一种氢型FER分子筛及其制备方法和应用与流程
一种氢型fer分子筛及其制备方法和应用
技术领域
1.本发明涉及催化剂技术领域,尤其涉及一种氢型fer分子筛及其制备方法和应用。
背景技术:
2.乙醇是重要的溶剂和化工原料,因具有较高的辛烷值,被公认为优良的汽油添加剂。煤经合成气间接法生产乙醇中的二甲醚羰基化路线在设备材质、催化剂成本、产物分离等方面优势突出,而实现该路径高效运行的关键技术在于二甲醚羰基化制乙酸甲酯催化剂的研制。
3.研究表明,分布在分子筛催化剂中八元环(8-mr)孔道中的b酸位点是二甲醚(dme)羰基化反应的主要活性中心。同时研究表明,限域在分子筛8-mr孔道内的b酸较10元环(10-mr)或12元环(12-mr)内的b酸强,即分子筛孔道内的强酸对二甲醚羰基化反应起到关键作用。具有8-mr和12-mr结构的丝光沸石在dme羰基化反应中表现出优异的催化活性。但是,其12-mr内的b酸位点很容易诱导烃类副产物的生成,并进一步沉积而造成催化剂快速积碳失活。目前关于丝光沸石的研究多集中于后处理改性方面,例如专利cn101613274a公开了采用吡啶预吸附的方式提高丝光沸石稳定性的方法,但吡啶在反应过程中会发生脱附,故其稳定性也很难长时间保持,且吡啶会在一定程度上堵塞孔道,影响反应物和产物的扩散。因此,丝光沸石的孔道特点在一定程度上限制了其在二甲醚羰基化反应中的应用。同样具有8-mr孔道结构的fer分子筛因其10-mr孔道较小,可以有效抑制积碳的生成,在dme羰基化反应中表现出优异的催化稳定性。
4.fer分子筛为二维片层状结构,属于中孔分子筛,包含沿c轴方向的10-mr孔道(5.4
×
4.2
å
),与沿b轴方向的8-mr孔道(4.8
×
3.5
å
)垂直交叉,6-mr(六元环)孔道平行于c轴,8-mr孔道与6-mr孔道交叉形成镁碱沸石笼[82686458]。由于其独特的孔道结构、优异的水热/热稳定性及酸性可调性,被广泛应用于烃类的异构化、裂化、芳构化以及二甲醚羰基化等反应。目前,关于水热法合成fer分子筛常见的有机模板剂有:环己胺(ca1401570a和ca1749164a)、乙二胺或吡咯烷(usp4016245和usp4107125)、1,4-二甲基哌嗪(usp5288475)、六亚甲基亚胺(usp4925548)、正丁胺(cn101468809a)等,采用这些模板剂合成fer分子筛不仅晶化周期长而且酸性较弱,8-mr b酸含量低,这在一定程度上限制了fer分子筛在二甲醚羰基化反应中的应用。
技术实现要素:
[0005]
本发明的目的在于提供一种氢型fer分子筛及其制备方法和应用,本发明制备的氢型fer分子筛8-mr b酸含量高,用于催化二甲醚羰基化反应具有高活性。
[0006]
为了实现上述发明目的,本发明提供以下技术方案:本发明提供了一种氢型fer分子筛的制备方法,包括以下步骤:将naoh、铝源、硅源、水和模板剂混合,得到反应混合物;以摩尔比计,所述反应混合物中si/al=6~18,naoh/sio2=0.1~0.9,h2o/sio2=10~50,模板剂/sio2=0.2~0.9;所述模板
剂为吗啉;将所述反应混合物进行水热晶化,得到晶化产物;将所述晶化产物焙烧脱除模板剂,得到fer分子筛;将所述fer分子筛进行nh
4+
交换,然后焙烧铵解,得到氢型fer分子筛。
[0007]
优选的,所述水热晶化包括:依次进行第一动态晶化、升温和第二动态晶化;所述第一动态晶化的温度为100~130℃,时间为30~50h;所述第二动态晶化的温度为160~180℃,时间为12~36h。
[0008]
优选的,所述焙烧脱除模板剂的温度为440~570℃,保温时间为14~35h。
[0009]
优选的,所述焙烧脱除模板剂包括:依次进行第一焙烧、升温和第二焙烧;所述第一焙烧的温度为440~480℃,保温时间为4~20h;所述第二焙烧的温度为520~570℃,保温时间为10~15h;所述升温的速率为1~2℃/min。
[0010]
优选的,所述进行nh
4+
交换包括:将fer分子筛在含nh
4+
的溶液中进行回流。
[0011]
优选的,所述回流的温度为70~90℃,时间为3~8h。
[0012]
优选的,所述焙烧铵解包括依次进行第ⅰ焙烧、升温和第ⅱ焙烧;所述ⅰ焙烧的温度为440~480℃,保温时间为4~20h;所述第ⅱ焙烧的温度为520~570℃,保温时间为10~15h;所述升温的速率为1~2℃/min。
[0013]
本发明提供了上述方案所述制备方法制备得到的氢型fer分子筛。
[0014]
本发明提供了上述方案所述氢型fer分子筛作为催化剂在催化二甲醚和一氧化碳进行羰基化反应中的应用。
[0015]
优选的,所述羰基化反应的温度为180~240℃,空速为800~6000ml/(g
cat
·
h),二甲醚和一氧化碳的进料体积比为1:10~50。
[0016]
本发明提供了一种氢型fer分子筛的制备方法,包括以下步骤:将naoh、铝源、硅源、水和模板剂混合,得到反应混合物;以摩尔比计,所述反应混合物中si/al=6~18,naoh/sio2=0.1~0.9,h2o/sio2=10~50,模板剂/sio2=0.2~0.9;所述模板剂为吗啉;将所述反应混合物进行水热晶化,得到晶化产物;将所述晶化产物焙烧脱除模板剂,得到fer分子筛;将所述fer分子筛进行nh
4+
交换,然后焙烧铵解,得到氢型fer分子筛。
[0017]
本发明利用吗啉作为模板剂,在fer分子筛的合成中,质子化的吗啉可以起到平衡骨架负电荷的作用,且吗啉的分子尺寸与fer分子筛椭圆形的十元环孔道尺寸相当,可以促使al向八元环孔道内富集。其结构中的氧原子可以有效减弱它与fer分子筛八元环孔道内骨架的作用力,故而不能进入八元环甚至六元环孔道,因此有利于na
+
位于这些小尺寸孔道结构中,在经过后续nh
4+
交换处理后使该处的b酸含量增高,最终提高fer分子筛8-mr b酸含量,进而提高了其催化二甲醚羰基化反应的活性。
[0018]
进一步的,分子筛在合成过程中低温有利于晶核的形成,高温有利于晶体的生长,本发明采用两步晶化的方式可以有效提高产物的晶化速率和结晶度。
[0019]
进一步的,本发明在模板剂的脱除过程中通过控制焙烧温度和升温速率有效降低了骨架al的迁移以及扭曲程度,较高含量的骨架铝可以进一步提高氢型fer分子筛的酸强度和酸密度。
[0020]
实施例的结果表明,本发明制备的氢型fer分子筛的强酸量最高达到1.185mmol/g,8元环b酸含量为1.037mmol/g,8元环b酸含量是采用常规模板剂合成分子筛的2~3倍。在
二甲醚羰基化反应中可使二甲醚(dme)的转化率最高达到66.87%,稳定运行至150h时转化率仍维持在52.17%,乙酸甲酯(ma)选择性大于99.3%。反应后的催化剂经过两次再生活性仍可以完全恢复。
附图说明
[0021]
图1为实施例1~5制备的氢型fer分子筛的xrd图谱;图2为对比例1~4和实施例8制备的氢型fer分子筛的xrd图谱;图3为实施例1、6、7、8以及对比例5制备的氢型fer分子筛的真空红外(羟基区)图谱;图4为实施例1、6、7、8和对比例5制备的氢型fer分子筛的
27
al核磁图;图5为部分实施例及对比例制备的氢型fer分子筛的nh
3-tpd曲线;图6为部分实施例及对比例制备的氢型fer分子筛的py-ir曲线;图7为部分实施例及对比例二甲醚羰基化反应评价图;图8为实施例7反应后的催化剂进行再生后的评价图。
具体实施方式
[0022]
本发明提供了一种氢型fer分子筛的制备方法,包括以下步骤:将naoh、铝源、硅源、水和模板剂混合,得到反应混合物;以摩尔比计,所述反应混合物中si/al=6~18,naoh/sio2=0.1~0.9,h2o/sio2=10~50,模板剂/sio2=0.2~0.9;所述模板剂为吗啉;将所述反应混合物进行水热晶化,得到晶化产物;将所述晶化产物焙烧脱除模板剂,得到fer分子筛;将所述fer分子筛进行nh
4+
交换,然后焙烧铵解,得到氢型fer分子筛。
[0023]
在本发明中,未经特殊说明,所用原料均为本领域熟知的市售商品。
[0024]
本发明将naoh、铝源、硅源、水和模板剂混合,得到反应混合物。
[0025]
在本发明中,所述铝源优选包括异丙醇铝、偏铝酸钠、十八水合硫酸铝和氯化铝中的一种或多种;所述硅源优选包括硅溶胶、硅胶粉和水玻璃中的一种;所述水优选为去离子水;所述模板剂为吗啉(缩写为mor)。本发明采用氢氧化钠为碱源能够确保得到结晶度良好的fer分子筛。采用氢氧化钾、氢氧化锂都得不到纯相的分子筛,原因是氢氧化锂遇到铝源会形成铝酸锂沉淀,而氢氧化钾的碱性不如氢氧化钠,用氢氧化钾做碱源的时候成胶困难,且最终产物的结晶度很差。
[0026]
在本发明中,所述混合优选包括:将naoh溶解于水中,搅拌加入铝源,溶解完全后加入硅源,成胶结束加入模板剂吗啉。本发明加入模板剂吗啉后优选在室温下搅拌2h得到反应混合物。
[0027]
在本发明中,以摩尔比计,所述反应混合物中si/al=6~18,naoh/sio2=0.1~0.9,优选为0.2~0.6;h2o/sio2=10~50,优选为20~30;模板剂/sio2=0.2~0.9,优选为0.3~0.5。
[0028]
得到反应混合物后,本发明将所述反应混合物进行水热晶化,得到晶化产物。
[0029]
在本发明中,所述水热晶化优选包括:依次进行第一动态晶化、升温和第二动态晶化。在本发明中,所述第一动态晶化的温度优选为100~130℃,更优选为115~125℃,进一步
优选为120℃;所述第一动态晶化的时间优选为30~50h,更优选为36~48h,进一步优选为40~42h;所述第二动态晶化的温度优选为160~180℃,更优选为165~175℃,进一步优选为170℃;时间优选为12~36h,更优选为20~30h,进一步优选为24h。本发明对升温至第二动态晶化的温度的速率没有特殊要求。在本发明中,所述水热晶化优选在高压反应釜中进行。分子筛在合成过程中低温有利于晶核的形成,高温有利于晶体的生长,本发明采用两步晶化的方式可以有效提高产物的晶化速率和结晶度。
[0030]
晶化结束后,本发明优选将高压反应釜放在自来水槽内淬冷,将物料进行固液分离,80℃烘干,得到晶化产物。
[0031]
得到晶化产物后,本发明将所述晶化产物焙烧脱除模板剂,得到fer分子筛。
[0032]
在本发明中,所述焙烧脱除模板剂前,本发明优选将晶化产物分子筛放于马弗炉中按照1~3℃/min升温至120℃并维持1~2h以脱除表面吸附的水分子。
[0033]
在本发明中,所述焙烧的温度优选为440~570℃,更优选为470~550℃;保温时间优选为14~35h,更优选为15~20h。
[0034]
在本发明中,所述焙烧脱除模板剂优选包括:依次进行第一焙烧、升温和第二焙烧;所述第一焙烧的温度优选为440~480℃,更优选为450~470℃,保温时间优选为4~20h,更优选为8~15h;所述第二焙烧的温度优选为520~570℃,更优选为530~560℃,保温时间优选为10~15h,更优选为12~14h;所述升温的速率优选为1~2℃/min,更优选为1~1.5℃/min,最优选为1℃/min。本发明通过控制焙烧的条件保证模板剂完全脱除。并且,通过控制焙烧温度和升温速率有效降低了骨架al迁移以及扭曲程度,较高含量的骨架铝可以进一步提高氢型fer分子筛的酸强度和酸密度。
[0035]
得到fer分子筛后,本发明将所述fer分子筛进行nh
4+
交换,然后焙烧铵解,得到氢型fer分子筛。
[0036]
在本发明中,所述进行nh
4+
交换优选包括:将所述fer分子筛在含nh
4+
的溶液中进行回流。在本发明中,所述nh
4+
优选由硝酸铵或氯化铵提供。所述含nh
4+
的溶液中nh
4+
的浓度优选为0.5~1mol/l,ph值优选为5;所述fer分子筛与含nh
4+
的溶液的固液比优选为1g:(20~60)ml,更优选为1g:(30~40)ml。在本发明中,所述回流的温度优选为70~90℃,更优选为80℃;时间优选为3~8h,更优选为4~6h。本发明优选重复进行nh
4+
交换,重复的次数优选为2~3次。nh
4+
交换过程中,具体是分子筛中的na
+
与溶液中的nh
4+
发生离子交换。
[0037]
完成nh
4+
交换后,本发明优选对交换后的分子筛进行洗涤烘干,然后除去烘干后的分子筛表面吸附的水分子,最后进行焙烧铵解。
[0038]
在本发明中,所述除去烘干后的分子筛表面吸附的水分子优选包括:将烘干后的分子筛放于马弗炉中按照1~3℃/min升温至120℃并维持1~2h。
[0039]
在本发明中,所述焙烧铵解优选包括依次进行第ⅰ焙烧、升温和第ⅱ焙烧;所述ⅰ焙烧的温度优选为440~480℃,更优选为450~470℃,保温时间优选为4~20h,更优选为8~15h;所述第ⅱ焙烧的温度优选为520~570℃,更优选为530~560℃,保温时间优选为6~8h,更优选为6.5~7.5h;所述升温的速率优选为1~2℃/min。
[0040]
本发明在所述焙烧铵解过程中,nh
4+
分解形成h
+
,得到氢型fer分子筛。
[0041]
本发明提供了上述方案所述制备方法制备得到的氢型fer分子筛。
[0042]
本发明提供了上述方案所述氢型fer分子筛作为催化剂在催化二甲醚和一氧化碳
进行羰基化反应中的应用。
[0043]
在本发明中,所述催化剂的粒径优选为20~40目。本发明对所述应用的方法没有特殊要求,采用本领域熟知的应用方法即可。在本发明的实施例中,具体是将催化剂装填于固定床反应管中间恒温段,在n2吹扫下经过180min升温至500℃并维持2h以除去成型过程中催化剂表面吸附的水,然后在n2氛围下降温至二甲醚羰基化反应的温度,通入二甲醚和一氧化碳,进行羰基化反应。
[0044]
在本发明中,所述羰基化反应的温度优选为180~240℃,更优选为190~230℃,进一步优选为200~220℃;空速优选为800~6000ml/(g
cat
·
h),更优选为1000~5000ml/(g
cat
·
h),进一步优选为2000~4000ml/(g
cat
·
h);二甲醚和一氧化碳的进料体积比优选为1:10~50,更优选为1:20~40。
[0045]
下面结合实施例对本发明提供的氢型fer分子筛及其制备方法和应用进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
[0046]
实施例1将2.71g naoh溶解于58.50g去离子水中,搅拌加入4.33g十八水合硫酸铝,溶解完全后缓慢加入19.50g硅溶胶,成胶结束加入4.00g mor(吗啉),反应混合物摩尔组成为:si/al=10,naoh/sio2=0.5,h2o/sio2=30,mor/sio2=0.35,室温搅拌2h装入高压反应釜于120℃动态晶化48h,升温至170℃继续动态晶化24h。晶化结束后,反应釜放在自来水槽内淬冷,将物料进行固液分离,80℃烘干,得到晶化产物。
[0047]
模板剂的脱除:将所述晶化产物放于马弗炉中按照1℃/min升温至120℃并维持2h以脱除表面吸附的水分子,之后经过270min升温至470℃并维持5h,再以1℃/min升温至570℃,并维持10h保证模板剂完全脱除,得到fer分子筛。
[0048]
氢型fer分子筛的制备:将所述fer分子筛进行nh
4+
交换,采用的固液比为1g:40ml,80℃回流4h,重复3次,洗涤烘干,将烘干后的分子筛放于马弗炉中按照2℃/min升温至120℃并维持2h以脱除表面吸附的水分子,之后经过270min升温至470℃并维持5h,再以1℃/min升温至570℃,并维持6h,得到氢型fer分子筛。
[0049]
氢型fer分子筛催化剂的二甲醚羰基化性能评价:将氢型fer分子筛压片后破碎成20~40目小颗粒,取2g催化剂颗粒装填于固定床反应管中间恒温段,在n2吹扫下经过180min升温至500℃并维持2h以除去成型过程中催化剂表面吸附的水等杂质,然后在n2氛围下降温至反应温度210℃。调节反应压力至3mpa进行活性评价,进料体积比为dme:co=1:30,空速为2000ml/(g
cat
·
h)。
[0050]
实施例2将1.53g naoh溶解于33.11g去离子水中,搅拌加入2.55g十八水合硫酸铝,溶解完全后缓慢加入18.39g硅溶胶,成胶结束加入8.09g mor,反应混合物摩尔组成为:si/al=16,naoh/sio2=0.3,h2o/sio2=20,mor/sio2=0.75,室温搅拌2h装入高压反应釜于120℃动态晶化48h,升温至170℃继续动态晶化36h,晶化结束后,反应釜放在自来水槽内淬冷,将物料进行固液分离,80℃烘干,得到晶化产物。
[0051]
后续模板剂焙烧、氢型fer分子筛的制备及催化剂性能评价同实施例1。
[0052]
实施例3将2.96g naoh溶解于31.97g去离子水中,搅拌加入5.63g十八水合硫酸铝,溶解完
全后缓慢加入17.76g硅溶胶,成胶结束加入5.21g mor,反应混合物摩尔组成为:si/al=7,naoh/sio2=0.6,h2o/sio2=20,mor/sio2=0.5,室温搅拌2h装入高压反应釜于120℃动态晶化48h,升温至170℃继续动态晶化24h,晶化结束后,反应釜放在自来水槽内淬冷,将物料进行固液分离,80℃烘干,得到晶化产物。
[0053]
后续模板剂焙烧、氢型fer分子筛的制备及催化剂性能评价同实施例1。
[0054]
实施例4将1.12g naoh溶解于36.58g去离子水中,搅拌加入2.76g异丙醇铝,溶解完全后缓慢加入20.32g硅溶胶,成胶结束加入4.17g mor,反应混合物摩尔组成为:si/al=10,naoh/sio2=0.2,h2o/sio2=20,mor/sio2=0.35,室温搅拌2h装入高压反应釜于120℃动态晶化48h,升温至170℃继续动态晶化24h,晶化结束后,反应釜放在自来水槽内淬冷,将物料进行固液分离,80℃烘干,得到晶化产物。
[0055]
后续模板剂焙烧、氢型fer分子筛的制备及催化剂性能评价同实施例1。
[0056]
实施例5将0.64g naoh溶解于57.97g去离子水中,搅拌加入2.00g偏铝酸钠,溶解完全后缓慢加入24.15g硅溶胶,成胶结束加入3.54g mor。反应混合物摩尔组成为:si/al=10,naoh/sio2=0.26,h2o/sio2=20,mor/sio2=0.25,室温搅拌2h装入高压反应釜于120℃动态晶化48h,升温至170℃继续动态晶化24h,晶化结束后,反应釜放在自来水槽内淬冷,将物料进行固液分离,80℃烘干,得到晶化产物。
[0057]
后续模板剂焙烧、氢型fer分子筛的制备及催化剂性能评价同实施例1。
[0058]
实施例6晶化产物制备同实施例1;模板剂的脱除:将所述晶化产物放于马弗炉中按照1℃/min升温至120℃并维持2h以脱除表面吸附的水分子,之后经过270min升温至470℃并维持5h,再以1℃/min升温至520℃,并维持13h保证模板剂完全脱除,得到fer分子筛;氢型fer分子筛的制备:将所述fer分子筛进行nh
4+
交换,采用的固液比为1g:40ml,80℃回流4h,重复3次,洗涤烘干,将烘干后的分子筛放于马弗炉中按照1℃/min升温至120℃并维持2h以脱除表面吸附的水分子,之后经过270min升温至470℃并维持5h,再以1℃/min升温至520℃,并维持6h,得到氢型fer分子筛。
[0059]
催化剂性能评价同实施例1。
[0060]
实施例7晶化产物制备同实施例1;模板剂的脱除:将所述晶化产物放于马弗炉中按照1℃/min升温至120℃并维持2h以脱除表面吸附的水分子,之后经过270min升温至470℃并维持15h保证有机模板剂完全脱除,得到fer分子筛;氢型fer分子筛的制备:将所述fer分子筛进行nh
4+
交换,采用的固液比为1g:40ml,80℃回流4h,重复3次,洗涤烘干,将烘干后的分子筛放于马弗炉中按照1℃/min升温至120℃并维持2h以脱除表面吸附的水分子,之后经过270min升温至470℃并维持5h,再以1℃/min升温至520℃,并维持8h,得到氢型fer分子筛。
[0061]
催化剂性能评价同实施例1。
[0062]
实施例8与实施例1的不同之处仅在于模板剂的脱除过程为:将按照实施例1合成得到的晶化产物由120℃经过270min升温至570℃并维持10h,降至室温,得到fer分子筛,其余步骤同实施例1。
[0063]
将该样品按照与实施例1相同的二甲醚羰基化评价方式进行活性评价。
[0064]
对比例1将2.43g naoh溶解于58.50g去离子水中,搅拌加入4.33g十八水合硫酸铝,溶解完全后缓慢加入19.5g硅溶胶,成胶结束加入2.76g乙二胺(en)作为模板剂,反应混合物摩尔组成为:si/al=10,naoh/sio2=0.45,h2o/sio2=30,en/sio2=0.35,室温搅拌2h装入高压反应釜于120℃动态晶化48h,升温至170℃继续动态晶化72h。晶化结束后,反应釜放在自来水槽内淬冷,将物料进行固液分离,80℃烘干,得到晶化产物。
[0065]
其余步骤同实施例1。
[0066]
对比例2将2.71g naoh溶解于58.50g去离子水中,搅拌加入4.33g十八水合硫酸铝,溶解完全后缓慢加入19.50g硅溶胶,成胶结束加入4.67g吡咯烷(pyrr)作为模板剂,反应混合物摩尔组成为:si/al=10,naoh/sio2=0.5,h2o/sio2=30,pyrr/sio2=0.35。室温搅拌2h装入高压反应釜于120℃动态晶化48h,升温至170℃继续动态晶化72h。晶化结束后,反应釜放在自来水槽内淬冷,将物料进行固液分离,80℃烘干,得到晶化产物。
[0067]
其余步骤同实施例1。
[0068]
对比例3将2.71g naoh溶解于58.50g去离子水中,搅拌加入4.33g十八水合硫酸铝,溶解完全后缓慢加入19.50g硅溶胶,成胶结束加入3.54g哌啶(pi)作为模板剂,反应混合物摩尔组成为:si/al=10,naoh/sio2=0.5,h2o/sio2=30,pi/sio2=0.35,室温搅拌2h装入高压反应釜于120℃动态晶化48h,升温至170℃继续动态晶化72h。晶化结束后,反应釜放在自来水槽内淬冷,将物料进行固液分离,80℃烘干,得到晶化产物。
[0069]
其余步骤同实施例1。
[0070]
对比例4将2.71g naoh溶解于58.50g去离子水中,搅拌加入4.33g十八水合硫酸铝,溶解完全后缓慢加入19.50g硅溶胶,成胶结束加入5.34g1,6-己二胺(dah)作为模板剂,反应混合物摩尔组成为:si/al=10,naoh/sio2=0.5,h2o/sio2=30,dah/sio2=0.35,室温搅拌2h装入高压反应釜于120℃动态晶化48h,升温至170℃继续动态晶化72h,晶化结束后,反应釜放在自来水槽内淬冷,将物料进行固液分离,80℃烘干,得到晶化产物。
[0071]
其余步骤同实施例1。
[0072]
对比例5与实施例1的不同之处仅在于模板剂的脱除过程为:将按照实施例1合成得到的晶化产物由120℃经过270min升温至600℃并维持10h,降至室温,得到fer分子筛,其余步骤同实施例1。
[0073]
该样品按照与实施例1相同的二甲醚羰基化评价方式进行活性评价。
[0074]
结构表征:
1、对实施例1~5制备的氢型fer分子筛进行xrd表征,结果见图1。由图1可知,采用本发明实施例中的合成条件均可得到纯相高结晶度的fer分子筛。
[0075]
2、图2为对比例1~4和实施例8制备的氢型fer分子筛的xrd图谱。由图2可知,对比例1~4中改变了合成所用的模板剂种类,采用常规的模板剂合成出了纯相fer分子筛,实施例8将两步焙烧脱除模板剂改为一步焙脱除模板剂,仍得到了纯相fer分子筛。
[0076]
3、图3为实施例1、6、7、8以及对比例5制备的氢型fer分子筛的真空红外(羟基区)图谱,图3中3650cm-1
的峰代表非骨架,非骨架铝一般呈l酸性;3600cm-1
的峰代表桥羟基(sio(h)al),是分子筛的b酸位点,也是二甲醚羰基化反应的活性位点。3600cm-1
的峰强说明分子筛的b酸含量高,对应的催化剂活性也高,而3650cm-1
的峰含量高则说明非骨架铝含量高,对应的骨架铝含量则降低。表现在酸量上为分子筛的弱酸含量高,强酸含量低,b酸含量低,l酸含量高,具有此特点的催化剂活性较差。因此,由真空红外羟基区的表征结果可知,实施例7拥有最多的骨架铝和最少的非骨架铝,而对比例5中的催化剂因焙烧过程升温速率的较快而造成骨架铝的脱除,从而形成了大量的非骨架铝。这说明升温速率会影响骨架铝的配位状态。
[0077]
4、图4为实施例1、6、7、8以及对比例5制备的氢型fer分子筛的
27
al核磁图;
27
al核磁图中50~60ppm代表四配位的骨架铝,大约40ppm处存在宽的肩峰归属于扭曲的骨架铝,大约30ppm的特征峰归属于五配位铝,约0ppm的特征峰归属于六配位的非骨架铝。扭曲的铝原子一般不显示b酸性,其含量越高,对应的b酸含量越低。由图4可知,实施例1制备的氢型fer分子筛中扭曲的骨架铝的含量为19.23%;实施例6制备的氢型fer分子筛中扭曲的骨架铝的含量为16.23%;实施例7制备的氢型fer分子筛中扭曲的骨架铝的含量为12.21%;实施例8制备的氢型fer分子筛中扭曲的骨架铝的含量为22.33%;对比例5制备的氢型fer分子筛中扭曲的骨架铝含量为25.19%。从图4中可以看出,升高焙烧温度骨架铝扭曲比例以及非骨架铝的比例都增加,而在焙烧温度仅为570℃时,提高焙烧时的升温速率骨架铝的扭曲程度显著,说明焙烧升温速率对骨架铝的扭曲影响也较大。
[0078]
5、图5为部分实施例及对比例制备的氢型fer分子筛的nh
3-tpd曲线。nh3程序升温脱附主要是表征催化剂中的弱酸量和强酸量(10-mr和8-mr b酸总量),其中200℃左右的脱附峰代表弱酸,350℃以上的脱附峰代表分子筛中的强酸;图6为部分实施例及对比例制备的氢型fer分子筛的py-ir(吡啶红外)曲线,可以表征催化剂中10-mr的b酸量,nh
3-tpd测得的强酸量减去10-mr的b酸量即为8-mr b酸量。对于二甲醚羰基化反应,起主要作用的是中强酸。从图5中可以看出,实施例1、5、6和7中强酸含量较高,尤其是实施例7强酸量最多,达到1.185 mmol/g,远远超出了常规报道的酸量。这主要是因为实施例7中的焙烧温度和升温速率最佳,分子筛的骨架铝扭曲量最少,非骨架铝含量最低,因而骨架铝含量最高,强酸含量最高。再与对比例1~4中进行对比可以发现,采用吗啉合成的fer分子筛在相同的焙烧条件下比用常规模板剂(乙二胺、哌啶、1,6-己二胺、吡咯烷)合成的fer分子筛的强酸量高,说明吗啉在fer分子筛合成过程中具有较其它模板剂较强的模板剂效应,可以诱导更多的铝形成骨架铝。图5和图6得到的实施例和对比例的酸分布情况见表1。由表1可知,本发明利用吗啉作为模板剂,能够提高fer分子筛的强酸量和8-mr b酸含量,进而提高了其催化二甲醚羰基化反应的活性。
[0079]
6、图7为部分实施例及对比例二甲醚羰基化反应评价图。由评价结果可以看出,实
施例7具有最优异的二甲醚羰基化催化活性和稳定性,二甲醚的最高转化率达到66%以上,且稳定性较好。实施例1、6、7和8都是采用吗啉合成的样品,只是焙烧温度和升温速率不同,可以看出,实施例8的催化活性明显低于实施例1、6、7,说明较快的升温速率对催化剂的活性提高不利。对比例1~4是采用常规模板剂合成的fer分子筛催化剂,即便与实施例1采用相同的焙烧条件,它们的催化活性也远低于采用吗啉合成的样品的催化活性,甚至低于实施例8在苛刻的焙烧条件的样品,通过对比说明采用吗啉合成的fer分子筛性能明显优于采用常规模板剂合成的fer分子筛催化性能。
[0080]
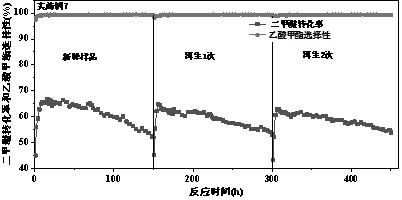
表1 各实施例和对比例所得样品的酸性表征结果以及反应10h的催化评价结果7、8、将实施例7反应后的催化剂进行再生:将经过二甲醚羰基化反应之后的催化剂在反应管内进行原位再生,再生时采用o2/n2混合气(其中o2体积含量分别为5%、10%),首先在氧气浓度为5%的条件下以1℃/min升温至470℃并维持5 h,然后在此温度下切换10%的o2/n2混合气并维持10h即可将催化剂表面的积碳物种除去,之后用氮气吹扫反应管并在氮气氛围下降温至反应温度进行二甲醚羰基化反应,再生评价结果见图8。由图8可知,采用实施例7中的焙烧条件所得到的催化剂具有优异的dme羰基化反应活性,dme的最高转化率达到66.6%,稳定运行至150h时转化率仍维持在52.17%,乙酸甲酯选择性大于99.3%,具有优异的催化活性和较高的乙酸甲酯选择性以及较高的稳定性。反应后的催化剂经过两次再生后催化活性仍可以完全恢复。
[0081]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应
视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1