一种富锂锰基层状正极材料的原位表面改性方法
1.本发明涉及锂离子电池技术领域,具体涉及一种富锂锰基层状正极材料的表面原位改性方法。
背景技术:
2.锂离子电池是便携式电子产品和新能源汽车的主要电源,由于续航里程的需求,目前对锂离子电池的高能量密度和高安全性能不断提出更高的要求。富锂层状正极材料xli2mno3·
(1-x)limo2(0《x《1,m=ni,co,mn),以其更高的工作电压、更多的锂含量、以及阴离子氧化还原超额容量等特性,是高容量正极材料的有力备选;相对于钴酸锂(175mah g-1
),磷酸铁锂(140mah g-1
)等传统正极材料,富锂正极材料的比容量可以超过250mah g-1
。然而该材料存在首周充放电效率低(《80%)、倍率性能差、容量损失大、电压衰减严重且电压滞后明显等问题,难以满足电动车的寿命和功率要求,且导致电池管理更为复杂。这需要在近年来取得重大突破,包括平衡阴离子氧化还原的超额容量极限,以及循环容量和电压稳定性(tarascon等nature energy,2018,3(5),373-386)。
3.早在2007年,利用li2mno
3-mo
2-litmo2三元相图分析发现该材料在充电至大于4.4v时可获得超额容量,该容量来源于阴离子氧化还原(thackeray等journal of materials chemistry,2007,17(30),3112-3125);而目前的方法中,通常需要将该材料在首次充放电循环中将其充电至高电压(≥4.8v)来激活阴离子容量(dahn等electrochemical and solid-state letters,2001,4(11),a191-a194),然而这将造成表面的不可逆相变,具有难以忽略的失氧、导致过渡金属扩散和还原,进一步造成表面持续发生副反应、容量持续损失、以及电压持续衰减(zhang等chemical reviews,2022,122(6),5641-5681)。
4.电化学性能衰减的根本原因在于高电压下的充放电,伴随着锂离子脱出,电荷重新分布,脱出电子能级更深,当电子的减少发生在氧的p轨道,则发生o-o键缩短、倾向于形成二聚结构,且其具有更强的氧化性,若不能将其固定在体相晶格内,将与电解质发生不期望的副反应或以氧气分子形式流失(lee,e等advanced energy materials,2014,4,1400498),并造成了mn
3+/4+
氧化还原对的激活,最终致使电压衰减;同时,过渡金属向内部扩散,会引起表面持续相变,并导致形貌变化(例如形成孔隙、开裂并最终造成粉化),电池容量损失。
5.综上,当前富锂锰基正极的应用问题来源于:超额的阴离子氧化还原容量,常利用高电压(》4.8v)进行首周激活。然而电极颗粒中,电位从表面到体相降低(gao等chinese physics b,2016,25,018210),高电压激活时,电压从表面到体相降低趋势过于剧烈,引起局域过大的过电位,从而导致不可逆的相变、并造成活性正极失效分解和容量损失;同时,表面的副反应,也会在后续电池工作的充放电循环中,造成显著的容量损失和电压衰减。
技术实现要素:
6.针对目前现有技术中的不足,本发明要解决的技术问题在于克服富锂锰基层状正极材料由于首周激活电压过高而导致的容量损失和电压衰减等缺陷,从而提供一种富锂锰基层状正极材料的原位表面改性方法、正极材料和二次电池。
7.本发明提供一种富锂锰基层状正极材料的原位表面改性方法即一种富锂锰基层状正极材料的近平衡电位本征原位激发形成表面稳定包覆层的方法,其特征在于,富锂锰基层状正极材料制备成正极,在近平衡电位下进行小倍率预充放电循环从而在富锂锰基层状正极材料表面激发本征分解反应,并原位生成稳定包覆层。所述富锂锰基层状正极材料为xli2mno3·
(1-x)limeo2组成的层状材料,其中0<x<1,me为mn,co,ni,al,ti,zr,fe,cr,mo,w,tc,re,ge,ru,os,rh,ir,ni,pd,pt中的至少一种。
8.近平衡条件,即将截止电压限制在可逆阴离子氧化还原反应的平衡电位,并将激活反应控制在低倍率条件下,进行预充放电循环激发表面相生成,可逆阴离子氧化还原反应的平衡电位低于传统的激活电压;本征,即该反应为电极材料自身在控制条件下本征分解、不需要其它添加剂、也非与电解质反应;原位,即反应实在预充放电过程中,在电极活性颗粒表面激发的。
9.上述预充放电循环过程利用的是正极材料在近平衡条件下本征分解反应。其中,本征反应是指,该反应不需要电解质或者其它添加剂参与到反应当中。为了证明电极发生了本征的分解反应,本发明优先采用在电化学循环条件下,既不会本身反应、也不与电极发生反应的稳定电解质体系,且不采用添加剂。同时,本发明设置可发生反应的电解质体系作为对比例,证明本发明所声明方法,对于非稳定电解质体系,同样可以起到正面效果。
10.表面稳定包覆层是在阴离子可逆氧化还原近平衡电位的条件下、利用预充放电循环,激发电极材料本征的分解反应,并在电极材料表面原位生成;表面包覆层主要为具有一定有序度的类岩盐相结构,厚度为2~10nm,主要为具有一定有序度的类岩盐相结构,氧原子以o3形式密堆积,对于完全无序的情况,li,me以无序形式随机占据其中的八面体空位,组成式为li
x
me
1-x
o;对于一定有序度情况,li,me并非完全随机占据,而是li以更高的概率占据富锂层,以更低的概率占据贫锂层;其中0《x《1,me为mn,co,ni,al,ti,zr,fe,cr,mo,w,tc,re,ge,ru,os,rh,ir,ni,pd,pt的至少一种。
11.为了将反应控制在近平衡态附近,用富锂正极材料阴离子可逆氧化还原平衡电位作为激活过程的截止电压上限,根据材料组分不同截止电压上限设置在4.3~4.6v,循环电压范围为2v~截止电压上限之间。
12.为了在动力学缓慢的近平衡条件下充分激活表面反应,预充放电循环的条件:其预循环的截止周数,以阴离子超额容量的充分激活为节点,预充放电循环圈数至5~30圈,优选为10~20圈;预充放电循环温度至40~70℃,优选为50~60℃;倍率设置在0.01~0.1c,优选倍率为0.05c;此外,为了加快近平衡条件下的反应速率,优选正极活性颗粒形貌为粒径0.2~2μm的一次单晶颗粒。
13.本发明目的在于提高富锂锰基层状正极材料体系的循环稳定性,抑制容量损失和电压衰减。其原理在于:通过将电压截止在阴离子可逆氧化还原的热力学平衡电位,将不可逆的相变局限在更薄表面,并在相平衡条件下,原位分解为更为稳定的混合物,作为致密且薄的包覆相,原位保护正极活性颗粒表面。其中,平衡电位的选择依据是:阴离子氧化并提
供超额容量、且并未氧化成氧气并造成不可逆氧流失的电压区间。
14.相对于传统高电压激活阴离子容量的方法相比(激活电压≥4.8v),本发明采用更低的可逆阴离子氧化还原平衡电压作为截止电位。充电条件下氧流失造成的表面分解反应,在4.8v传统激活条件下,以类尖晶石相为主,而在本发明所提出的近平衡激活条件下,以类岩盐相为主。本方法不仅充分利用了首周激活容量,同时从根本上抑制了不可逆氧流失带来的一系列不可逆副反应。即,传统方法在4.8v存在一个氧流失的电压平台,导致不可逆相变、并带来不可逆的容量损失和电压衰减;相对于将激活电压截止在4.8v的传统方式而言,当表面相激活电位截止在4.5v时,该不可逆氧流失平台消失。
15.相对于通常采用的表面包覆与表面掺杂等表面改性手段而言,本发明的改性方法不需要额外设计电解质和添加剂的反应,降低了成本;同时,原位本征激发表面相的方式,提高了表面与体相的适配度,更加薄、致密且稳定,进一步有助于电池的稳定循环。
16.本发明通过以下技术方案实现:选用富锂正极材料阴离子可逆氧化还原平衡电位作为激活过程的截止电压上限,根据材料组分不同截止电压上限设置在4.3~4.6v(不同于传统激活条件4.8v),例如对于常见组分li
1.2
ni
0.2
mn
0.6
o2(1.25li
1.2
ni
0.2
mn
0.6
o2=0.5li2mno3·
0.5lini
0.5
mn
0.5
o2),优选4.5v作为的平衡电位。该电压介于所选择组分的阴离子氧化还原电位与不可逆氧流失电位之间。放电的截止电压设为2v即循环电压范围为2v-截止电压上限之间,倍率设置在0.01~0.1c,优选倍率为0.05c。
17.由于近平衡条件下动力学缓慢,可采取以下方法令近平衡电位条件下阴离子容量充分激活(相对而言,当不采取促进手段时,该活化过程并不能进行完全,在后续附图中说明):
18.进一步,为了证明正极活性颗粒发生本征分解,而非与电解质或者添加剂发生非本征反应,并生成表面包覆相,选取在预充放电循环步骤中选取与正极不发生失氧副反应的稳定的电解质体系,包括但不限于离子液体电解质,无机陶瓷固体电解质,改性后耐高压液体电解质(例如高浓度水系电解液)等。优选离子液体电解质(1m litfsi-pyr
14
fsi)和无机固体电解质。同时,本发明所采取的表面保护方法,对于非稳定电解质体系也同样有促进作用。
19.本发明的有益之处在于:在近平衡电位下进行小倍率充放电预循环,可以激发正极活性颗粒表面本征、原位分解,并生成稳定、致密、均匀且薄的包覆相。该方法在满足阴离子容量激活的同时,从源头抑制了不可逆氧流失和不可逆容量损失、以及过多氧空位导致的过渡金属扩散和电压衰减。本发明利用了正极活性颗粒的本征分解反应原位形成类岩盐结构的稳定相,而非与电解质反应,因而生成的表面包覆相与体相具备更好的匹配,致密度更高、均匀度更好;此外,由于电位在电极颗粒从外到内逐渐降低,表面相先于体相发生相变,因此可以将本征热力学反应限制在表面,表面相更薄。本发明的电极活性颗粒的新型包覆方法,可以显著提高工作电压下容量和电压的循环稳定性。与现有二次锂离子电池和二次锂电池的正极相比,本发明具有高容量保持率,以及长循环稳定性,例如,50ma g-1
电流密度下循环150圈的容量为227.9mah/g,保持率为94.76%;与传统方法(即不进行近平衡电位循环预处理、直接利用4.8v进行工作的方法,并认为4.8v首周充电的时候为激活步骤)高上限截止电压激活相比,容量提升了64.5mah/g,保持率提升了18.13%。
20.本发明提供的表面包覆改性正极颗粒,可原位进行电池的工作循环,也可拆卸后
应用于正极材料,匹配传统电解质材料和负极材料,应用于二次锂电池和二次锂离子电池,并应用于电动车和便携电子设备。
附图说明
21.图1为本发明原位表面改性方法制备的实施例1在55℃,0.2c,4.8~2.0v的库伦效率与循环效率图
22.图2为本发明原位表面改性方法制备的实施例1和对比例1,在55℃,0.05c,4.5~2.0v(实施例),4.8~2.0v(对比例)的激活过程电压和倍率下的充放电曲线
23.图3为本发明原位表面改性方法制备的实施例1和对比例1的dq/dv图
24.图4为本发明原位表面改性方法制备的实施例1和对比例1,在55℃,0.2c,4.8~2.0v的工作循环过程电压和倍率下的电化学性能图
25.图5为本发明原位表面改性方法制备的实施例1和对比例1,在55℃,0.2c,4.8~2.0v的工作循环过程电压和倍率下的电压衰减图
26.图6为本发明原位表面改性方法制备的实施例2和对比例2,在55℃,0.05c,4.5~2.0v(实施例),4.8~2.0v(对比例)的激活过程电压和倍率下的充放电曲线
27.图7为本发明原位表面改性方法制备的实施例3,在55℃,0.05c,4.5~2.0v(激活循环过程),4.8~2.0v(工作循环过程)的充放电曲线
28.图8为本发明原位表面改性方法制备的实施例8,在25℃,0.05c,4.5~2.0v(激活循环过程),4.8~2.0v(工作循环过程)的充放电曲线
29.图9为本发明原位表面改性方法制备的实施例1,进行拆卸收集正极,重装电池后在25℃,0.05c,4.8~2.0v的放电电压容量曲线图
30.图10为本发明原位表面改性方法表面相变过程示意图
31.图11为本发明原位表面改性方法制备的实施例1在激活完成后的高分辨透射电子显微镜图,表面为类岩盐结构
32.图12为本发明原位表面改性方法制备的实施例1在激活完成并工作循环200圈后的高分辨透射电子显微镜图。
具体实施方式
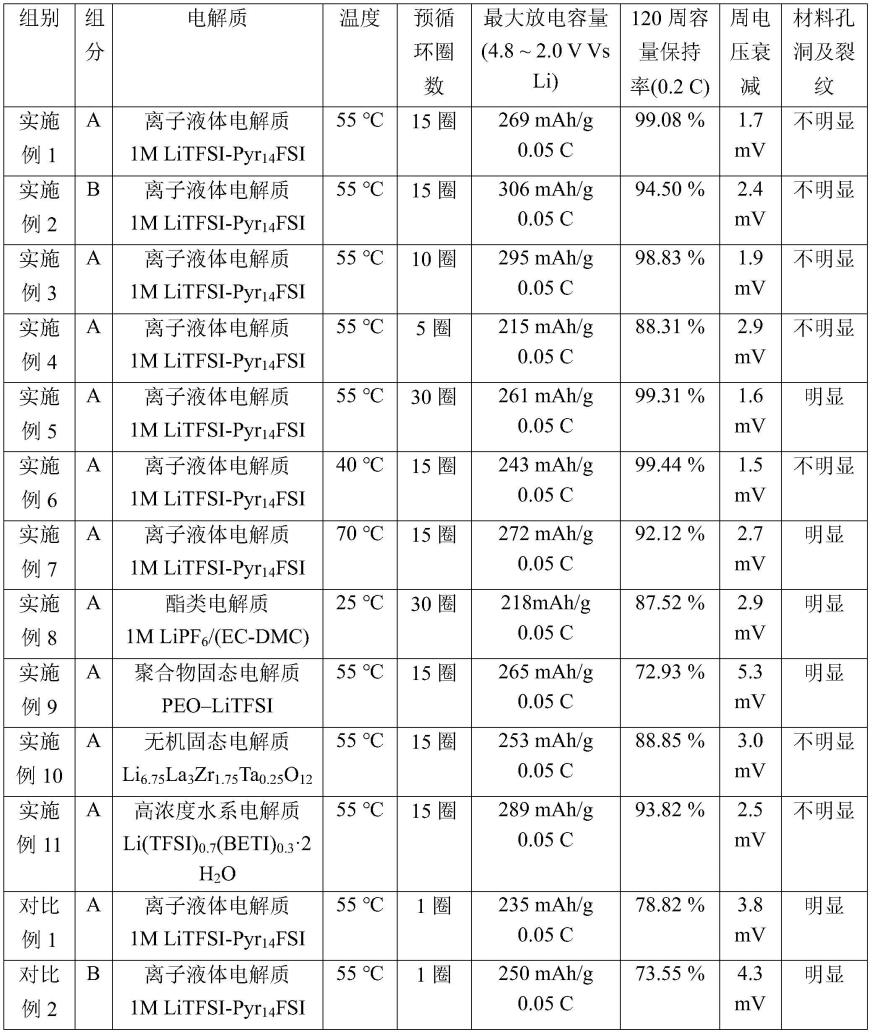
33.下面结合具体的实施例,对本发明进行进一步介绍。应当理解这些实施例是为了更好地进一步理解本发明,并不局限于所述最佳实施方式,不对本发明的内容和保护范围构成限制,任何人在本发明的启示下或是将本发明与其他现有技术的特征进行组合而得出的任何与本发明相同或相近似的产品,均落在本发明的保护范围之内。
34.实施例中未注明具体实验步骤或条件者,按照本领域内的文献所描述的常规实验步骤的操作或条件即可进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规试剂产品。其中xli2mno3·
(1-x)limeo2材料为自行制备获得。
35.结合下列具体实例对本发明做进一步详细的阐述。
36.实施例1
37.1.按照3:1的摩尔比称取mnso4和niso4盐溶于去离子水配置2mol/l的混合溶液;浓度为1.2mol/l的氨水络合剂,沉淀剂为2mol/l的na2co3溶液;通过蠕动泵将混合溶液与
na2co3溶液同时滴加至剧烈搅拌的反应釜中,反应釜中预先加入适量的络合剂溶液,同时控制ph值在7.8左右,水浴加热,温度为60℃;滴加完毕后,陈化10h;反应产物过滤,洗涤,在90℃真空烘箱中干燥12h得到前驱体;
38.2.按照li
1.2
ni
0.2
mn
0.6
o2的比例称取过量5%的li2co3作为锂源,充分混合后置于马弗炉中,以5℃/min的升温速率,在500℃下保温5h,后升至900℃12h煅烧,冷却至室温后得到富锂正极材料li
1.2
ni
0.2
mn
0.6
o2(1.25li
1.2
ni
0.2
mn
0.6
o2=0.5li2mno3·
0.5lini
0.5
mn
0.5
o2);其为粒径为2μm左右的二次颗粒;
39.3.将富锂正极材料与导电炭黑以及溶解在n-甲基吡咯烷酮中的聚偏二氟乙烯以80:10:10的质量比例均匀混合,涂覆于铝箔上,在65℃真空干燥箱中干燥12h,冲压为直径12mm的电极片,活性物质负载量为3.2~4.2mg/cm2;
40.4.将双三氟甲磺酰亚胺锂(litfsi)溶于1-丁基-1-甲基吡咯烷双(三氟甲磺酰)亚胺盐(pyr
14
tfsi))中,摩尔比2:8,在h2o<0.1ppm,o2<0.1ppm手套箱中搅拌24h,温度为30℃,得到高温稳定的离子液体电解质;
41.5.将锂金属负极,富锂正极,以离子液体为电解液组装半电池;
42.6.电池转移至55℃下静置10h,在55℃下使用0.05c倍率电流在4.5~2.0v区间循环15圈,完成激活循环过程(以阴离子容量不再增加为节点),并可用于后续在0.2c倍率电流下4.8v~2v的工作循环;
43.实施例2
44.与实施例1的区别仅在于,富锂正极材料组分改为li
1.2
ni
0.13
co
0.13
mn
0.54
o2;
45.实施例3
46.与实施例1的区别仅在于,形貌控制为粒径为1μm左右的一次颗粒,预循环10圈即可充分激活阴离子容量、并完成激活过程;
47.实施例4、5
48.与实施例1的区别仅在于,将预循环分别设定为5圈和30圈;
49.实施例6、7
50.与实施例1的区别仅在于,激活循环过程的温度分别改为40℃和70℃;
51.实施例8、9、10、11
52.与实施例1的区别为,匹配电解质分别替换为酯类电解质1m lipf6/(ec-dmc)(此时充放电温度改为25℃)、无机固态电解质li
6.75
la3zr
1.75
ta
0.25o12
、聚合物固态电解质peo
–
litfsi、高浓度水系电解质li(tfsi)
0.7
(beti)
0.3
·
2h2o,来代替实施例1中的离子液体电解质;
53.对比例1
54.与实施例1的区别仅在于,激活过程截止电压取为传统4.8v,即在0.05c倍率电流、4.8~2.0v区间,单圈常规激活;
55.对比例2
56.与实施例3的区别仅在于,激活过程截止电压取为传统4.8v,即在0.05c倍率电流、4.8~2.0v区间,单圈常规激活;
57.对比例3、4
58.与实施例8、9的区别仅在于,激活过程截止电压取为传统4.8v,即在0.05c倍率电
流、4.8~2.0v区间,单圈常规激活;
59.对比例5、6
60.与实施例10、11的区别仅在于,激活过程截止电压取为传统4.8v,即在0.05c倍率电流、4.8~2.0v区间,单圈常规激活;
61.电化学性能测试:电化学性能测试设备为武汉蓝电电子股份公司所售蓝电测试系统,恒温设备为天津泰斯特电池恒温测试箱,均可于市面购买得到。电池电化学性能测试步骤为,将上述扣式电池置于恒温箱中,在测试温度下恒温以蓝电静置程序静置10h,充放电程序为恒电流充/放电式,恒流充电至所设置截止电压上限,静置2min,再以恒流放电至设置截止电压下限,静置2min,完成一次循环。容量保持率计算方式为,电池第120周上述循环后的放电容量与首周放电容量的比值;电压衰减计算方式为:以放电容量为总容量一半时的对应的电压,即放电中压,作为判断标准。
62.图1到图9为实施例与对比例的电化学性能测试图
63.实施例1在55℃,0.05c,4.5~2.0v,循环15圈之后,达到最大放电容量为269mah/g,并完成激活循环过程。如图1所示,在55℃,0.2c,4.8~2.0v区间循环,首周放电比容量为269mah/g,150圈之后,比容量为235.3mah/g,库伦效率保持在97.83%。从现有的相关文献统计,当前常规4.8v激活方法li
1.2
ni
0.2
mn
0.6
o2的最大放电比容量通常不高于250mah/g,百圈后比容量保持率通常仅为80%左右;在对比例1和图4中也有佐证。这证明了本发明的富锂正极材料的近平衡电位本征原位激发表面稳定保护层的方法,对于循环稳定性方面的提升是有效的。
64.如图2所示,实施例1在4.5v的低截止电压下,首圈充电曲线仍表现出阴离子容量激活的长平台,且首周没有氧气释放的斜坡区域;预循环15圈之后,其激活后的最高放电比容量为268.9mah/g,并从第16圈开始在工作电压2~4.8v区间循环。相比较,对比例1为不进行预循环处理的正极材料,通过常规高电压激活阴离子容量;其首圈充电曲线4.6~4.8v区间表现为氧气释放的斜坡区域,对应着不可逆的氧气流失,并造成阴离子可逆容量的流失,其激活后的放电比容量为234.8mah/g。对比而言,本发明的预激活方法,首周工作放电的可逆比容量提升了34.2mah/g,如实施例1以及图1所示;这证明,本发明抑制了氧气释放,保留了更多的可逆阴离子容量,并形成稳定保护层,提升了后续工作循环的容量保持率。
65.如图3所示,为实施例1和对比例1在经过激活循环过程后,首圈工作循环的dq/dv图;mn
3+/4+
充电特征峰位于2.8~3.2v,如图所示,实施例1此处的峰明显弱于对比例1,这表明实施例1的阴离子流失得到了有效的抑制,抑制了mn
4+/3+
氧化还原对的生成;位于3.3v附近的o
α-/2-放电特征峰,实施例1相比于对比例1也有显著的增强,进一步证明了阴离子容量的保留率的提高。
66.如图4所示,为实施例1和对比例1在0.2c工作循环120圈时,比容量的变化趋势。经历过本发明方法进行表面保护的实施例1,在120圈后的放电比容量为238.1mah/g,容量保持率为99.0%。相对而言,传统方法进行激活的对比例1,其相同工作循环条件下,120圈后比容量下降为163.4mah/g,容量保持率为78.8%。
67.如图5所示,实施例1与对比例1的放电中压趋势表明,本发明的激活循环方法,可以有效抑制工作循环过程中的电压衰减。
68.为证明本发明方法对富锂锰基层状正极材料具有一定的普适性,将实施例2与对
比例2的组分修改为li
1.2
ni
0.13
co
0.13
mn
0.54
o2,其电压曲线如图6所示。实施例2在4.5~2v电压、0.05c倍率电流,进行15圈激活循环处理之后,在4.8~2.0v的电压区间的首圈工作循环的放电比容量为305.5mah/g,与对比例2的250mah/g相比,提升了55.5mah/g,证明本发明方法对该组分具有同样的提升效果。
69.实施例3通过降低前驱体合成过程中的络合剂浓度,降低高温煅烧段的温度,减小了材料颗粒粒径,形貌控制为粒径1μm左右的一次颗粒单晶。其电压曲线如图7所示,与实施例1相比,充分的激活循环(4.5~2.0v)从15圈降低为10圈,这证明,减小颗粒的粒径,提高了激活反应动力学。此外,实施例3的首圈激活循环充电容量为284.9mah/g,放电容量216.3mah/g,高于实施例1的175.5mah/g;且实施例3的首圈工作循环(4.8~2.0v),放电容量为294.5mah/g,高于实施例1的269mah/g。
70.如图8所示,实施例8将55℃的温度条件,改变为常温环境下,此时为了减小低温极化,匹配电解质为酯类电解质1m lipf6/(ec-dmc);采用相同激活循环截止电压上限4.5v,首圈充电曲线的平台区域表现为显著的减少。且在0.05c电流倍率下激活循环30圈后,首次工作循环充电至4.8v时,充电曲线仍然存在4.5~4.8v的平台区,证明激活反应不完全。与实施例1对比,当温度降低时,阴离子氧化还原的预激活过慢,在4.5v近平衡条件下,即使预循环30圈,仍没有激活出全部阴离子超额容量;相对而言,实施例1经过15圈预循环即激发出阴离子的超额容量。也就是说,在近平衡电位激活方式下,由于电位条件温和,降低温度大大阻碍了超额阴离子氧化还原容量的充分激活。
71.如图9所示,将本发明表面改性得到的实施例1,在完成激活循环阶段后(4.5~2v,0.05c,15圈),拆卸电池,收集正极粉末,将电解质更换为商用酯类电解液1m lipf
6-(ec:dmc=1:1),按照上述电池组装步骤,重新装配电池,在25℃恒温下,以0.05c倍率电流进行循环。重装电池在小倍率电流下,经过30圈循环后,放电容量与放电电压平台表现出良好的可逆性。小倍率电流下循环时,由于一圈循环充放电时间的大幅增加,使得电化学反应充分发生,不期望的副反应也会充分反应,且材料结构变化更加显著,对电极材料的稳定性要求更高,因此,发明原位表面改性方法改性后的正极,在商用电解液表现出良好的结构稳定性,再次证明本方法生成的表面包覆相对于富锂正极材料的电化学性能提升效果。
72.其它扣式电池电化学性能测试数据,如表1所示。
73.表1、扣电电池电化学性能数据
[0074][0075][0076]
表1所示为实施例与对比例的电化学性能,实施例均通过本发明方法完成本征表面改性,对比例均未改性,使用常规高电压手段激活。其中组分a为li
1.2
ni
0.2
mn
0.6
o2,组分b
为li
1.2
ni
0.13
co
0.13
mn
0.54
o2。与对比例相比,实施例对于电池的最大放电容量、120周容量保持率(0.2c)以及周电压衰减,均具有改善效果。
[0077]
实施例4、5,将实施例1中的预循环圈数15圈,分别改为5圈和30圈,证明预循环圈数过少,则阴离子容量激活不完全,容量较低,且表面氧空位过少,难以形成均匀表面稳定相层,在高电压下不能实现对材料的保护与氧流失的抑制,循环性能较差;预循环圈数过多,则在小倍率电流下循环次数过多,由于一圈循环中的充放电时间较长,在30圈后电池高电压高温环境循环时间过长,不可预期的副反应不断累计,最终影响电池性能,实施例5相对实施例1的容量有所下降;且阴离子容量在15圈附近激活完成后,在后续的循环中容量也不再增加,后续预循环过程失去意义,且低倍率循环非常耗时。
[0078]
实施例6、7,将实施例1中的预循环环境温度55℃,分别改为55℃和70℃,证明低温下由于激活过程动力学性能缓慢,在循环相同圈数时,阴离子容量难以在低电压下实现完全激活,在以高电压循环时,仍出现阴离子激活特征平台,氧流失副反应的抑制效果有限;然而,不能一味提高环境温度,在过高的温度下,电极材料动力学性能过高,从表面到体相发生不可逆的相变反应,加速了氧流失和过渡金属扩散,导致更为剧烈的容量损失和电压损失。
[0079]
实施例1与实施例8、9的区别在于,所用电解质不同;由于酯类与聚合物电解质在高温高电压下的分解,改性的效果降低。实施例1中采用离子液体,与正极材料不反应,也不会高温分解,证明本发明方法为材料的本征反应过程,非与电解质的副反应产物。实施例8、9采用有机电解质,高温分解,且与正极材料发生反应。比较而言,实施例8、9的循环性能均变差,说明电解质副反应程度的增加会降低该方法表面相重建结构的稳定性。然而,与传统高压激活方法相比,本发明所采用的低电压预激活的处理,除了对于耐高压和高温的离子液体以外,对于其它耐高压高温的电解质体系有效果(实施例10和对比例5,无机固体电解质高压和高温稳定性佳;实施例11和对比例6,高浓度水系电解质有一定的温度和电压耐受性),其对于高电压不稳定的传统酯类电解质和聚合物电解质,也有一定改善效果(实施例8和对比例3,其中酯类电解质在高电压下不稳定;实施例9和对比例4,其中聚合物电解质在高温下不稳定)。
[0080]
实施例1与实施例10、11的区别在于,所用电解质不同;采用宽电压窗口的无机陶瓷与高浓度盐电解质,降低了电解质副反应程度,通过本发明方法改性显著提升了电化学性能。
[0081]
实施例与同组别对比例相比较,均有明显的电化学性能提升效果,说明本发明近平衡条件下循环激活表面稳定包覆相的方法具有一定的普适性,且为材料的本征反应过程,不需要额外设计电解质和添加剂的反应,在正极表面原位形成了更加薄、致密且稳定保护层,抑制了容量损失和电压衰减,显著提升了其循环稳定性。
[0082]
图10为原位表面改性方法的示意图,阴离子容量充电激活后,近表面产生氧空位和锂空位,并促使过渡金属扩散、表面相重建,最终形成类岩盐相的表面包覆层。
[0083]
图11为经历激活循环(4.5~2v,0.05c,15圈)后的实施例1表面区域的高分辨率透射电镜,经过原位表面重建后,富锂正极材料表面形成了一层均匀、致密、厚度为3~4nm的类岩盐相。
[0084]
图12为经历激活循环处理的实施例1,再进行200圈工作循环(4.8~2v,0.2c),其
表面区域的高分辨率透射电镜,此时厚度为3~4nm的类岩盐相主体得以保留,表面1nm区域变得更无序,未出现氧流失造成的孔洞、以及应力变化造成的裂痕。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1