池的单独的系统。可选地,两种类型的池均可流出相同的溶液,因此所述方法可在任何需要的ph下运行,并且可以例如通过改变一个池或另一个池所处理的百分比来增加或降低溶液的ph。例如,如果需要降低ph,则可以增加双室池的电流密度和/或可以降低三室池的电流密度。在用fumatechfab膜产生硫酸的情况下,ph将会被控制在1.5附近,例如以维持fab膜导电并将质子运送最小化。在用astomaha产生硫酸铵的情况下,之前的研究中报道的问题之一是阻止给料ph的增加,因为苛性碱的电流效率比硫酸根移出的电流效率低得多。本方法中使用的双室池可被用于将整体给料ph维持在更低的ph下。两种方法(即双室方法和三室方法)的组合还可允许更好地利用给料溶液,因为更大量的水被从给料中去除,这可能允许更连续的操作。已经关于具体实例来描述本公开。该描述意图帮助理解本公开,而非限制它的范围。对本领域技术人员而言显而易见的是,在不脱离如本文所述的本公开的范围的情况下,可对本公开进行各种修改,并且本文件意图涵盖这样的修改。因此观察到,通过利用高电流效率以及需要低的总的池面积,本公开的方法和系统对于以低成本将li2so4和/或lihso4转化为lioh是有效的。发现通过将双室单极或双极膜电解方法与双室单极或双极膜电解方法组合,此类较高的电流效率是可能的,由此导致这样的在电流和空间方面的节约。尽管具体参考特定的实施方案而做出描述,但是将会理解,本领域技术人员会想到对其的多种修改。因此,上述描述和附图应被作为特定实例,并且不具有限制意义。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1