一种仿生孔道结构SAPO-34分子筛的制备方法
本发明属于沸石分子筛合成技术领域,具体为一种仿生孔道结构sapo-34分子筛的制备方法。
背景技术:
石油化工的许多催化反应过程,如甲苯择形歧化、甲醇制烯烃(mto/mtp)以及今年来受到关注的重芳烃脱烷基或烷基转移等催化反应,往往要求催化剂具有亚纳米孔/纳米孔多级复合孔道结构,以适应择形催化反应的要求。因此,针对材料多级孔道构建方法与技术的研究非常重要。
在多孔催化材料中,孔道性质,包括孔道类型、大小、形状等,与催化反应过程中分子的吸附、扩散及择形筛分作用等密切相关,从而影响材料的催化活性、选择性以及积碳失活行为。因此,选择、构造或调变多孔材料的孔道性质是提高多孔催化材料性质的重要手段。多级孔材料也有叫梯度孔、等级孔材料,是具有两种或两种以上孔径的材料。微孔分子筛通常作为催化剂用于化学反应中,由于其孔道太小,一些大分子反应物无法进入,而大分子的生成物在生成之后又无法扩散出。根据孔径结构类型,多级复合孔材料大致可分成三类:①微孔-微孔复合分子筛,如两相共结晶分子筛、核壳结构分子筛等;这类材料由两种或两种以上微孔孔道复合,形成了特殊孔结构性质和特殊的酸性,有可能带来某些特异的催化性能。②介孔-微孔复合孔分子筛,也称介孔沸石。这类材料同时具有微孔和介孔两种孔道体系,可大大提高材料的扩散(物质传输)性能,改善材料的催化性能,同时介孔的存在有望提高大分子的催化转化能力,并抑制结焦失活。③微孔-大孔以及微孔-介孔-大孔复合材料。即在材料的制备过程中加入一些造孔剂,经过处理使其同时拥有微孔、介孔和/或大孔。这种具有不同尺度多级孔道系统的复合材料有利于提高物质的传输与扩散性能,便于进行改性处理,从而在催化转化中可能表现出独特的催化性能。
自从1992年由mobil公司的研究人员利用表面活性剂的超分子自组装作用合成m41s系列介孔分子筛(wo91/11390,1991)以来,目前己报道的有mcm-41和mcm-48等类型,该介孔材料(mcm-41和mcm-48)以其可调的规整孔道(其孔径可在1.5-10nm范围内调变,打破了常规分子筛孔径不能超过l.2nm的局限。然而,这些硅酸铝介孔材料的无定形孔壁使其稳定性和热稳定性较差,表面酸性也较低,因而不利于其实际应用。为了提高介孔材料的性能,人们试图进行具有介孔的沸石材料的构建(liuy.,etal.,j.am.chem.soc.,2000,122:8791;zhangz.t.,etal.,j.am.chem.soc.,2001,123,5014)。尽管人们相信所谓的介孔沸石的骨架可以由沸石晶体来构建,但实际中的材料仍是无定形的,因而酸性和水热稳定性仍没有提高。
尽管具有规则孔道结构的多级孔材料的合成在某些材料中取得了一定成果,但是其制备方法的推广与应用依然存在诸多困难,目前人们广泛合成的多级孔材料往往是具有不规则介孔结构的分子筛,其常规方法有刻蚀法、硬模板法、软模板法、固相合成法、原位合成法等。这些制备多级孔分子筛的方法中,刻蚀法属于后处理的方法,需要先合成微孔分子筛再以酸或碱溶液对其进行刻蚀,制备过程繁琐,还会造成原料浪费、环境污染等。硬模板法需要考虑硬模板及与分子筛结合的问题以确保分子筛能在硬模板上生长。软模板法中介孔模板剂和微孔模板剂存在竞争关系,而且往往需要特殊的软模板剂和用于合成分子筛的有机原料配合,两者都价格昂贵。介孔模板剂的作用条件较为温和,高温下其形成的胶束难以稳定存在,而分子筛的结晶温度一般较高,这也使得软模板法合成多级孔分子筛受到极大的限制。固相合成法只是利用原料在晶化过程中难以扩散的性质产生部分多级孔结构,其多级孔结构的状态和含量并不理想。原位合成法是利用特殊原料的稳定的初始结构来构造多级孔分子筛。
以分形结构为基础的仿生孔道结构具有贯通性好,扩散效率高的特点(何立群等,自然科学进展,2002,12:1167;陈景帝等,稀有金属材料与工程,2009,38:271.)水结晶为冰时形成的天然仿生分形结构,而且冷冻干燥也一直作为一种优秀的制备多级孔材料的方法而广受关注(nishiharahetal,journalofmaterialschemistry,2006,16(31):3231-3236;morihetal.journalofmaterialschemistry,2011,21(15):5677-5681;杨苗.冰模板法制备长程有序多孔材料.浙江:浙江大学,2017;胡钟元,化学工程与装备,2016,1:177.)。为了得到更合理的孔道分布,我们采取了冰晶造孔的方法,以期利用冰晶的天然分形结构构建分子筛的多级孔孔道。
技术实现要素:
为解决现有技术的缺点和不足之处,本发明的目的在于提供一种仿生孔道结构sapo-34分子筛的制备方法,用于解决现有技术中的问题。
本发明的技术方案是:一种仿生孔道结构sapo-34分子筛的制备方法,其特征在于,包括以下步骤:
(1)sapo-34分子筛初始凝胶的配制:将铝源、磷源、硅源及水混合均匀得到反应混合物,该反应混合物中nal2o3:np2o5:nsio2:nh2o=1:(0.1~1):(0.1~1):(5~130),制备得到晶种凝胶前驱体,将晶种凝胶前驱体置于-50~-30℃的温度下使其迅速冻结,待冻结完成后抽真空干燥,至样品温度与室温相同时取出样品并置于马弗炉中在300~700℃条件下焙烧1~5h,然后冷却至室温,获得焙烧后固体;
(2)仿生孔道结构sapo-34分子筛的合成:以导向剂和水配成导向剂水溶液,将焙烧后固体置于高压反应釜中,加入导向剂水溶液并搅拌使其分散均匀,静置12h后晶化处理,晶化温度为180℃,晶化时间为24~72h,经洗涤、干燥、焙烧后得到仿生孔道结构sapo-34分子筛样品。
本发明方法所制备的一种仿生孔道结构sapo-34分子筛具有层序状的介孔结构是根据冰晶的花样形状形成的,孔径可从6nm至20nm可调。
步骤(1)中所述晶种凝胶前驱体的制备过程为:称取铝源加水搅拌均匀,滴加质量浓度为10-60%的磷酸溶液,滴加完成后搅拌均匀滴入硅溶胶,之后补足所需水分进行充分搅拌,即得到晶种凝胶前驱体。
步骤(2)中导向剂为四丙基氢氧化铵。
铝源与导向剂的摩尔比为:1:(1~16)。
优选的,铝源与导向剂的摩尔比为:1:(1~6)。
本发明方法大大的减少了有机溶剂导向剂的用量,仅为现有技术制备方法中导向剂用量的20-50%,具有环境保护的效果。
步骤(2)干燥温度为90~150℃,干燥时间为5~20h;焙烧温度为300~550℃,焙烧时间为1~5h。
步骤(1)中铝源选自拟薄水铝石、偏铝酸钠、异丙醇铝或硫酸铝中的至少一种,磷源选自正磷酸、偏磷酸或亚磷酸中的至少一种,硅源选自硅溶胶、正硅酸甲酯、正硅酸乙酯、白炭黑或硅酸钠中的至少一种。
优选的,步骤(1)中的铝源为拟薄水铝石,磷源为正磷酸,硅源为硅溶胶。
步骤(1)中晶种凝胶前驱体在25~80℃条件下陈化6~8h后,再将晶种凝胶前驱体置于-50~-30℃的温度下使其迅速冻结。
晶种凝胶前驱体的陈化处理可以显著提高分子筛的结晶度,应是由于陈化处理的过程中使合成分子筛的原料进行了充分的反应,并初步聚合,在经焙烧后非常有利于sapo-34分子筛的晶化。
步骤(1)中晶种凝胶前驱体在80℃条件下陈化6h后,再将晶种凝胶前驱体置于-50~-30℃的温度下使其迅速冻结。
步骤(1)中将晶种凝胶前驱体在室温条件下缓慢升温陈化,陈化过程中保持升温速率为15~20℃/h,升温至80℃后保持3~4h,然后将晶种凝胶前驱体置于-50~-30℃的温度下使其迅速冻结。
本发明研究人员发现,采用逐步升温方法和80℃恒温陈化获得的样品晶化程度远远高于室温条件下陈化获得的样品,这是由于陈化温度的提高有利于磷酸和拟薄水铝石的充分反应生成凝胶,也使硅溶胶能更好的参与凝胶生成,凝胶体系更加均匀。而室温陈化由于温度较低使得磷酸与拟薄水铝石难以充分反应,在后续的冷冻干燥、焙烧过程中反应也无法继续进行,这势必造成部分原料无法晶化,使sapo-34分子筛结晶度降低。
在升温的过程中,逐步调整升温速率,发现采用15~20℃/h的升温速率陈化,能够更好的使磷酸和拟薄水铝石的充分反应生成凝胶,显著的提高最终仿生孔道结构sapo-34分子筛样品的结晶度。
本发明的另一目的在于提供一种权力要求1-9所述方法制备的仿生孔道结构sapo-34分子筛的用途,作为固体催化剂和吸附剂用于重油有机分子的催化转换和分离提纯。
与现有技术相比,本发明具有以下特点:
(1)本发明通过低温冷冻技术使水分形成冰晶,以水结晶为冰时形成的天然仿生分形结构为模板合成可工业应用的多级复合孔结构分子筛。由于冰晶的天然分形结构,本方法合成的多级孔分子筛具有更合理的孔道分布,孔容较高,提高了分子筛的总吸附量,且催化寿命长。在许多与传统分子筛有关的过程,如多相催化、吸附、分离以及离子交换,特别是用于大的有机分子的化学转化,如重油处理,具有超过传统分子筛的诸多优点,如低的扩散阻力。
(2)本发明合成分子筛的过程中,结构导向剂的用量大幅度地降低。该产品性能指标稳定、可实现持续批量生产。
本发明方法所制备的仿生孔道结构sapo-34分子筛的介孔孔体积为0.018-0.023m3/g,微孔孔体积为0.207-0.215m3/g,总的孔体积为0.230-0.233m3/g。
该技术具有很大的技术、环保、成本等优势,具有广阔的市场前景,经济效益、社会效益巨大。
附图说明
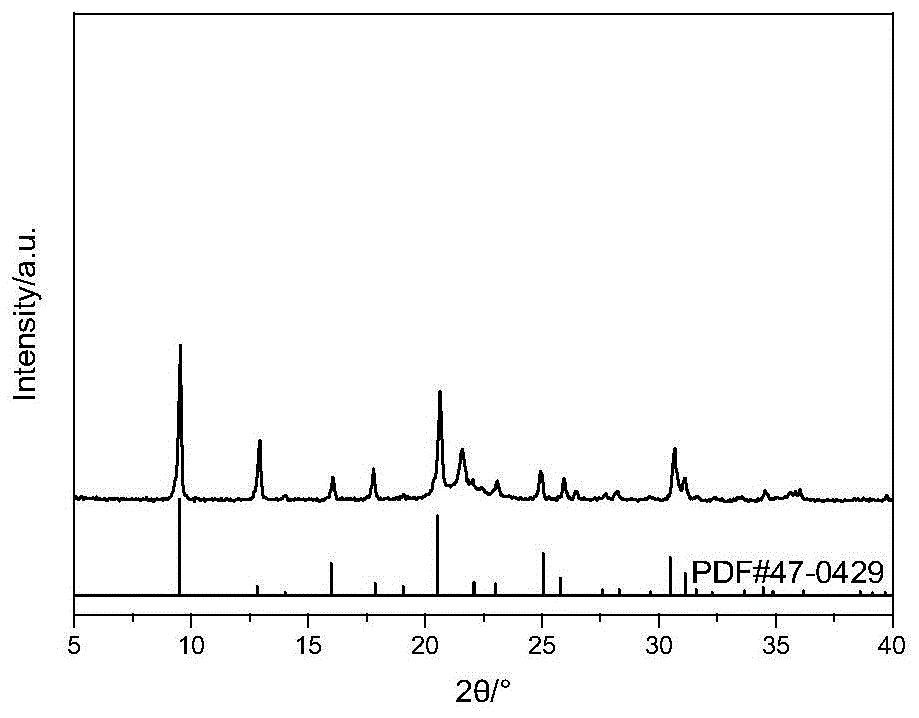
图1-3是本发明实施例1中不同方法合成sapo-34分子筛xrd图谱
图4是实施例1-3中不同晶化时间的仿生孔道结构sapo-34分子筛的n2吸附-脱附等温线
图5是实施例1-3中不同晶化时间的仿生孔道结构sapo-34分子筛孔分布
图6是实施例1-3中晶化之前的样品以及晶化不同时间仿生孔道结构sapo-34分子筛的sem照片a)未晶化样品,b)晶化12h样品,c,d)晶化24h样品,e)晶化36h样品,f)晶化48h样品
具体实施方式
下面通过实施例对本发明作进一步说明,但并不因此而限制本发明的内容。
试剂名称规格生产厂家
拟薄水铝石69.0wt%(al2o3)山东铝业集团
磷酸85.0wt%(h3po4)国药集团化学试剂有限公司
硅溶胶25.5wt%(sio2)青岛海洋化工有限公司
四丙基氢氧化铵99.0wt%(tpaoh)国药集团化学试剂有限公司
纯化水青岛成达蒸馏水有限公司
实施例l
sapo-34分子筛初始凝胶的配制:称取14.78g69.0wt%的拟薄水铝石,加入85ml纯化水搅拌均匀。称取4.61g85.0wt%的磷酸,并用5ml纯化水进行稀释,将稀磷酸滴入快速搅拌下的拟薄水铝石悬浊液中,进行充分搅拌后均匀滴入4.71g25.5wt%的硅溶胶,搅拌2h后得到晶种凝胶前驱体,将其置于冷冻干燥机冷冻室内于-30℃下冻结。待冻结完成后抽真空干燥24h,至样品温度与室温相同时取出样品置于马弗炉中300℃焙烧1h,冷却至室温备用。
仿生孔道结构sapo-34分子筛的合成:称取32.54gtpaoh,用100ml纯化水溶解得到导向剂溶液。将焙烧后的固体装入带聚四氟乙烯内衬的高压反应釜中,取上述导向剂溶液加入其中搅拌使其分散均匀。静置12h后于180℃下晶化24h,经洗涤、90℃干燥5h、300℃焙烧1h后得到sapo-34分子筛样品。
对照例1:
传统水热法合成sapo-34分子筛:称取14.78g69.0wt%的拟薄水铝石,加入25ml纯化水搅拌均匀。称取4.61g85.0wt%的磷酸,并用5ml纯化水进行稀释,将稀磷酸滴入快速搅拌下的拟薄水铝石悬浊液中,搅拌40min得到混合溶液。称取4.71g25.5wt%的硅溶胶滴入上述混合溶液中,搅拌1h后得到溶液a。取100ml纯化水、32.54gtpaoh及3.64gctab(十六烷基三甲基溴化铵)溶解后得到溶液b,将a与b两者混合,搅拌状态下老化2h,装入具有聚四氟乙烯内衬的不锈钢高压反应釜中,在180℃条件下晶化48h。晶化完成后经过洗涤、干燥、焙烧得到sapo-34分子筛。
对照例2
原位晶化法合成仿生孔道结构sapo-34分子筛:称取14.78g69.0wt%的拟薄水铝石,加入85ml纯化水搅拌均匀。称取4.61g85.0wt%的磷酸,并用5ml纯化水进行稀释,将稀磷酸滴入快速搅拌下的拟薄水铝石悬浊液中,进行充分搅拌后均匀滴入4.71g25.5wt%的硅溶胶,搅拌2h后得到晶种凝胶前驱体,将其置于-30℃条件下冻结。待冻结完成后抽真空干燥24h,至样品温度与室温相同时取出样品置于马弗炉中300℃焙烧1h,冷却至室温备用。取100ml纯化水、32.54gtpaoh及3.64gctab(十六烷基三甲基溴化铵)溶解后得到溶液a,称取相同质量的焙烧后固体同时装入具有聚四氟乙烯内胆的不锈钢高压反应釜中,静置12h后,于180℃下晶化48h,经洗涤、干燥、焙烧后得到sapo-34分子筛样品。
由图1xrd图谱可知原位合成法得到的样品xrd衍射峰与传统水热法得到的衍射峰强度差别不大,最终形成的分子筛应为微孔sapo-34分子筛。浸渍法在2θ=22°左右存在的衍射峰说明尚有未能晶化的alpo4存在,但已经晶化的原料得到的是纯相sapo-34分子筛。
实施例2
sapo-34分子筛初始凝胶的配制:称取14.78g69.0wt%的拟薄水铝石,加入115ml纯化水搅拌均匀。称取11.53g85.0wt%的磷酸,并用10ml纯化水进行稀释,将稀磷酸滴入快速搅拌下的拟薄水铝石悬浊液中,进行充分搅拌后均匀滴入14.14g25.5wt%的硅溶胶,搅拌2h后得到晶种凝胶前驱体,将其置于冷冻干燥机冷冻室内于-35℃下冻结。待冻结完成后抽真空干燥24h,至样品温度与室温相同时取出样品置于马弗炉中400℃焙烧3h,冷却至室温备用。
仿生孔道结构sapo-34分子筛的合成:称取65.08gtpaoh,用260ml纯化水溶解得到导向剂溶液。将焙烧后的固体装入带聚四氟乙烯内衬的高压反应釜中,取上述导向剂溶液加入其中搅拌使其分散均匀。静置12h后于180℃下晶化36h,经洗涤、120℃干燥10h、400℃焙烧3h后得到sapo-34分子筛样品。
实施例3
sapo-34分子筛初始凝胶的配制:称取14.78g69.0wt%的拟薄水铝石,加入165ml纯化水搅拌均匀。称取16.14g85.0wt%的磷酸,并用15ml纯化水进行稀释,将稀磷酸滴入快速搅拌下的拟薄水铝石悬浊液中,进行充分搅拌后均匀滴入14.14g25.5wt%的硅溶胶,搅拌2h后得到晶种凝胶前驱体,将其置于冷冻干燥机冷冻室内于-40℃下冻结。待冻结完成后抽真空干燥24h,至样品温度与室温相同时取出样品置于马弗炉中500℃焙烧3.5h,冷却至室温备用。
仿生孔道结构sapo-34分子筛的合成:称取93.55gtpaoh,用380ml纯化水溶解得到导向剂溶液。将焙烧后的固体装入带聚四氟乙烯内衬的高压反应釜中,取上述导向剂溶液加入其中搅拌使其分散均匀。静置12h后于180℃下晶化48h,经洗涤、130℃干燥15h、500℃焙烧3.5h后得到sapo-34分子筛样品。
实施例4
sapo-34分子筛初始凝胶的配制:称取14.78g69.0wt%的拟薄水铝石,加入200ml纯化水搅拌均匀。称取20.75g85.0wt%的磷酸,并用20ml纯化水进行稀释,将稀磷酸滴入快速搅拌下的拟薄水铝石悬浊液中,进行充分搅拌后均匀滴入18.85g25.5wt%的硅溶胶,搅拌2h后得到晶种凝胶前驱体,将其置于冷冻干燥机冷冻室内于-50℃下冻结。待冻结完成后抽真空干燥24h,至样品温度与室温相同时取出样品置于马弗炉中550℃焙烧5h,冷却至室温备用。
仿生孔道结构sapo-34分子筛的合成:称取122.02gtpaoh,用610ml纯化水溶解得到导向剂溶液。将焙烧后的固体装入带聚四氟乙烯内衬的高压反应釜中,取上述导向剂溶液加入其中搅拌使其分散均匀。静置12h后于180℃下晶化72h,经洗涤、150℃干燥15h、550℃焙烧5h后得到sapo-34分子筛样品。
实施例5
sapo-34分子筛初始凝胶的配制:称取14.78g69.0wt%的拟薄水铝石,加入85ml纯化水搅拌均匀。称取4.61g85.0wt%的磷酸,并用5ml纯化水进行稀释,将稀磷酸滴入快速搅拌下的拟薄水铝石悬浊液中,进行充分搅拌后均匀滴入4.71g25.5wt%的硅溶胶,搅拌2h后得到晶种凝胶前驱体,将晶种凝胶前驱体室温条件下陈化6h后,将其置于冷冻干燥机冷冻室内于-30℃下冻结。待冻结完成后抽真空干燥24h,至样品温度与室温相同时取出样品置于马弗炉中300℃焙烧1h,冷却至室温备用。
仿生孔道结构sapo-34分子筛的合成:称取32.54gtpaoh,用100ml纯化水溶解得到导向剂溶液。将焙烧后的固体装入带聚四氟乙烯内衬的高压反应釜中,取上述导向剂溶液加入其中搅拌使其分散均匀。静置12h后于180℃下晶化24h,经洗涤、90℃干燥5h、300℃焙烧1h后得到sapo-34分子筛样品。
实施例6
sapo-34分子筛初始凝胶的配制:称取14.78g69.0wt%的拟薄水铝石,加入85ml纯化水搅拌均匀。称取4.61g85.0wt%的磷酸,并用5ml纯化水进行稀释,将稀磷酸滴入快速搅拌下的拟薄水铝石悬浊液中,进行充分搅拌后均匀滴入4.71g25.5wt%的硅溶胶,搅拌2h后得到晶种凝胶前驱体,将晶种凝胶前驱体80℃条件下陈化6h后,将其置于冷冻干燥机冷冻室内于-30℃下冻结。待冻结完成后抽真空干燥24h,至样品温度与室温相同时取出样品置于马弗炉中300℃焙烧1h,冷却至室温备用。
仿生孔道结构sapo-34分子筛的合成:称取32.54gtpaoh,用100ml纯化水溶解得到导向剂溶液。将焙烧后的固体装入带聚四氟乙烯内衬的高压反应釜中,取上述导向剂溶液加入其中搅拌使其分散均匀。静置12h后于180℃下晶化24h,经洗涤、90℃干燥5h、300℃焙烧1h后得到sapo-34分子筛样品。
实施例7
sapo-34分子筛初始凝胶的配制:称取14.78g69.0wt%的拟薄水铝石,加入200ml纯化水搅拌均匀。称取20.75g85.0wt%的磷酸,并用20ml纯化水进行稀释,将稀磷酸滴入快速搅拌下的拟薄水铝石悬浊液中,进行充分搅拌后均匀滴入18.85g25.5wt%的硅溶胶,搅拌2h后得到晶种凝胶前驱体,将晶种凝胶前驱体置于加热箱内缓慢升温陈化,陈化过程中保持升温速率为15-20℃/h,升温至80℃后保持3-4h,将其置于冷冻干燥机冷冻室内于-50℃下冻结。待冻结完成后抽真空干燥24h,至样品温度与室温相同时取出样品置于马弗炉中550℃焙烧5h,冷却至室温备用。
仿生孔道结构sapo-34分子筛的合成:称取65.08gtpaoh,用260ml纯化水溶解得到导向剂溶液。将焙烧后的固体装入带聚四氟乙烯内衬的高压反应釜中,取上述导向剂溶液加入其中搅拌使其分散均匀。静置12h后于180℃下晶化72h,经洗涤、150℃干燥15h、550℃焙烧5h后得到sapo-34分子筛样品。
图4bet可以看出样品均含有从介孔到大孔的多级孔结构。由图5孔径图可知本发明方法合成仿生孔道结构sapo-34分子筛时,晶体在成长过程中虽然没有介孔模板剂的支撑,却也可以较好的保持介孔结构。由图6sem可知图a中样品是层状结构通道,图b中样品是小颗粒堆砌而成的不规则多级孔通道,晶化24h后继续延长晶化时间,样品的宏观形貌变化不大,均为多级孔结构。
以上对本发明的实施例进行了详细说明,但所述内容仅为本发明的较佳实施例,不能被认为用于限定本发明的实施范围。凡依本发明申请范围所作的均等变化与改进等,均应仍归属于本发明的专利涵盖范围之内。
- 还没有人留言评论。精彩留言会获得点赞!