一种氮化物纤维增强复合材料及其制备方法和应用与流程
1.本发明涉及纤维复合材料技术领域,尤其涉及一种氮化物纤维增强复合材料及其制备方法和应用。
背景技术:
2.高速飞行器的发展对耐更高温度的高性能透波材料提出了更高的要求。纤维增强硅硼氮透波复合材料中,因基体中硼元素的加入,提高了基体的耐温性,得到的复合材料具有良好的高温力学性能、稳定的介电性能及较好的抗烧蚀性能,是新一代飞行器天线罩的理想材料。目前,制备硅硼氮、氮化硅等氮化物为基体的复合材料主要工艺方法为pip工艺。
3.在纤维增强复合材料中,纤维是复合材料强度和韧性的主要来源。由于此类复合材料中的基体由前驱体通过有机
‑
无机转化形成,具有较高的活性和侵蚀性,在界面处容易与纤维发生界面反应而导致结合过强,纤维无法拔出而发挥增强增韧的作用,发生类似脆性断裂的现象,导致复合材料的力学性能较低;因此,通过一定的方式弱化界面结合强度,进而改善复合材料的力学性能,对复合材料的界面结合状态的改善具有重要作用。
4.通过在纤维和基体间引入间隙界面相的方法可以更好的实现纤维的增强增韧效果。目前常用的界面相为碳涂层和氮化硼涂层,由于碳会影响介电性能不适合用于透波陶瓷基复合材料中,因此一般选择氮化硼涂层作为界面相。化学气相沉积法是制备均匀光滑的氮化硼涂层的常用方法。氮化硼涂层的层状结构可以促进裂纹在涂层内部的偏转,弱化界面结合强度,进而改善复合材料的力学性能,对复合材料的界面结合状态的改善具有重要作用。
5.因此,改善氮化硅纤维与基体间的界面结合状态,可以更好地发挥纤维的增强增韧的作用,进而提高复合材料的力学性能,有益于复合材料应用范围的扩展,对于纤维增强氮化物复合材料的发展具有重要意义。
技术实现要素:
6.本发明要解决的技术问题在于氮化物前驱体与纤维之间因界面结合过强导致的纤维增韧效果不明显,导致复合材料力学性能较低的问题,针对现有技术中的缺陷,提供一种氮化物纤维增强复合材料及其制备方法和应用。
7.为了解决上述技术问题,第一方面本发明提供了一种氮化物纤维增强复合材料的制备方法,所述制备方法包括如下步骤:
8.(1)采用氮化硅纤维通过三维编织制备得到预制体;
9.(2)通过化学气相沉积对步骤(1)得到的预制体进行处理,得到表面具有氮化硼(bn)界面层的纤维预制体;
10.(3)将步骤(2)得到的具有界面层的纤维预制体进行氮化物前驱体的浸渍,然后进行交联固化和裂解;
11.(4)重复步骤(3)至少一次得到所述氮化物纤维增强复合材料。
12.本发明提供的制备方法可制得具有更好的界面结合状态、前驱体与纤维预制体之间的交联较充分、综合性能较好的复合材料。本发明尤其利用化学气相沉积法处理与前驱体浸渍固化裂解的方式,解决了界面结合的问题,使得基体与纤维的界面结合更适宜,纤维增强作用更显著,复合材料强度更高。
13.优选地,步骤(1)中所述预制体的纤维提及含量为30%~50%,例如可以是30%、35%、40%、45%或50%等。
14.在本发明中,步骤(1)中优选采用纤维体积含量为30%~50%的预制体,兼顾较好的性能与可操作性。对于预制体来说,一般纤维含量越高,经过致密化后最终材料的强度越高,但编织难度也越大,且孔隙小导致浸渍难度也越大。同时,本发明所述预制体的长度和宽度可根据实际需要确定,一般控制在100mm至400mm的范围内,厚度一般在5mm至20mm的范围内。
15.优选地,所述化学气相沉积为双组元化学气相沉积法;化学气相沉积法中,根据氮化硼产物中氮和硼两种元素是否来源于同一种化合物,可以分为双组元和单组元,氮源和硼源为不同物质时为双组元体系。由于单组元化学气相沉积法合成难度大,价格昂贵难以广泛应用,因此一般选择双组元化学气相沉积法作为试验方法。通过调节反应室气体、温度、时间等控制涂层厚度,其中添加氢气用来抑制化学反应式(1)的反应程度,添加氮气作为稀释气。具体化学反应式如下:
16.nh3+bcl3=3hcl+bn
ꢀꢀꢀ
(1)
17.在本发明中,化学气相沉积处理的次数为1次,并且沉积处理的参数可根据需要进行优化:通过选择合适的沉积温度和沉积时间,以完成纤维预制体的表面涂层沉积,这是改善界面结合状态、提高复合材料力学性能的重要途径。
18.化学气相沉积法,相比现有的加热氧化处理方法的操作更为可控,可以在纤维和基体之间形成更好的界面结合状态,且界面状态稳定性较好,对复合材料力学性能的提高和稳定性均有重要作用。
19.在本发明中,化学气相沉积在沉积炉中进行,并且一般在惰性的气氛中进行。
20.优选地,双组元法化学气相沉积中的双组元为nh3和bcl3。
21.优选地,所述nh3和bcl3的体积比为1:1~4:1,例如可以是1:1、 2:1、3:1或4:1等。它们分别作为氮源和硼源,而过量nh3用来中和 hcl,化学反应式如下:
22.nh3+hcl=nh4cl
ꢀꢀꢀ
(2)
23.化学反应式(1)和(2)都完全反应时,nh3和bcl3的体积比为4:1。但在氢气的抑制下,化学反应式(1)并不会完全反应,因此nh3和bcl3的体积比例不大于4:1。另一方面,由于需要过量nh3用来中和hcl,避免过多hcl具有腐蚀性,因此nh3和bcl3的体积比例不小于1:1,一般选取2:1~3:1,最优选3:1。
24.优选地,所述双组元法化学气相沉积中还包括氢气。
25.优选地,所述nh3和bcl3的体积之和,与氢气的体积比一般为 1:2~2:1,例如可以是1:2、1:1或2:1等等。由于氢气又抑制反应(1)的作用,如果氢气过少会导致需要更多的nh3且生成过多hcl具有腐蚀性,而如果氢气过多会导致生成氮化硼偏少。
26.优选地,所述双组元法化学气相沉积中以氮气作为稀释气。
27.优选地,所述化学气相沉积的温度为600~1000℃,例如可以是 600℃、700℃、800
℃、900℃或1000℃等。
28.对不同温度制备的涂层进行分析得知,600到900℃时,氮化硼涂层的xrd图谱为较宽的衍射峰,说明其中的氮化硼的晶体结构缺乏三维有序,结晶度较差,主要为涡轮层状结构;随着沉积温度的升高,大于1000℃时,氮化硼涂层呈现明显的结晶趋势,其xrd图谱的衍射峰逐渐变得尖锐,主要呈现六方氮化硼结构。沉积温度对氮化硼涂层的晶体结构具有较大影响,随着温度的升高,结晶度呈现增加趋势,对复合材料的界面结合状态的改善更明显,但对纤维的损伤也逐渐增加。综合以上因素,本文选取的沉积温度为600~1000℃。
29.优选地,所述化学气相沉积的沉积时间为4~10h,例如可以是4h、 5h、6h、7h、8h、9h或10h等。
30.沉积时间的选择主要和需要的界面厚度相关,一般来说,沉积时间越长,界面层的厚度越大。涂层的厚度可能会改变纤维
‑
基体界面的断裂模式,预计对复合材料的力学性能影响较大:由于此类复合材料中的前驱体为有机
‑
无机转化形成,具有较高的活性和侵蚀性,在界面处容易与纤维发生界面反应而导致结合过强,纤维无法拔出而发挥增强增韧的作用,导致复合材料的力学性能较低;纤维容易如若涂层太薄可能不能有效降低界面结合力,而涂层太厚可能导致界面结合过弱,使得界面无法有效传递载荷。另一方面,由于不同预制体的厚度有一定差异,由外向内的沉积难度增大,沉积厚度降低,考虑沉积时间还需要兼顾预制体内外不均匀的问题,才能提高复合材料整体的力学性能。综合以上因素,本文选取的沉积时间为4~10h。
31.优选地,所述化学气相沉积的升温速率为5℃/min~20℃/min。
32.在本发明中,升温速率一般指的是从室温上升至沉积温度的速率。
33.优选地,步骤(3)中所述氮化物前驱体为硅硼氮前驱体或氮化硅前驱体。
34.优选地,所述硅硼氮前驱体为聚硅硼氮烷类聚合物陶瓷前驱体,使用硅氮烷、硅烷和三氯化硼为原材料,主要通过共氨解的方式制备而成。
35.优选地,所述氮化硅前驱体为聚硅氮烷类聚合物陶瓷前驱体,使用硅氮烷、硅烷为原材料主要通过共氨解的方式制备而成。
36.优选地,步骤(3)中所述浸渍为真空吸注方法浸渍。在本发明中,通过真空吸注方法浸渍,可以实现复合材料的致密。
37.优选地,步骤(3)中所述交联固化的温度为100~300℃,例如可以是100℃、120℃、150℃、180℃、200℃、240℃、260℃或300℃等。
38.优选地,步骤(3)中所述交联固化的时间为5~20h,例如可以是 5h、7h、9h、13h、15h、18h或20h等。
39.在本发明中,调控温度可以引发交联固化反应,并且固化过程中向固化装置中进行通入氮气打压操作,提供惰性氛围。
40.优选地,步骤(3)中所述裂解的温度为400~1000℃,例如可以是 400℃、500℃、600℃、700℃、800℃、900℃或1000℃等。
41.在本发明中,对于加入前驱体的坯体,裂解过程中向裂解装置中进行通氨气操作,确保裂解装置通风良好,从而减少前驱体反应产物在坯体的残留,减少残碳含量,优化复合材料的透波性能。
42.优选地,步骤(3)中所述裂解的时间为3~9h,例如可以是3h、4h、5h、6h、7h、8h或9h
等。
43.优选地,步骤(4)中重复步骤(3)的次数为3~6次,例如可以是3次、4次、5次或6次。
44.第二方面,本发明提供了一种如第一方面所述的制备方法制备得到的氮化物纤维增强复合材料。
45.优选地,所述氮化物纤维增强复合材料的密度为1.72~1.74g/cm3。
46.优选地,所述氮化物纤维增强复合材料的拉伸强度为48~63mpa。本发明的拉伸强度一般指的是室温时的拉伸强度,温度具体为25℃。
47.在本发明中,当氮化物前驱体为硅硼氮前驱体时,复合材料的拉伸强度一般为51~63mpa;而当氮化物前驱体为氮化硅前驱体时,复合材料的一般为48~61mpa。
48.本发明提供的氮化物纤维增强复合材料具有良好的强度和韧性。
49.第三方面,本发明提供了一种如第二方面所述的氮化物纤维增强复合材料在飞行器透波系统中的应用。
50.实施本发明的,具有以下有益效果:
51.(1)本发明提供的制备方法可制得具有更好的界面结合状态、前驱体与纤维预制体之间的交联较充分、综合性能较好的复合材料。本发明利用化学气相沉积法处理与前驱体浸渍固化裂解的方式,解决了界面结合的问题,使得基体与纤维的界面结合更适宜,纤维增强作用更显著,复合材料强度更高。
52.(2)本发明制备了常温下密度1.72~1.74g/cm3、拉伸强度48~63mpa 的复合材料,具有良好的强度和韧性。
具体实施方式
53.为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都属于本发明保护的范围。
54.本发明以下实施例1
‑
12中所使用到的氮化硅纤维的密度约为 2.3g/cm3,所使用到的前驱体为聚硅硼氮烷。
55.实施例1
56.通过三维编织方式将氮化硅纤维加工成纤维体积含量为40%的预制体,然后不进行纤维涂层沉积处理,直接进入后续步骤。通过真空吸注方法采用硅硼氮前驱体浸渍预制体,再进行氮气气氛下交联固化反应(温度为220℃,固化时间为10h),然后进行氨气气氛下裂解反应(温度为900℃,时间为3h),重复以上致密化过程4次,得到常温下密度 1.71g/cm3、拉伸强度30mpa的复合材料。
57.实施例2
58.通过三维编织方式将氮化硅纤维加工成纤维体积含量为40%的预制体。然后通过双组元化学气相沉积法对纤维预制体进行处理,设定温度为600℃,沉积时间为7h,由升温速率为10℃/min,气体比例为: nh3与bcl3的体积比为3:1,nh3与bcl3体积之和与h2的体积比为1:1, n2作为稀释气,沉积得到bn界面层。
59.最后通过真空吸注方法采用硅硼氮前驱体浸渍预制体,再进行氮气气氛下交联固
化反应(固化温度220℃,固化时间10h),然后进行氨气气氛下裂解反应(温度为900℃,时间为3h),重复以上致密化过程4 次,得到常温下密度1.72g/cm3、拉伸强度51mpa的复合材料。
60.实施例3
61.通过编织方式将氮化硅纤维加工成纤维体积含量为40%的预制体。然后通过双组元化学气相沉积法对纤维预制体进行处理,设定温度为700℃,沉积时间为7h,升温速率为10℃/min,气体比例为:nh3与bcl3的体积比为3:1,nh3与bcl3体积之和与h2的体积比为1:1, n2作为稀释气,沉积得到bn界面层。
62.最后通过真空吸注方法采用硅硼氮前驱体浸渍预制体,再进行氮气气氛下交联固化反应(温度220℃,时间10h),然后进行氨气气氛下裂解反应(温度900℃,时间3h),重复以上致密化过程4次,得到常温下密度1.73g/cm3、拉伸强度57mpa的复合材料。
63.实施例4
64.通过编织方式将氮化硅纤维加工成纤维体积含量为40%的预制体。然后通过双组元化学气相沉积法对纤维预制体进行处理,设定温度为800℃,沉积时间为7h,升温速率为10℃/min,气体比例为:nh3与bcl3的体积比为3:1,nh3与bcl3体积之和与h2的体积比为1:1, n2作为稀释气,沉积得到bn界面层。
65.最后通过真空吸注方法采用硅硼氮前驱体浸渍预制体,再进行氮气气氛下交联固化反应(温度220℃,时间10h),然后进行氨气气氛下裂解反应(温度900℃,时间3h),重复以上致密化过程4次,得到常温下密度1.73g/cm3、拉伸强度63mpa的复合材料。
66.实施例5
67.通过编织方式将氮化硅纤维加工成纤维体积含量为40%的预制体。然后通过双组元化学气相沉积法对纤维预制体进行处理,设定温度为900℃,沉积时间为7h,升温速率为10℃/min,气体比例为:nh3与bcl3的体积比为3:1,nh3与bcl3体积之和与h2的体积比为1:1, n2作为稀释气,沉积得到bn界面层。
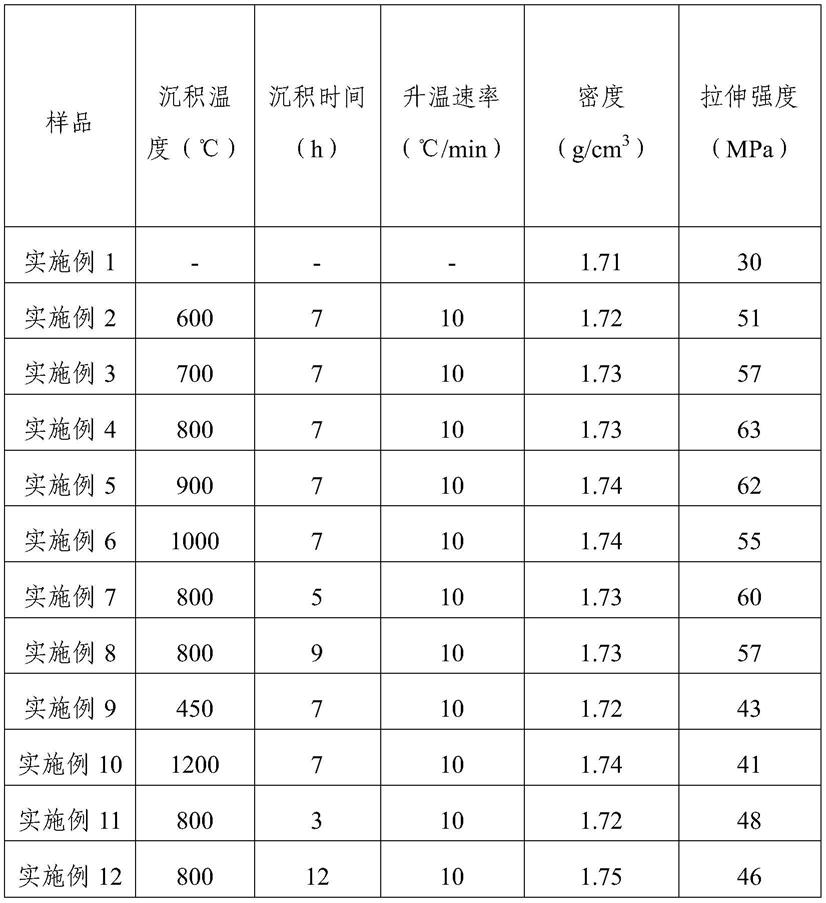
68.最后通过真空吸注方法采用硅硼氮前驱体浸渍预制体,再进行氮气气氛下交联固化反应(温度220℃,时间10h),然后进行氨气气氛下裂解反应(温度900℃,时间3h),重复以上致密化过程4次,得到常温下密度1.74g/cm3、拉伸强度62mpa的复合材料。
69.实施例6
70.通过编织方式将氮化硅纤维加工成纤维体积含量为40%的预制体。然后通过双组元化学气相沉积法对纤维预制体进行处理,设定温度为1000℃,沉积时间为7h,升温速率为10℃/min,气体比例为:nh3与bcl3的体积比为3:1,nh3与bcl3体积之和与h2的体积比为1:1, n2作为稀释气,沉积得到bn界面层。
71.最后通过真空吸注方法采用硅硼氮前驱体浸渍预制体,再进行氮气气氛下交联固化反应(温度220℃,时间10h),然后进行氨气气氛下裂解反应(温度900℃,时间3h),重复以上致密化过程4次,得到常温下密度1.74g/cm3、拉伸强度55mpa的复合材料。
72.实施例7
73.通过编织方式将氮化硅纤维加工成纤维体积含量为40%的预制体。然后通过双组元化学气相沉积法对纤维预制体进行处理,设定温度为800℃,沉积时间为5h,升温速率为10℃/min,气体比例为:nh3与bcl3的体积比为3:1,nh3与bcl3体积之和与h2的体积比为1:1, n2作为稀释气,沉积得到bn界面层。
74.最后通过真空吸注方法采用硅硼氮前驱体浸渍预制体,再进行氮气气氛下交联固化反应(温度220℃,时间10h),然后进行氨气气氛下裂解反应(温度900℃,时间3h),重复以上致密化过程4次,得到常温下密度1.73g/cm3、拉伸强度60mpa的复合材料。
75.实施例8
76.通过编织方式将氮化硅纤维加工成纤维体积含量为40%的预制体。然后通过双组元化学气相沉积法对纤维预制体进行处理,设定温度为800℃,沉积时间为9h,升温速率为10℃/min,气体比例为:nh3与bcl3的体积比为3:1,nh3与bcl3体积之和与h2的体积比为1:1, n2作为稀释气,沉积得到bn界面层。
77.最后通过真空吸注方法采用硅硼氮前驱体浸渍预制体,再进行氮气气氛下交联固化反应(温度220℃,时间10h),然后进行氨气气氛下裂解反应(温度900℃,时间3h),重复以上致密化过程4次,得到常温下密度1.73g/cm3、拉伸强度57mpa的复合材料。
78.实施例9
79.本实施例与实施例4的区别仅在于,本实施例化学气相沉积设定温度为450℃,制备得到复合材料。
80.实施例10
81.本实施例与实施例4的区别仅在于,本实施例化学气相沉积设定温度为1200℃,制备得到复合材料。
82.实施例11
83.本实施例与实施例4的区别仅在于,本实施例化学气相沉积设定时间为3h,制备得到复合材料。
84.实施例12
85.本实施例与实施例4的区别仅在于,本实施例化学气相沉积设定时间为12h,制备得到复合材料。
86.记录上述各个实施例1
‑
12使用的工艺条件及所制得的复合材料的性能,其结果见表1。
87.表1
[0088][0089]
本发明以下实施例13
‑
22中所使用到的氮化硅纤维的密度约为 2.3g/cm3,所使用到的前驱体为聚硅氮烷。
[0090]
实施例13
[0091]
通过编织方式将氮化硅纤维加工成纤维体积含量为40%的预制体,然后不进行纤维涂层沉积处理,直接进入后续步骤。通过真空吸注方法采用氮化硅前驱体浸渍预制体,再进行氮气气氛下交联固化反应(温度200℃,时间为10h),然后进行氨气气氛下裂解反应(温度为 900℃,时间为3h),重复以上致密化过程4次,得到常温下密度 1.70g/cm3、拉伸强度27mpa的复合材料。
[0092]
实施例14
[0093]
通过编织方式将氮化硅纤维加工成纤维体积含量为40%的预制体。然后通过双组元化学气相沉积法对纤维预制体进行处理,设定温度为600℃,沉积时间为7h,升温速率为10℃/min,气体比例为:nh3与bcl3的体积比为3:1,nh3与bcl3体积之和与h2的体积比为1:1, n2作为稀释气,沉积得到bn界面层。
[0094]
最后通过真空吸注方法最后通过真空吸注方法采用氮化硅前驱体浸渍预制体再
进行氮气气氛下交联固化反应(温度200℃,时间10h),然后进行氨气气氛下裂解反应(温度900℃,时间3h),重复以上致密化过程4次,得到常温下密度1.72g/cm3、拉伸强度48mpa的复合材料。
[0095]
实施例15
[0096]
通过编织方式将氮化硅纤维加工成纤维体积含量为40%的预制体。然后通过双组元化学气相沉积法对纤维预制体进行处理,设定温度为700℃进,沉积时间为7h,升温速率为10℃/min,气体比例为: nh3与bcl3的体积比为3:1,nh3与bcl3体积之和与h2的体积比为1:1, n2作为稀释气,沉积得到bn界面层。
[0097]
最后通过真空吸注方法采用最后通过真空吸注方法采用氮化硅前驱体浸渍预制体,再进行氮气气氛下交联固化反应(温度200℃,时间 10h),然后进行氨气气氛下裂解反应(温度900℃,时间3h),重复以上致密化过程4次,得到常温下密度1.72g/cm3、拉伸强度55mpa的复合材料。
[0098]
实施例16
[0099]
通过编织方式将氮化硅纤维加工成纤维体积含量为40%的预制体。然后通过双组元化学气相沉积法对纤维预制体进行处理,设定温度为800℃,沉积时间为7h,升温速率为10℃/min,气体比例为:nh3与bcl3的体积比为3:1,nh3与bcl3体积之和与h2的体积比为1:1, n2作为稀释气,沉积得到bn界面层。
[0100]
最后通过真空吸注方法采用最后通过真空吸注方法采用氮化硅前驱体浸渍预制体,再进行氮气气氛下交联固化反应(温度200℃,时间 10h),然后进行氨气气氛下裂解反应(温度900℃,时间3h),重复以上致密化过程4次,得到常温下密度1.73g/cm3、拉伸强度61mpa的复合材料。
[0101]
实施例17
[0102]
通过编织方式将氮化硅纤维加工成纤维体积含量为40%的预制体。然后通过双组元化学气相沉积法对纤维预制体进行处理,设定温度为900℃,沉积时间为7h,升温速率为10℃/min,气体比例为:nh3与bcl3的体积比为3:1,nh3与bcl3体积之和与h2的体积比为1:1, n2作为稀释气,沉积得到bn界面层。
[0103]
最后通过真空吸注方法采用最后通过真空吸注方法采用氮化硅前驱体浸渍预制体,再进行氮气气氛下交联固化反应(温度200℃,时间 10h),然后进行氨气气氛下裂解反应(900℃/3h),重复以上致密化过程 4次,得到常温下密度1.73g/cm3、拉伸强度59mpa的复合材料。
[0104]
实施例18
[0105]
通过编织方式将氮化硅纤维加工成纤维体积含量为40%的预制体。然后通过双组元化学气相沉积法对纤维预制体进行处理,设定温度为1000℃,沉积时间为7h,升温速率为10℃/min,气体比例为:nh3与bcl3的体积比为3:1,nh3与bcl3体积之和与h2的体积比为1:1,n2作为稀释气,沉积得到bn界面层。
[0106]
最后通过真空吸注方法采用最后通过真空吸注方法采用氮化硅前驱体浸渍预制体,再进行氮气气氛下交联固化反应(温度200℃,时间 10h),然后进行氨气气氛下裂解反应(温度900℃,时间3h),重复以上致密化过程4次,得到常温下密度1.74g/cm3、拉伸强度52mpa的复合材料。
[0107]
实施例19
[0108]
通过编织方式将氮化硅纤维加工成纤维体积含量为40%的预制体。然后通过双组元化学气相沉积法对纤维预制体进行处理,设定温度为800℃,沉积时间为5h,升温速率为10℃/min,气体比例为:nh3与bcl3的体积比为3:1,nh3与bcl3体积之和与h2的体积比为1:1, n2作为稀释气,沉积得到bn界面层。
[0109]
最后通过真空吸注方法采用最后通过真空吸注方法采用氮化硅前驱体浸渍预制体,再进行氮气气氛下交联固化反应(温度200℃,时间 10h),然后进行氨气气氛下裂解反应(温度900℃,时间3h),重复以上致密化过程4次,得到常温下密度1.72g/cm3、拉伸强度58mpa的复合材料。
[0110]
实施例20
[0111]
通过编织方式将氮化硅纤维加工成纤维体积含量为40%的预制体。然后通过双组元化学气相沉积法对纤维预制体进行处理,设定温度为800℃,沉积时间为9h,升温速率为10℃/min,气体比例为:nh3与bcl3的体积比为3:1,nh3与bcl3体积之和与h2的体积比为1:1, n2作为稀释气,沉积得到bn界面层。
[0112]
最后通过真空吸注方法采用最后通过真空吸注方法采用氮化硅前驱体浸渍预制体,再进行氮气气氛下交联固化反应(温度200℃,时间 10h),然后进行氨气气氛下裂解反应(温度900℃,时间3h),重复以上致密化过程4次,得到常温下密度1.73g/cm3、拉伸强度55mpa的复合材料。
[0113]
实施例21
[0114]
本实施例与实施例16的区别仅在于,本实施例化学气相沉积设定温度为450℃,制备得到复合材料。
[0115]
实施例22
[0116]
本实施例与实施例16的区别仅在于,本实施例化学气相沉积设定温度为1200℃,制备得到复合材料。
[0117]
实施例23
[0118]
本实施例与实施例16的区别仅在于,本实施例化学气相沉积设定时间为3h,制备得到复合材料。
[0119]
实施例24
[0120]
本实施例与实施例16的区别仅在于,本实施例化学气相沉积设定时间为12h,制备得到复合材料。
[0121]
记录上述各个实施例13
‑
24使用的工艺条件及所制得的复合材料的性能,其结果见表2。
[0122]
表2
[0123][0124][0125]
实施例1至实施例8,实施例13至实施例20所用的制备复合材料的原料和成型工艺完全相同,预制体纤维含量、前驱体浸渍固化以及裂解工艺均相同,只是沉积处理方式不同。实施例1不对纤维进行处理,实施例2至实施例8的升温速率均相同(同理,实施例13不对纤维进行处理,实施例14至实施例20的升温速率均相同)。实施例2(实施例14)至实施例6(实施例18)的保温时间均相同,反应温度逐渐升高。实施例4(实施例16)、实施例7(实施例19)及实施例8(实施例20)的反应温度相同,保温时间不同。从表1和表2的结果可以看出,上述各个实施例制得的复合材料,具有较低的密度和较高的拉伸强度。
[0126]
实施例9
‑
12以及实施例21
‑
24,可以看出,沉积温度和时间对复合材料密度的影响不明显。沉积温度过低时,拉伸强度较低,应该是由于低温下反应不充分难以形成足够的界面层;沉积温度过高时,拉伸强度也较低,应该是由于高温对纤维的损伤;沉积时间过低时,形成的界面较薄或者状态不好,导致界面处不能实现裂纹偏转;沉积时间过高时,形成的界面较厚,难以实现裂纹偏转的增韧机制;综合来看,沉积温度和时间对复合材料的拉伸强度影响较大。
[0127]
通过观察表1和表2中的数据结果可以看出,通过化学气相沉积处理制得氮化硼涂
层作为复合材料的纤维与基体间的界面后,对复合材料的拉伸强度有一定提升,说明界面结合状态得到了一定的改善。不同沉积温度对复合材料的密度影响很小,但对拉伸强度的影响较大:采用较低的沉积温度,形成的界面层结构有序度较差,会导致效果有限;采用较高的沉积温度,形成的界面结构中三维有序逐渐增多,但对纤维的损伤加剧,也会导致效果降低;因此选择合适的沉积温度较为重要。另一方面,沉积时间对效果也有一定的影响,时间过短或者过长都对效果有一定的影响,应该是沉积时间影响界面厚度导致界面层的效果发生变化,进而影响了复合材料的力学性能。
[0128]
综合以上结构,通过使用本发明的制备方法制备得到的复合材料,在具有较低密度的同时,具有较好的力学性能,综合性能优势明显。
[0129]
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1