用于高效乳酸生产的材料和方法与流程
用于高效乳酸生产的材料和方法本申请是申请日2006年8月10日、标题为“用于高效乳酸生产的材料和方法”申请号200610109332.9的分案申请。相关申请的交叉引用本申请要求于2005年8月10日提交的美国临时申请系列号第60/706,887号、2006年1月24日提交的系列第60/761,576号、以及2006年5月11日提交的系列号第60/799,619号的权益。政府支持本发明是在政府的支持下完成的,其由能源部资金支持,批准号为USDOE-DEFG02-96ER20222;以及由能源部联合美国农业部资金支持,批准号为USDA&DOEBiomassRDIDEFG36-04GO14019。本发明还得到来自美国农业部的政府支持,批准号为01-35504-10669和00-52104-9704。政府对本发明具有一些权利。
背景技术:
乳酸通常用作用于保存、增加风味和酸味的食品添加剂。乳酸还用于制造可生物降解塑料,即聚乳酸(PLA)。PLA作为石油基产品的可更新替代品的应用迅速扩大(Agrawal,2003)。PLA的物理性能和生物降解速率可通过调整手性底物(D-乳酸和L-乳酸)的比率加以控制(Narayananetal.,2004)。估计每年全球乳酸市场超过100,000吨,并且有望在接下来的几年内随着新的PLA设备投入运行会有显著增长。例如,对可生物降解的溶剂乳酸乙酯(一种乳酸衍生物)的需求有望在近年内有显著增长。据估计,乳酸酯有可能代替美国每年使用的380万吨溶剂的80%。这种溶剂无毒并且具有许多有益的应用,包括用于电子制造、油漆和涂料、纺织品、清洁剂和脱脂剂、粘合剂、印刷、以及脱墨。生产乳酸时,人们常常优选发酵方法而非化学合成,因为其得到的是D和L异构体的混合物。微生物发酵的产物依赖于所使用的有机体。微生物发酵可产生两种异构体的混合物或者立体专一形式的光学纯乳酸。希望的产物的立体专一性依赖于其计划的用途。采用乳酸杆菌(Lactobacilli)的细菌发酵通常用于乳酸的工业生产,但是这些发酵很少产生光学纯产物。另外,这些细菌需要复杂营养的性质要求将相当大量的补充营养素添加到生长培养基,增加了额外费用并且使纯化更加困难。此外,用于生产乳酸的发酵方法效率严重不足,必须加以改进以确保上述预期市场扩张的经济可行性。尽管描述过重组酿酒酵母菌(Saccharmycescerevisiae)菌株含有来自乳酸杆菌或者牛源(bovineorigins)乳酸脱氢酶(ldh)基因(PatentWO99/14335andAdachietal.,1998),但是酵母菌不能生产可观水平的乳酸。虽然能够生产达2-4%(w/v)的乳酸,但是这些菌株显示出较差的生产能力并且相当一部分葡萄糖转化成乙醇。丝状真菌米根霉(Rhizopusoryzae)(也称作少根霉(R.arrhizus))也用于乳酸的工业生产。米根霉能够在化学成分确知的培养基(chemicallydefinedmedium)中将葡萄糖有氧地(aerobically)转化为大量的光学纯L-(+)-乳酸。之所以持续研究通过根霉的乳酸生产,主要是因为在基本生长培养基中容易进行产物纯化以及真菌利用复合碳水化合物和戊糖的能力(美国专利第4,963,486号)。这使得可利用真菌将低价值的农业生物质转化为乳酸。遗憾的是,通过对根霉和其它真菌的遗传修饰来改变乳酸生产的能力比较有限。基于大肠杆菌K-12的生物催化剂已经被改造用于D-(-)-乳酸(盐)(lactate)生产,但是不能将复合或基本培养基中10%的葡萄糖或蔗糖发酵完全(Changetal.,1999;Dienetal.,2001;Zhouetal.,2003;ZhuandShimizu2004)。人们开发了一种大肠杆菌生物催化剂,即SZ63(pLOI3501),其通过在质粒上功能化地表达来自大肠杆菌B的cscR’cscA’cscKB’基因而用于蔗糖发酵(Shuklaetal.2004)。尽管能够有效地发酵5%的葡萄糖或蔗糖,但是这种生物催化剂不能彻底代谢更高的糖浓度并且为了质粒的维持需要连续进行抗生素选择。由大肠杆菌菌株B衍生的其他生物催化剂,如KO11(保藏编号:ATCC55124),具有天生的发酵蔗糖的能力(Moniruzzamanetal.,1997)。同菌株SZ63一样,这种生物催化剂不能完全地代谢更高糖浓度并且为了质粒的维持需要连续进行抗生素选择。因此,为了减少与大量生产型化学品的基于生物生产相关的费用,对于具有增加发酵速度、产物效价和产率的改进的乳酸生物催化剂仍然存在需求(Arntzenetal.,1999;Chotanietal.,2000;Dattaetal.,1995;HofvendahlandHahn-Hagerdal,2000;andOharaetal.,2001)。
技术实现要素:
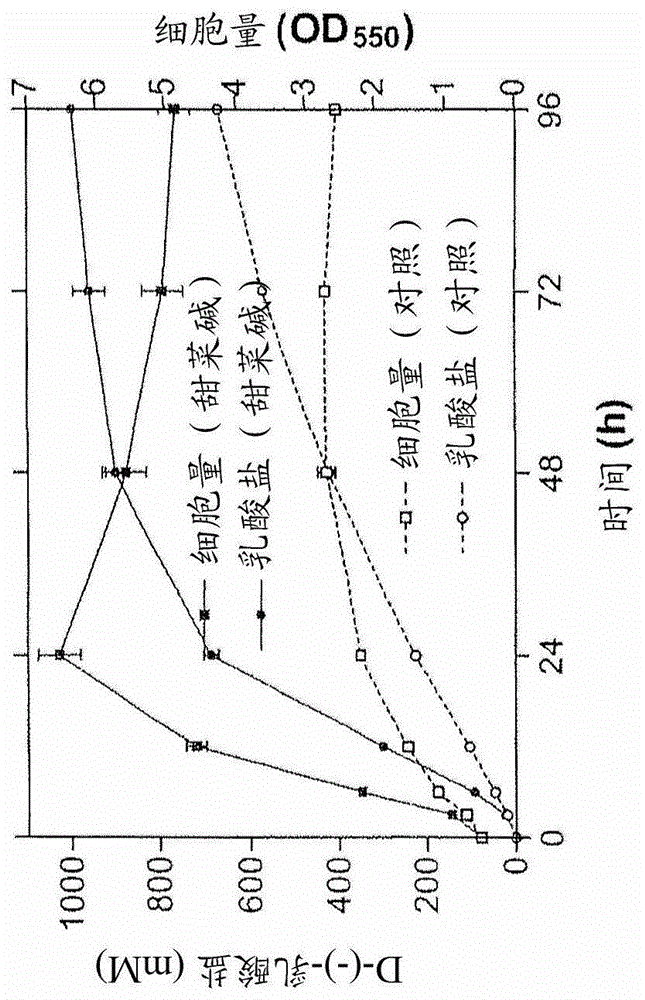
本主题发明提供了在乳酸生产中非常有用的新型微生物。另外,本主题发明还提供了新型构建体,用于转化大量寄主生物中的任何一种,优选大肠杆菌,使该寄主生物在可发酵培养基中培养时表达和(或)抑制一些基因,从而产生乳酸。因此,本主题发明的材料和方法可用来提高寄主生物中的乳酸生产,从而增加用于食品和工业应用中的乳酸供应。在一些具体实施方式中,构建了产乙醇(ethanologenic)的大肠杆菌(本文中也称为大肠杆菌E.coli)KO11的衍生物用于D-(-)-乳酸(盐)(lactate)的生产。在本发明的另外的具体实施方式中,E.coli根据本主题发明进行改造,用于L-(+)-乳酸(盐)的生产。根据本主题发明,通过去掉编码竞争途径的基因接着通过基于生长选择发酵葡萄糖和(或)蔗糖的性能改善的突变体,可以制得基于E.coliKO11的新型生物催化剂。本发明改造的微生物优选含有利用蔗糖的天然基因。本发明的一些改造的E.coli菌株可发酵10%的葡萄糖或蔗糖,而生产超过1摩尔D-(-)-乳酸盐/l的发酵液,根据发酵液的不同,基于代谢的糖的产率在约88%到约95%的范围内。本发明的其他改造的E.coli菌株可发酵葡萄糖或蔗糖而生产L-(+)-乳酸盐。本发明其他的优点根据随后的描述将变得显而易见。附图说明图1A-1F用图示阐明了本发明一个具体实施方式与菌株SZ63相比之间活性的差异。图2A-2D图解阐明了发酵条件下在NBS无机盐培养基中通过本发明各种具体实施方式的乳酸生产进程(图2A:10%葡萄糖(NBS,SZ132);+和-1mM甜菜碱;图2B:10%蔗糖(NBS,SZ132);+和-1mM甜菜碱;图2C:SZ186NBS10%葡萄糖;+和-1mM甜菜碱;图2D:SZ186NBS10%蔗糖;+和-1mM甜菜碱;SZ186是改进的菌株,更纯净的产物,更高的产率。)。图3A-3B图解阐明了通过本发明一个具体实施方式的乳酸生产过程中甜菜碱增加耐酸性和耐糖性的能力(图3A:甜菜碱增强乳酸盐耐性;图3B:甜菜碱增强葡萄糖耐性;甜菜碱增加耐酸性和耐糖性)。图4A-4B图解阐明了本发明的一个具体实施方式适应无机培养液(mineralmedia)的能力(图4A:细胞生长(OD550nm);图4B:生产的乳酸盐。SZ186对无机(盐)培养基的适应,得到菌株SZ194。)。图5A-5G图解阐明了通过本发明的另一个具体实施方式在各种发酵条件下乳酸生产的进程(图5A:SZ194在含10%葡萄糖的NBS(1mM甜菜碱)中的发酵;SZ194比SZ186快;图5B:LB10%葡萄糖(SZ194);图5C:LB10%葡萄糖(SZ194)。pH7.5和pH8.0优于pH6.5和pH6.0。图5D:SZ194NBS葡萄糖10-14%(乳酸盐);图5E:SZ194NBS葡萄糖10-14%(OD);高于10%糖,培养物较慢地复制(takeoff)。图5F:分批(大肠杆菌SZ194NBS12%葡萄糖);图5G:补料分批(大肠杆菌SZ194NBS12%葡萄糖);补料分批有助于改进速率,72-96h后培养液中>1M乳酸盐)。图6图解阐明了SZ132在10%蔗糖(pH7.0)中用于改善生长以及乳酸盐生产的代谢演化。将菌株SZ132在含有10%(w/v)蔗糖的无机盐培养基中进行连续转移,以富集那些在没有甜菜碱时生长和乳酸盐生产改善的自然突变体。克隆体SZ136从最后的转移中分离出来。图6A示出了细胞量(cellmass)。图6B示出了总有机酸(碱消耗)。图7图解阐明了SZ186在10%葡萄糖(pH7.0)中用于改善生长和乳酸盐生产的代谢演化。将菌株SZ186在含有10%(w/v)葡萄糖和1mM甜菜碱的无机盐培养基中的进行连续转移,以富集生长和乳酸盐生产改善的自然突变体。一种克隆,SZ136,从最后的转移中分离出来。图7A示出了细胞量。图7B示出了总有机酸(保持pH的碱消耗)。图8图解阐明了通过SZ194以分批和补料分批发酵方式生产乳酸盐的比较。发酵在含有1mM甜菜碱(pH7.5)的无机盐培养基中进行。图8A示出了含有12%(w/v)葡萄糖的简单分批发酵;图8B示出了补料分批发酵。通用符号:□,细胞质量;●,总有机酸(保持pH的碱消耗);○,葡萄糖。图9A-9B示出了E.coli中乳酸盐生产的天然途径。图9A为二羟基丙酮-P节点。图9B为丙酮醛支路。缩写词:DHAP,磷酸二羟丙酮;Gly3P,3-磷酸甘油醛。图10A-10F示出了通过重组E.coli将12%(w/v)的葡萄糖(NBS无机盐培养基+1mM甜菜碱)发酵为乳酸盐的过程。图10A示出了通过D-(-)-乳酸盐菌株的有机酸生产。图10B示出了D-(-)-乳酸盐菌株的生长。图10A和10B的符号:Δ,SZ194;○,TG112(10%w/v葡萄糖);◇,TG113;□,TG114。图10C示出了mgsA缺失对通过L-(+)-乳酸盐菌株(10%葡萄糖+1mM甜菜碱)有机酸生产的影响。图10D示出了mgsA缺失对L-(+)-乳酸盐菌株(10%葡萄糖)生长的影响。图10C和10D的符号:●,TG102(mgsA+,10%w/v葡萄糖,不含甜菜碱);○,TG103(mgsA+,10%w/v葡萄糖,不含甜菜碱);■,TG105(mgsA缺失,1mM甜菜碱);*,TG103(mgsA+,1mM甜菜碱)。图10E示出了通过L-(+)-乳酸盐菌株的有机酸生产。图10F示出了L-(+)-乳酸盐菌株的生长。图10E和10F的符号:■,TG105;▲,TG106;●,TG107;◆(□),TG108。图11A-11B示出了用于改善D-(-)-乳酸盐生产和生长的代谢演化。将含有10%或12%(w/v)葡萄糖和1mM甜菜碱的无机盐培养基中的E.coli菌株TG112每24或48小时进行连续转移,以选择生长、抗性和乳酸盐生产改善的自然突变体。将一个克隆TG113从TG112转移第28号(第29天)分离作为中间体,并且将另一种克隆体TG114在第81天从最后的培养物中分离出来。为清晰起见,示出了每4次24小时转移和每隔48小时转移的曲线图。作为比较还包括了亲代SZ194。图11A示出了根据为保持pH值为7的碱消耗而计算出的总有机酸(mM)。图11B示出了细胞量(g/L)。符号:▲,SZ194,12%(w/v)葡萄糖;○,TG112,10%(w/v)葡萄糖,1:100稀释,24小时转移;●,TG112,12%(w/v)葡萄糖,1:100稀释,24小时转移;□,TG113,12%(w/v)葡萄糖,1:350稀释,24小时转移;以及◆(□),TG113,12%(w/v)葡萄糖,1:100稀释,48小时转移。图12A-C示出了pLOI4417的产生。pLOI4417通过在pLOI4411上进行PCR而生成,一引物在frdA的3’端退火、向上游延伸,另一引物在frdC内部退火、向下游延伸(图12A)。将得到的4263bpPCR产物用限制性内切酶DpnI处理以消化天然pLOI4411质粒(图12B)。消化的PCR产物随后进行自身连接,以产生新的质粒pLOI4417(图12C)。序列说明SEQIDNO:1是用于frdBC缺失的有义引物的核苷酸序列。SEQIDNO:2是用于frdBC缺失的反义引物的核苷酸序列。SEQIDNO:3是用于adhE缺失的有义引物的核苷酸序列。SEQIDNO:4是用于adhE缺失的反义引物的核苷酸序列。SEQIDNO:5是用于celY缺失的有义引物的核苷酸序列。SEQIDNO:6是用于celY缺失的反义引物的核苷酸序列。SEQIDNO:7是用于mgsA缺失的有义引物的核苷酸序列。SEQIDNO:8是用于mgsA缺失的反义引物的核苷酸序列。SEQIDNO:9是用于FRT-cat-sacB扩增的有义引物的核苷酸序列。SEQIDNO:10是用于FRT-cat-sacB扩增的反义引物的核苷酸序列。SEQIDNO:11是用于带有5’NheI位点的cat-sacB扩增的有义引物的核苷酸序列。SEQIDNO:12是用于带有5’NheI位点的cat-sacB扩增的反义引物的核苷酸序列。SEQIDNO:13是用于克隆frdABCD的有义引物的核苷酸序列。SEQIDNO:14是用于克隆frdABCD的反义引物的核苷酸序列。SEQIDNO:15是用于克隆ackA的有义引物的核苷酸序列。SEQIDNO:16是用于克隆ackA的反义引物的核苷酸序列。SEQIDNO:17是用于克隆lacZ-cynX的有义引物的核苷酸序列。SEQIDNO:18是用于克隆lacZ-cynX的反义引物的核苷酸序列。SEQIDNO:19是用于克隆mgsA的有义引物的核苷酸序列。SEQIDNO:20是用于克隆mgsA的反义引物的核苷酸序列。SEQIDNO:21是用于克隆focA-pflB的有义引物的核苷酸序列。SEQIDNO:22是用于克隆focA-pflB的反义引物的核苷酸序列。SEQIDNO:23是用于克隆adhE的有义引物的核苷酸序列。SEQIDNO:24是用于克隆adhE的反义引物的核苷酸序列。SEQIDNO:25是用于缺失frdBC的有义引物的核苷酸序列。SEQIDNO:26是用于缺失frdBC的反义引物的核苷酸序列。SEQIDNO:27是用于缺失ackA的有义引物的核苷酸序列。SEQIDNO:28是用于缺失ackA的反义引物的核苷酸序列。SEQIDNO:29是带有5’NheI位点的lacZ反义引物的核苷酸序列。SEQIDNO:30是带有5’NheI位点的cynX反义引物的核苷酸序列。SEQIDNO:31是用于缺失mgsA的有义引物的核苷酸序列。SEQIDNO:32是用于缺失mgsA的反义引物的核苷酸序列。SEQIDNO:33是用于缺失focA-pflB的有义引物的核苷酸序列。SEQIDNO:34是用于缺失focA-pflB的反义引物的核苷酸序列。SEQIDNO:35是用于缺失adhE的有义引物的核苷酸序列。SEQIDNO:36是用于缺失adhE的反义引物的核苷酸序列。SEQIDNO:37-46描述了其中FRTscar被去除的遗传区的部分序列。以粗体示出了该基因的5’和3’区的部分序列,斜体用于标明FRTscar,并且下划线区在菌株TG128中被去除。具体实施方式本发明提供的新型微生物当在多种发酵条件中生长时均能够生产乳酸。本发明还提供了用于改造这样的微生物的方法以及利用本发明的新型微生物有效地和稳定地生产乳酸的方法,以便可从相对低廉的初级产品诸如葡萄糖或蔗糖提供高产率的乳酸。本申请中的术语“乳酸(lacticacid)”指的是游离酸形式或者盐形式的2-羟基丙酸。乳酸的盐形式称作“乳酸盐(lactate)”,不管中和剂是碳酸钙还是氢氧化铵。本文所提到的乳酸可指乳酸两种立体异构形式中任何一种(L-(+)-乳酸或D-(-)-乳酸)。术语乳酸盐可指乳酸盐两种立体异构形式中任何一种(L-(+)-乳酸盐或D-(-)-乳酸盐)。本发明提供了生产乳酸或乳酸盐的单一立体异构体的微生物。根据本发明的一些方面生产的乳酸立体异构体或乳酸盐立体异构体是“手性纯”的。术语“手性纯(chirallypure)”表示没有可检测到的乳酸或乳酸盐的一种立体异构形式与另一种立体异构形式之间的污染(特定的立体异构体的手性纯度(chirallypurity)至少高于(或者高于或等于)99.9%)。本文披露的已将丙酮醛途径(mgsA)缺失或失活的遗传修饰微生物能够生产“手性纯”的D-(-)-乳酸或L-(+)-乳酸。在本发明的一些具体实施方式中,利用本发明改造的微生物生产L-(+)-乳酸。在本发明的其他具体实施方式中,利用本发明改造的微生物生产D-(-)-乳酸。在一个具体实施方式中,本发明利用大肠杆菌(或E.Coli)作为生物催化剂,用于提高葡萄糖和/或蔗糖向乳酸的转化。根据本发明,微生物的代谢可通过引入并表达多种基因进行改性。根据本主题发明,可将多种新型质粒引入E.Coli,以使转化的(transformed)微生物可在多种发酵条件下生产大量的乳酸。优选将本发明的重组E.Coli进行改性,以便当在包含葡萄糖和/或蔗糖的培养基上生长时,可稳定地以高产率生产乳酸。含有本主题发明的质粒的E.Coli寄主由美国61604伊利诺斯州皮奥里亚市大学街1815N美国农业研究机构保藏中心(AgriculturalResearchServiceCultureCollection,1815N.UniversityStreet,Peoria,Illinois,61604U.S.A.)保藏。入藏登记号和保藏日期如下:已将本主题培养物在如下条件下保藏,该条件确保在本专利申请未决期间由专利和商标局根据37CFR1.14和35USC122确定的人被授权可以获得这些培养物。在本主题申请的优先权文本或其分案提交的国家中,按照国外专利法的要求可以获得这些保藏物。但是,应该理解,保藏物的可获得性不构成在由政府行为授予的专利权部分失效时对实施本主题发明的许可。而且,将该主题培养保藏物按照布达佩斯微生物保藏条约的规定进行储存并且对于公众是可以获得的,即,将它们在所有必要的管理下进行储存,使其保持存活并且不被污染,持续时间为最近一次要求提供保藏物的样品后至少5年期间,并且在任何情况下,持续时间为在保藏日期之后的至少30(三十)年或任何可以提出披露培养物的专利的可实施期。如果由于保藏条件在需要时保藏处不能提供样品,保藏人则有义务替换保藏物。公众对本主题培养保藏物可获得性的所有限制在披露它们的专利授权后将不能取消的去除。本发明的一个具体实施方式提供了一种E.ColiB菌株(“SZ132”;保藏编号NRRLB-30861),其被改造以在多种培养基中提高细胞生长和乳酸盐生产。在一个具体实施方式中,可将SZ132菌株通过下面的步骤进行构建以生产D-乳酸盐(乳酸):(a)通过将用于纤维二糖利用的产酸克雷伯氏菌(保藏编号ATCC68564)casAB基因整合到lacY的终止密码子后以及将编码内切葡聚糖酶的菊欧文氏菌(保藏编号为ATCC55124)celY基因整合将到frdA基因中,可从E.ColiKO11构建菌株LY52(Ohtaetal.,1991;Moniruzzamanetal.,1997;ZhouandIngram,1999);(b)依次将E.ColiK-12(菌株W3110)的一些突变转导入LY52,其用于构建菌株SZ63(Zhouetal.,2003);(c)通过从菌株SZ31(菌株W3110)ΔfocA-pflB缺失的P1转导而去除用于乙醇生产的运动发酵单胞菌基因;(d)通过从菌株TC20adhE突变的P1转导使天然E.Coli醇脱氢酶生产失活(Zhouetal.,2003);(e)利用从菌株SZ61(菌株W3110)的突变去除乙酸激酶(acetatekinase)(ackA)(Zhouetal.,2003);(f)通过FLP重组酶去除用于选择的抗生素基因(Zhouetal.,2003);以及(g)在步骤(f)的抗生素基因去除过程中,去除casAB的内部片段(或在去除抗生素基因步骤之后去除casAB基因的内部片段)。本发明的相关具体实施方式还涉及到转化菌株SZ132以去除所有外源基因。在一个具体实施方式中,通过利用线性DNA的同源重组将产酸克雷伯氏菌casAB和菊欧文氏菌(Erwiniachrysanthemi)celY基因从菌株SZ132缺失;然后利用FLP重组酶消除在菌株SZ132的构建过程中所用的抗生素基因。所得菌株SZ186(保藏编号NRRLB-30862)只含有天然E.Coli基因。本发明另一个具体实施方式提供了E.ColiB菌株(“TG103”),其被改造以在多种培养基中提高细胞生长和乳酸的L异构体的生产。在一个具体实施方式中,TG103菌株可通过下面的步骤进行构建:(a)利用本文所述的方法改造菌株SZ186;(b)将菌株SZ186进行系列培养,以选择生长和/或乳酸生产提高的菌株,其中所选菌株为菌株SZ194;(c)将异源L-乳酸基因和卡那霉素标记物整合到SZ194的天然ldhA(D-乳酸脱氢酶)基因中并去除编码区的中心部分以产生TG101;(d)通过FLP重组酶去除抗生素基因,以产生TG102;以及(e)将菌株TG102进行若干天(如从约7到21天,优选13天)的系列转移,其中对每天预先接种培养物的培养液(broth)进行1:100稀释,并且选择那些能显示出生长提高和/或L-乳酸生产增加的菌株,其中所选菌株为TG103。根据本主题发明的一些具体实施方式,加快的生长和增加的乳酸生产是联系的。对于能改善生长和乳酸生产的菌株的共选择过程称为“代谢演化(Metabolicevolution)”。本发明的一些具体实施方式还提供使本申请提供的遗传修饰微生物中的一些基因失活或缺失。在本发明的一方面,去除基因是为了使期望的活性失活。缺失能提供最大的稳定性,因为没有发生可逆突变而导致功能恢复的机会。可选地,可以通过插入能够破坏基因功能和/或表达的核酸序列而使基因失活(例如,P1转导或其他本领域已知的方法)。本文讨论的一种或多种特殊多核苷酸序列的失活或缺失也可称为“改性(modification)”。用于将特异性基因引入到寄主微生物中的载体可以是任何载体,只要它能够在寄主微生物中进行复制。本发明的载体可在所选寄主细胞中作为克隆载体或表达载体进行操作。许多载体为本领域的技术人员所通晓,选用合适的载体和寄主细胞只是选择的问题。例如,载体可以是噬菌体、质粒、病毒、或其混合物,如在Maniatisetal.,1989、Ausubeletal.,1995、Miller,J.H.,1992、SambrookandRussell,2001中描述的那些载体。而且,本发明的载体可以是非融合载体或融合载体。在每个具体载体中,可选择不同部位用于插入感兴趣的核苷酸序列。这些部位通常通过切开它们的限制性内切酶或核酸内切酶指定。载体可用匹配基因末端序列的限制性内切酶进行消化并且该序列可以被连接。通常连接通过利用诸如T4DNA连接酶的连接酶实现。选择用来将所选核苷酸片段插入载体以形成重组载体的特殊位点由多种因素决定。这些因素包括要表达的多肽的大小和结构、预期多肽对由寄主细胞组分的酶的降解及其蛋白质污染的敏感性、诸如起始和终止密码子的部位的表达特性、以及由本领域技术人员公认的其他因素。这些因素没有一个单独能够完全控制特殊多肽的插入位点的选择。确切地说,所选择的部位反映了这些因素的平衡,并且对特定蛋白质来说,并非所有部位同样有效。可使用多种载体-寄主细胞表达系统来实施本发明。细菌菌株如E.Coli在本发明的实施中用来生产乳酸特别有用。但是,这里描述的新发明可通过对各种乳酸生产方案适合的多种寄主应用。寄主菌株可以是细菌、真菌、或酵母菌来源的。选择寄主菌株可考虑的因素包括底物范围(substraterange)、抗性、糖耐性、盐耐性、温度耐性、pH耐性以及乳酸盐耐性。确定最合适的寄主-载体系统在本领域技术人员的能力范围内。用于染色体缺失、整合、以及可去除抗生素抗性基因的方法以前曾描述过(Causeyetal.,2004;DatsenkoandWanner,2000;Martinez-Moralesetal.,1999;Zhouetal.,2003)。这些已知方法中的任何一个或其组合可用来实施本发明。由于用于基因表达的启动子将存在于寄主微生物中(例如,产酸克雷伯氏菌casAB或菊欧文氏菌celY基因),所以当对于基因特异性的启动子在寄主细胞中起作用,即可采用该启动子。可选地,还可以将外源启动子连接到编码该基因的DNA,以便在该启动子的控制下获得表达。当将埃希氏菌(Escherichiabacterium)用作寄主时,lac启动子、trp启动子、trc启动子、tac启动子、λ噬菌体的PR启动子和PL启动子、tet启动子、amyE启动子、spac启动子等等均可作为这样的启动子使用。而且,还有可能利用含有如pUC19等启动子的表达载体,并将编码例如产酸克雷伯氏菌casAB(K.oxytocacasAB)的DNA片段插入该载体中,以使该片段可在该启动子的控制下进行表达。用于染色体DNA的制备、PCR、质粒DNA的制备、DNA的消化和连接、转化、用作引物的寡核苷酸的设计和合成等的方法可以是本领域技术人员熟知的常用方法。这些方法在例如Sambrook,J.etal.(1989)等中描述过。乳酸可通过如上所述的用转化的微生物将葡萄糖和/或蔗糖转化成乳酸并收集生成的乳酸来生产。在本发明的一个方面,当转化的微生物在无机盐培养基(如NBS无机盐培养基)中进行培养时,生产的乳酸的水平可高于0.5M。本发明的载体可自发地复制或整合到寄主的基因组中。整合常常通过同源重组(例如,精氨酸可选择性标记物整合到染色体精氨酸基因中)或者在与载体上任何基因都不相关的染色体部位发生。可通过单一或者双重跨接过程(singleordoublecross-overevent)进行整合。还有可能在同一个构建的微生物中存在任何数目的这些整合和复制类型。在一些具体实施方式中,将本发明改造的微生物的培养在需氧条件下进行约0.5到240小时。培养期间,培养温度优选控制在约25℃到45℃,pH值优选控制在5-8。无机或有机、酸性或碱性物质以及氨气或其类似物可用来调节pH。通过转化属于埃希氏菌属的细菌以表达表达一些在乳酸生产中有用的酶可以获得本发明的微生物。在优选的具体实施方式中,用于本发明的属于埃希氏菌属的细菌是大肠杆菌。在本发明的其他具体实施方式中,可用于本发明的细菌包括但不限于:氧化葡糖杆菌、浅井氏葡糖杆菌、德尔马瓦无色杆菌、粘无色杆菌、乳无色杆菌、根癌土壤杆菌、放射形土壤杆菌、粪产碱菌、柠檬节杆菌、肿大地杆菌(Arthrobactertumescens)、石蜡节杆菌、Arthrobacterhydrocarboglutamicus、氧化节杆菌、天牛金杆菌、印度固氮菌、产氨棒杆菌、离胺酸生产菌(divaricatum)、谷氨酸棒杆菌、黄色短杆菌、球状短杆菌(Brevibacteriumglobosum)、暗褐短杆菌(Brevibacteriumfuscum)、酮戊二酸短杆菌、微黄短杆菌(Brevibacteriumhelcolum)、极小短小杆菌、需土金杆菌、玫瑰色短杆菌、Brevibacteriumimmariophilium、扩展短杆菌、原玻璃蝇节杆菌(Brevibacteriumprotopharmiae)、嗜乙酰棒杆菌、谷氨酸棒杆菌、美棒杆菌、嗜乙酰乙酸棒杆菌、醋谷棒杆菌、产气肠杆菌、解淀粉欧文氏菌、胡萝卜软腐欧文氏菌、草生欧文氏菌、菊欧文氏菌、外来稳杆菌、染色假杆菌、橙色黄杆菌(Flavobacteriumaurantinum)、莱茵黄杆菌、塞沃尼假杆菌、短稳杆菌、脑膜脓毒性金黄杆菌、微球菌sp.CCM825(Micrococcussp.CCM825)、摩氏摩根氏菌、红串红球菌、粗糙诺卡氏菌、Planococcuseucinatus、雷氏普罗威登斯菌、谢氏丙酸杆菌、产黄假单胞菌(Pseudomonassynxantha)、产氮假单胞菌、荧光假单胞菌、卵状假单胞菌、施氏假单胞菌、食酸丛毛单胞菌(Pseudomonasacidovolans)、霉味假单胞菌、睾丸酮丛毛单胞菌、铜绿假单胞菌、红串红球菌、紫红红球菌、红球菌sp.ATCC15592(Rhodococcussp.ATCC15592)、红球菌sp.ATCC19070(Rhodococcussp.ATCC19070)、脲芽孢八叠球菌、金黄色葡萄球菌、梅氏弧菌(Vibriometschnikovii)、干酪弧菌(Vibriotyrogenes)、马杜拉马杜拉放线菌、紫产色链霉菌、Kitasatosporiaparulosa、天蓝色链霉菌、浅黄链霉菌(Streptomycesflavelus)、浅灰链霉菌、浅青紫链霉菌、橄榄链霉菌、田无链霉菌、弗吉尼亚链霉菌、抗生链霉菌、可可链霉菌、淡紫灰链霉菌、绿色产色链霉菌、杀鲑气单胞菌、短小芽孢杆菌、环状芽孢杆菌、解硫胺素芽孢杆菌、弗氏埃希氏菌、嗜氨微杆菌、粘质沙雷氏菌、鼠伤寒沙门氏菌、薛氏沙门氏菌(Salmonellaschottmulleri)、柑橘黄单胞菌等等。通过使根据本发明制备的含有转化微生物的培养物接触蔗糖和(或)葡萄糖,可在反应混合物中生产乳酸。在本发明的其他具体实施方式中,将细胞从转化微生物的培养物中分离并收集;将经过处理的转化微生物细胞进行丙酮处理或冷冻干燥;从这些转化的微生物细胞或者处理过的细胞中制备无细胞(cellfree)提取物;从这样的无细胞提取物中分离诸如膜片段的片段;或可通过固定转化的微生物细胞、处理过的细胞、无细胞提取物和片段产生固定物质,其中任何一种独立接触或联合接触蔗糖和(或)葡萄糖可以生产乳酸。微生物可包括一种微生物,或可用作两种或多种微生物的任意混合物。发酵参数依赖于用于产生重组酶的寄主微生物的类型。生长培养基可以是基本/合成(defined)或完全/复合培养基。可发酵碳源可包括己糖和戊糖、淀粉、纤维素、木聚糖、低聚糖及其组合。根据本主题发明可以使用的一种形式的生长培养基包括改性的由MillerJ.H.(1992)描述的Luria-Bertani(LB)液体培养基(每升具有10gDifco胰胨、5gDifco酵母提取物以及5g氯化钠)。在本发明的其他具体实施方式中,本发明构建的菌株的培养物可以在NBS无机盐培养基中生长(如由Causeyetal.,2004描述的)并补充了2%到20%的糖(w/v)或5%或者10%的糖(葡萄糖或蔗糖)。该微生物可在NBS无机盐培养基里面或上面生长。NBS无机盐培养基包括、主要组成为、或包含以下组分(每升):3.5gKH2PO4;5.0gK2HPO4;3.5g(NH4)2HPO4;0.25gMgSO4·7H2O;15mgCaCl2·2H2O;0.5mg硫胺;以及1mL的痕量金属成分(stock)、葡萄糖(例如,平皿中2%或培养液中3%)和1.5%的琼脂(用于平皿)。将痕量金属成分在0.1MHCl中制备,并且包括、主要组成为或包含(每升):1.6gFeCl3;0.2gCoCl2·6H2O;0.1gCuCl2;0.2gZnCl2·4H2O;0.2gNaMoO4;0.05gH3BO3。当需要进行pH控制时,可将4-吗啉代丙烷磺酸(0.1M,pH7.1)加入到液体和固体培养基(过滤除菌)(并且可选地包括在用于10升发酵的培养基中)。基本培养基也可通过将琥珀酸盐(1g/升)用作碳(不可发酵基质)的唯一来源进行制备并可在需要时作为补充成分加入葡萄糖-基本培养基中。在一些具体实施方式中,当菌株构建需要时还可包括抗生素。生长和乳酸盐的生产可用常规分批发酵、补料分批发酵(fed-batchfermentation)或连续发酵来进行。在一些具体实施方式中,在减氧或缺氧条件下进行发酵对于一些寄主是理想的。在另外的具体实施方式中,乳酸盐生产可在有氧条件下进行;并且,可选地使用气提式或类似的发酵罐。发酵的pH值应足够高以允许通过寄主的生长和乳酸盐生产。调节发酵液的pH可利用中和剂如碳酸钙或氢氧化钙来进行。可选地,可以在发酵期间,利用诸如膜技术、电渗析、溶剂萃取以及吸收树脂的方法持续移出乳酸。任何上述发酵方法的选择和结合很大程度上依赖于寄主菌株和优选的下游过程。本主题发明的各种非限制性具体实施方式包括:1.一种遗传修饰的E.Coli菌株,包括对E.Coli菌株KO11(ATCC55124)进行以下遗传修饰:a)在lacY的终止密码子之后插入产酸克雷伯氏菌casAB基因;b)将菊欧文氏菌celY基因整合到frdA基因中;c)使focA-运动发酵单胞菌pdc-adhB-pflB失活或缺失;d)使天然E.Coli醇脱氢酶基因失活或缺失;以及e)使乙酸激酶(ackA)基因失活或缺失;2.根据具体实施方式1的遗传修饰的E.Coli菌株,其中所述遗传修饰的E.Coli菌株还包括失活或缺失的抗生素抗性基因;3.根据具体实施方式1的遗传修饰的E.Coli菌株,其中产酸克雷伯氏菌casAB基因和菊欧文氏菌celY基因插入后在所述遗传修饰的E.Coli菌株中失活或缺失;4.根据具体实施方式2的遗传修饰的E.Coli菌株,其中产酸克雷伯氏菌casAB基因和菊欧文氏菌celY基因插入后在所述遗传修饰的E.Coli菌株中失活或缺失;5.根据具体实施方式2的遗传修饰的E.Coli菌株,其中所述遗传修饰的E.Coli菌株是代谢演化的;6.根据具体实施方式1或具体实施方式3的遗传修饰的E.Coli菌株,其中所述遗传修饰的E.Coli菌株是代谢演化的;7.根据具体实施方式4的遗传修饰的E.Coli菌株,其中所述遗传修饰的E.Coli菌株是代谢演化的;8.根据具体实施方式1、2、3、4、5、6或7的遗传修饰的E.Coli菌株,其中所述抗生素基因利用FLP重组酶进行缺失;9.根据具体实施方式7的遗传修饰的E.Coli菌株,其中所述遗传修饰的E.Coli菌株是SZ186;10.根据具体实施方式5的遗传修饰的E.Coli菌株,其中所述遗传修饰的E.Coli菌株是SZ132;11.根据具体实施方式1、2、3、4或5的遗传修饰的E.Coli菌株,其中在所述遗传修饰的E.Coli菌株中失活或缺失所述菌株的mgsA基因;12.一种遗传修饰的E.Coli菌株,包括已将mgsA基因失活或缺失的E.Coli菌株SZ194(NRRLB30863);13.根据具体实施方式12的遗传修饰E.Coli菌株,其中所述遗传修饰的E.Coli菌株是代谢演化的;14.根据具体实施方式13的遗传修饰E.Coli菌株,其中所述遗传修饰的E.Coli菌株是TG112、TG113或TG114;15.根据具体实施方式11或12的遗传修饰E.Coli菌株,其中所述遗传修饰的E.Coli菌株还包括失活或缺失的天然ldhA基因和重组插入的编码L-特异性乳酸脱氢酶的异源基因;16.根据具体实施方式15的遗传修饰的E.Coli菌株,其中所述异源L-特异性乳酸脱氢酶基因是ldhL基因(例如,从乳酸片球菌获得的ldhL基因);17.根据具体实施方式15或具体实施方式16的遗传修饰的E.Coli菌株,其中所述菌株是代谢演化的;18.一种遗传修饰的E.Coli菌株,选自TG112、TG113或TG114、SZ194、SZ132、SZ186或TG103;19.一种培养或培育遗传修饰的E.Coli菌株的方法,包括将一种或多种根据具体实施方式1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17或18中任一个的遗传修饰的E.Coli菌株接种培养基,以及培养或培育所述遗传修饰的E.Coli菌株;20.一种生产D-(-)-乳酸盐或D-(-)-乳酸的方法,包括在允许D-(-)-乳酸生产的条件下培养一种或多种根据具体实施方式1、2、3、4、5、6、7、8、9、10、11、12、13或14中任一个的遗传修饰的E.Coli菌株,以及可选地中和D-(-)-乳酸以形成D-(-)-乳酸盐;21.根据具体实施方式20的方法,其中所述一种或多种遗传修饰的E.Coli菌株选自TG112、TG113、TG114或SZ194;22.一种生产L-(+)-乳酸盐或L-(+)-乳酸的方法,包括在允许L-(+)-乳酸生产的条件下培养一种或多种根据具体实施方式15、16或17中任一个的遗传修饰的E.Coli菌株,以及可选地中和L-(+)-乳酸以形成L-(+)-乳酸盐;23.根据具体实施方式22的方法,其中所述一种或多种遗传修饰的E.Coli菌株是TG103、TG105、TG106、TG107或TG108;24.根据具体实施方式19、20、21、22或23的任一个的方法,其中所述遗传修饰的E.Coli菌株是在无机盐培养基中进行培养的;25.根据具体实施方式24的方法,其中无机盐培养基包括2%到20%(w/v)的糖;26.根据具体实施方式25的方法,其中无机盐培养基含有2%、2.5%、3%、3.5%、4%、4.5%、5%、5.5%、6%、6.5%、7%、7.5%、8%、8.5%、9%、9.5%、10%、10.5%、11%、11.5%、12%、12.5%、13%、13.5%、14%、14.5%、15%、15.5%、16%、16.5%、17%、17.5%、18%、18.5%、19%、19.5%或20%(w/v)的糖;27.根据具体实施方式25或26的方法,其中该糖是葡萄糖或蔗糖或葡萄糖和蔗糖的组合;28.根据具体实施方式20的方法,其中如具体实施方式11所述的遗传修饰的E.Coli菌株是在允许生产手性纯D-(-)-乳酸盐或D-(-)-乳酸的条件下进行培养的;29.根据具体实施方式22的方法,其中如具体实施方式15、16或17所述的遗传修饰的E.Coli菌株是在允许生产手性纯L-(+)-乳酸盐或L-(+)-乳酸的条件下进行培养的;30.根据具体实施方式21的方法,其中所述方法生产手性纯D-(-)-乳酸盐或D-(-)-乳酸;31.根据具体实施方式23的方法,其中所述方法生产手性纯L-(+)-乳酸盐或L-(+)-乳酸;32.根据具体实施方式28、29、30或31的方法,进一步包括纯化手性纯乳酸盐(L-(+)-乳酸盐或D-(-)-乳酸盐)的步骤;33.根据具体实施方式20、21、22、23、24、25、26、27、28、29、30、31或32中任一个的方法,其中生产的乳酸盐(例如,L-(+)-乳酸盐或D-(-)-乳酸盐)的浓度为至少0.5M;34.根据具体实施方式33的方法,其中培养基是化学成分确知(人工合成)的无机盐培养基如NBS无机盐培养基;35.根据具体实施方式20、21、22、23、24、25、26、27、28、29、30、31、32、33或34的方法,其中乳酸(L-(+)-乳酸或D-(-)-乳酸)的产率为至少或高于(或者高于或等于)90%;36.根据具体实施方式35的方法,其中该产率为至少90%、90.5%、91%、91.5%、92%、92.5%、93%、93.5%、94%、94.5%、95%、95.5%、96%、96.5%、97%、97.5%、98%、98.5%、或99%;37.根据具体实施方式29、30、31、32、33、34、35或36的方法,其中没有可检测的乳酸或乳酸盐的一种立体异构形式与另一种立体异构形式的污染(例如,特异性的立体异构体的手性纯度为至少、高于(或者高于或等于)99.9%);或38.一种组合物,包括一种或多种根据具体实施方式1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17或18中任一个的遗传修饰的E.Coli菌株以及培养基。以下是阐述用于实施本发明的方法的实施例。这些实施例不应解释为限制性的。除非另有说明,所有百分数均以重量计并且所有的溶剂混合物的比例均以体积计。实施例1E.Coli菌株SZ132和SZ186的制备与分析本实施例中使用的E.Coli菌株列在表1中。菌株SZ63是E.ColiK-12的衍生物(ATCC27325)。KO11(ATCC55124)是E.ColiB(ATCC11303)的衍生物,其含有整合到pflB基因中的运动发酵单胞菌乙醇生产基因。培养物在改性Luria-Bertani(LB)培养液(每升:10gDifco胰胨;5gDifco酵母提取物;5g氯化钠)或者在补充了5%或10%的糖(葡萄糖或蔗糖)的NBS无机盐培养基中、于37℃进行培养。菌株构建时需包括抗生素。质粒构建采用标准方法。染色体缺失、整合以及可去除抗生素抗性基因的方法之前已描述过了。菌株E.ColiDH5a和S17-1用作用于质粒构建的寄主。菌株NC3用作中间寄主以将在从K-12菌株到B菌株的P1转导过程中与限制性内切酶有关的问题减到最少(Dienetal.,2001)。种子培养物如之前由Shuklaetal.(2004)和Zhouetal.,(2003)描述过的方法制备,并用来接种小发酵容器(350mL工作体积,35℃,150rpm搅拌)。通过自动加入6N的KOH将培养液保持在pH7.0。标绘的数据代表2次或更多次重复的平均值。对于3次或更多次发酵的平均值还将表示平均值标准差的长条包括在内。乳酸盐的最大体积生产率从每个图表的最陡区进行估计。在种子培养物的生长过程中或发酵液中不包括抗生素。通过在550nm测量光密度(在1.0OD550nm处,330g干细胞wt/l)估计细胞量(cellmass)。总有机酸产量(主要是乳酸盐)通过碱(KOH)消耗进行测量。酸性产物在发酵结束时通过高性能液相色谱进行分析。乙醇通过气相色谱检测。菌株SZ132的构建菌株LY52由KO11通过将用于纤维二糖利用的产酸克雷伯氏菌casAB基因(Moniruzzamanetal.1997)整合到lacY的终止密码子之后以及将编码内切葡聚糖酶的菊欧文氏菌celY基因(ZhouandIngram,1999)整合到frdA基因中进行构建。将之前描述的用于构建用于乳酸盐生产的SZ63(Zhouetal.,2003)的E.ColiK-12(菌株W3110)中的突变连续地转导入LY52中。通过从SZ31的ΔfocA-pflB的缺失的P1转导消除了用于乙醇生产的运动发酵单胞菌基因。天然E.Coli醇脱氢酶通过从TC20的adhE突变的P1转导失活。利用SZ61中的突变去除乙酸激酶(ackA)。用于选择的抗生素基因通过FLP重组酶去除(Zhouetal.,2003)。用FLP重组酶去除抗生素基因的过程中,也缺失了casAB的内部片段。casAB和启动子的序列分析表明存在与FLP重组酶识别位点非常相似的DNA区,推测可能是造成这种缺失的原因。通过用1%的接种物连续进行传代培养而使所得菌株适应基本培养基。在这些转移过程中,细胞生长和乳酸盐生产得到了改善。来自最后一次转移的培养液在固体培养基上划线用于克隆的分离。一个克隆被选出并命名为SZ132。菌株SZ186的构建构建SZ132的进一步衍生物,其中去除所有外源基因。利用线性DNA通过同源重组去除产酸克雷伯氏菌casAB和菊欧文氏菌celY基因。使用FLP重组酶来消除在构建过程中使用的抗生素基因。所得菌株SZ186只含有天然E.Coli基因。乳酸生产的分析在E.ColiK-12和E.ColiB的衍生物中构建类似遗传突变,分别产生SZ63(Zhouetal.,2003)和SZ132。在细胞生长和乳酸盐(D异构体)生产方面,SZ132优于SZ63(参见图1A)。在丰富培养基中,SZ132在48h内完成10%葡萄糖的发酵,产生超过1mol乳酸盐/l发酵液,为以前对基于E.ColiK-12的菌株报道的两倍。用SZ63和10%葡萄糖的发酵在72h后停止,在120h之后只产生840mmol乳酸盐/l发酵液。在丰富培养基中,SZ132d的乳酸盐产率(95%)高于K-12菌株SZ63的乳酸盐产率(88%)。SZ132的最大体积生产率为75mmol/lh,比SZ63高76%(表2)。在含有5%葡萄糖的NBS无机盐培养基中,SZ132的细胞产率和体积生产率为SZ63的两倍(参见图1B)。菌株SZ132在36h内完成5%葡萄糖的发酵,少于SZ63所需的时间的1/4。在这些条件下,用NBS无机盐培养基发酵的细胞产率和体积生产率仅为用丰富培养基及10%葡萄糖的25-35%。在NBS无机盐培养基中,菌株SZ132和SZ63在144h内均不能完成10%葡萄糖的发酵(参见图1C)。基于代谢的糖,两种菌株的乳酸盐产率都超过90%。利用含有10%葡萄糖的NBS无机盐培养基,SZ132的细胞产率(0.83g/l)、乳酸盐生产(700mmol/l的发酵液)、以及体积生产率(11.2mmol乳酸盐/lh)比SZ63大约高两倍。利用NBS无机盐培养基和10%葡萄糖,SZ132的体积生产率低于在丰富培养基中观察到的体积生产率的20%。在丰富培养基和NBS无机盐培养基中,蔗糖发酵比葡萄糖慢。以前的研究表明,在丰富培养基中SZ63(pLOI3501)需要36h来完成5%蔗糖的发酵,但在这种培养基中不能完成10%蔗糖的发酵。在丰富培养基中SZ132在120h内完成10%蔗糖的发酵并产生超过1mol乳酸盐/l发酵液(参见图1D),几乎是以前报道的含有(harboring)蔗糖质粒的SZ63的两倍。SZ132在含有10%蔗糖(9.3mmol/l)的NBS无机盐培养基中的体积生产率为在含有蔗糖的丰富培养基中观察到的体积生产率的1/3,低于在含有10%葡萄糖的丰富培养基中观察到的体积生产率的1/8(表2)。尽管在72h内、在NBS无机盐培养基中将5%蔗糖发酵完全(参见图1E),但是在这种培养基中即使利延长孵育时间也不能充分代谢10%蔗糖。基于代谢的糖,在两种培养基中蔗糖的产率在从86%到93%的范围内变动。这些结果表明,高水平的有机酸可通过E.ColiB的改性菌株从葡萄糖和(或)蔗糖进行生产。估计丰富培养基(LB)中的最大体积生产率为75mmol乳酸盐/lh,比NBS无机盐培养基高3倍以上。存在许多用于改进的机会。在复合培养基中,完成10%蔗糖代谢所需的孵育时间比10%葡萄糖长3倍,在无机盐培养基中则更长。有必要在不增加昂贵的复合营养成分的情况缩短发酵时间。确定增加生长和生产率的组分可显著减少发酵相关的费用,而且还提供减少乳酸盐纯化和废物处理相关的费用的机会。图2-5图解阐述了在NBS无机盐培养基中,通过本发明的各种具体实施方式,即SZ132、SZ186以及SZ194(保藏号NRRLB-30863),于NBS无机盐培养基中在各种发酵条件下进行乳酸(D异构体)生产的进程。实施例2E.Coli菌株SZ194的制备菌株SZ194的制备中使用的菌株和质粒列在表3中。将培养物在37℃、改性Luria培养液(每升:10gDifco胰胨,5gDifco酵母提取物,5g氯化钠)(Miller,J.H.1992AShortCourseinBacterialGenetics:ALaboratoryManualandHandbookforEscherichiacoliandRelatedBacteria.ColdSpringHarborPress,ColdSpringHarbor,N.Y.)或补充了1mM甜菜碱和2%-14%(w/v)糖(葡萄糖或蔗糖)的NBS无机盐培养基(Causey,T.B.etal.,2004,“EngineeringEscherichiacoliforefficiientconversionofglucosetopyruvate,”Proc.Natl.Acad.Sci.USA101:2235-2240)中进行培育。如果需要,氨苄青霉素(50mg/L)和卡那霉素(50mg/L)也用于菌株构建。采用标准方法进行DNA扩增、酶消化、纯化以及质粒构建(Milleretal.,1992;SambrookandRussell,2001)。染色体基因缺失和整合的方法之前已经描述过(Causeyetal.,2004;DatsenkoandWanner,2000;Martinez-Moralesetal.,1999;Zhouetal.,2003a)。构建质粒pLOI3924以有利于casAB基因的缺失,可通过利用ORFmers(Sigma-Genosis,TheWoodlands,TX)克隆lacY和lacA基因以及在lacY的羧基端的NdeI位点和lacAN终端的ATG之间插入来自pLOI2511的FRT-kan-FRT盒(SmaI消化)而实现。来自这个质粒的扩增片段(对于lacY正向(forward)ORFmer;对于lacA反向ORFmer)用于染色体整合,去除casAB基因。在表4中描述了用于其他基因缺失的杂种引物(Hybridprimer)及包含的序列,这些序列对应于靶标基因的开始或结束的约45个碱基对以及对应于FRT侧面的卡那霉素盒的DNA序列的20个碱基对。除非另有说明,所有发酵(pH7.0)中均使用含有1mM甜菜碱和10%糖(葡萄糖或蔗糖)的NBS无机盐培养基(Causeyetal.,2004)。种子培养物如前所述(Zhouetal.,2003)进行制备并用于接种500ml发酵容器(350mL工作体积,35℃,150rpm搅拌)至33mgcdwl-1的初始密度。如有指明,再加入甜菜碱(1mM)。通过自动加入6NKOH而保持培养液的pH。在补料分批发酵(6%+3%+3%)中使用的总的12%(w/v)葡萄糖如下:308ml的NBS培养基含有21g的葡萄糖。在24h和48h之后,将21ml50%的葡萄糖缓慢加入到每个容器中。将来自有pH调控的发酵的细胞在不同时间(24或48h)进行连续转移,以利于通过基于生长的选择的代谢演化。将连续转移的培养物以33mgcdwl-1的初始密度进行接种。选择结束时分离的克隆则赋予新的菌株名称。利用Bausch&LombSpectronic70分光光度计在550nm处检测光密度而估计细胞量。通过用于pH维持的碱(KOH)消耗估计总的有机酸产量(主要是乳酸盐)。通过高性能液相色谱在发酵结束时分析酸性产物和手性纯度(Zhouetal.,2003a)。标绘的数据表示2次或更多次重复的平均值。对于3次或更多次发酵的平均值还将表示标准差的长条包括在内。乳酸生产的分析甜菜碱(一种保护性调渗剂(osmolyte))对由SZ132在含有高糖浓度的无机盐培养基中的细胞生长和乳酸盐生产非常有益,可能是由于参与(partitioninginto)诸如谷氨酸盐和海藻糖等天然调渗剂的生物合成的碳不足(Purvisetal.,2005)。SZ132的连续转移在含有10%蔗糖的无机盐培养基中进行,以选择在缺乏甜菜碱的情况下具有等价性能的菌株(图6A和6B)。用于菌株SZ132生长的能量产生依赖于利用乳酸盐作为NADH氧化主要途径的糖酵解代谢流(flux),提供基于生长的选择。在连续转移的过程中,生长和酸产量(碱消耗)稳定提高。在最后的转移中,不添加甜菜碱即可使10%蔗糖发酵完全。从这种培养物中分离出一种克隆并命名为菌株SZ136。这个菌株在96小时后产生母体SZ132两倍的细胞产率和3倍的更高效价的乳酸盐(图6)。然而,SZ136还产生比SZ132更高浓度的琥珀酸盐和乙醇(表5),降低了乳酸盐产率(基于代谢的糖)。a含有10%葡萄糖的NBS无机盐培养基(pH7.0),除非另有说明。如有指明,则再加入1mM甜菜碱。b产率是基于代谢的糖,假定每摩尔己糖有2摩尔乳酸盐的最大理论产率(等重量转化)。对SZ132在10%(w/v)蔗糖上的生长改善看来伴随一些突变,这些突变能够改善生长,但由于副产物途径也部分恢复了突变基因的功能。菌株SZ132中的延胡索酸盐还原酶的突变表征得较差,最初作为一种Tn5缺失而获得(Ohtaetal.,1991),随后在frdA和frdB之间插入celY(Zhouetal.,2005)。通过插入带侧翼FRT位点的抗生素标记而破坏醇脱氢酶基因。然后使用翻转酶(flippase)去除抗生素基因,只留下FRT区。进一步缺失产生SZ162的frdBC和adhE的编码区,消除了两种副产物,而且由于糖利用不完全降低了生长和最终乳酸盐效价(表5)。在这个菌株中还进行其他的缺失以消除之前整合的用于纤维素利用的外源基因(Zhouetal.,2005):来自产酸克雷伯氏菌的casAB(纤维二糖转运蛋白基因)和来自菊欧文氏菌的celY(内切葡聚糖酶)。在有或没有甜菜碱的无机盐培养基中,所得菌株SZ186产生可忽略浓度的副产物,但不能完全利用10%的糖(图7A;表5)。然而,SZ162和SZ186基于代谢糖的产率都很高(96%-99%)。SZ136中能够消除副产物途径(SZ186)的缺失也将细胞产率降低了几乎一半,对碳参与生物合成造成了不良影响。使用代谢演化来共选择(co-select)在1mM甜菜碱和10%葡萄糖存在时生长和发酵性能改善的SZ186衍生物(图7A和7B)。细胞产率和有机酸生产在最初几轮选择的过程中同时增加。从最后富集中分离出单个克隆体并命名为SZ194。菌株SZ194比SZ186生长更快并且达到更高的细胞产率。由SZ194在部分发酵中生产的乳酸盐浓度高达900mM,始终高于SZ186的乳酸盐浓度(表5)。两种菌株从葡萄糖生产乳酸的产率都接近理论产率并且副产物最少。但是,利用两种菌株,pH7.0时,即使在延长孵育时间糖仍不被利用。pH7.0时,由10%(w/v)葡萄糖生产的800-900mM乳酸盐大大超过了pH7.0时抑制生长所需的最大乳酸盐浓度并且还可能会抑制进一步的葡萄糖代谢。人们探索了可能引起SZ194对10%(w/v)糖不完全发酵的两个因素:未知的营养缺陷和乳酸盐耐受性。用pH7.0的Luria发酵液(丰富培养基)代替含有1mM甜菜碱的无机盐培养基对乳酸盐产率(表5)或乳酸盐生产(表6)几乎没有影响。乳酸盐毒性通过比较pH对发酵的影响进行检测(表5)。弱有机酸如乳酸盐的毒性部分与共轭中性形式的解偶联作用有关(Warneckeetal.,2005),解偶联作用又与pH反相关。发酵也观察到了相似的趋势。在Luria液体培养基中,最终乳酸盐效价在pH6.5时最低而在pH7.5和pH8.0时最高。pH的变化对细胞产率影响较小。在pH7.5时,利用Luria液体培养基在72h内将10%(w/v)的葡萄糖发酵完全。a为含有10%葡萄糖和1mM甜菜碱的NBS无机盐(培养基)(pH7.0),除非另有说明。b生产率最高的24h周期的平均值pH增加的好处在含有1mM甜菜碱的无机盐培养基中得到确认(图8;表5)。在pH7.5时,可将10%和12%(w/v)的葡萄糖发酵完全,产生超过1mol乳酸盐L-1并且只有痕量副产物(图3A)。基于加入发酵液中的总糖量,每g葡萄糖产生0.95g乳酸盐产物的产率代表95%的最大理论产率。加入更高浓度的糖不会进一步增加乳酸盐的最终效价。在补料分批实验过程中观察到相似的最终效价(图8B)且再加入糖时不会进一步增加。接近1.0-1.2M的最终乳酸盐效价似乎代表pH7.5时通过SZ194进行的糖代谢的上限(表5)。人们注意到pH7.5(发酵的最佳pH)非常接近报道的E.Coli细胞质的pH(Axeetal.,1995;Warneckeetal.,2005),这一事实非常有趣。尽管在E.Coli中还没有确定有乳酸盐转运蛋白基因,但是若干研究已提供了它们存在于E.Coli和乳酸菌中的证据(Axeetal.,1995;Konings,2002;Poolman,2002)。推测这些转运蛋白是乳酸盐/H+同向转运蛋白,可能会增加效率的活性伴随外部pH的增加。根据“能量循环模式”(Michelsetal.,1979),代谢终产物如乳酸盐的载体媒介流出物可导致跨膜的电化学质子梯度产生并有助于ATP生成。估计乳酸菌中的乳酸盐流出物贡献30%的总细胞能量(vanMarisetal.,2004a)。由于E.ColiSZ194和乳酸菌以相似途径代谢葡萄糖,所以E.Coli有可能通过E.Coli乳酸盐流出物来产生额外的ATP。对由SZ194生产的乳酸盐的手性纯度进行检测,发现比95%的D-乳酸盐更高。尽管较好,但是这种手性纯度低于母体菌株SZ132(99.5%的D-乳酸盐)(Zhouetal.,2005)。这种手征不纯性的来源还不清楚。根据本主题发明,超过1M的乳酸盐效价可通过pH7.5时E.ColiSZ194在补充了1mM甜菜碱的无机盐培养基中进行生产。这种培养基中SZ194的乳酸生产率和细胞产率相当于Luria培养基(表6)。菌株SZ194的110gL-1的最终乳酸盐效价和产率(0.95g乳酸盐g葡萄糖-1)不亚于诸如瑞士乳杆菌(L.helveticus)(Kyla-Nikkilaetal.,2000)和保加利亚乳杆菌(L.delbrueckii)(Demircietal.,1992)的乳酸菌,并且在丰富培养基和无机盐培养基中超过以前改造的生物催化剂的性能(Dienetal.,2001;Porroetal.,1999;Saitohetal.,2005;vanMarisetal.,2004b)。实施例3材料与方法菌株、质粒、培养基以及生长条件本研究中利用的E.Coli菌株、质粒和引物列在表7中。菌株SZ194从E.ColiB(ATCC11303)的衍生物预先构建并作为用于构建的起始点(参见实施例2和Zhouetal.,2006)。在菌株构建过程中,有氧条件下(aerobically)培养物在30°C、37°C、或39°C含有2%(w/v)葡萄糖或阿拉伯糖的Luria培养液(每升:10gDifco胰胨,5gDifco酵母提取物以及5g氯化钠)(Miller1992)中培养。需要时加入氨苄青霉素(50mg/L)、四环素(12.5或6.25mg/L)或卡那霉素(25或50mg/L)。为了进行发酵测试,37℃没有抗生素的条件下、将菌株在补充了1mM甜菜碱和2-12%(w/v)葡萄糖的NBS无机盐培养基(Causeyetal.,2004)中进行培养。在缺少pH控制(平皿、试管、摇瓶)的条件下,将MOPS缓冲液(100mM,pH7.4)加到固体和液体培养基中。遗传学方法DNA纯化(Qiagen)、限制性内切核酸酶消化(NewEnglandBiolabs)、DNA扩增(StratageneandInvitrogen)以及转化,依照厂商方案和标准方法(Miller1992,SambrookandRussell2001)。染色体缺失和整合的方法之前已经描述过(Causeyetal.,2004,Zhouetal.,2003,DatsenkoandWanner2000,Martinez-Moralesetal.,1999)。将含有与mgsA基因(斜体)的开始或结束的50个核苷酸以及对应于pKD4的FRT侧面的卡那霉素盒(加下划线的)的序列的20个核苷酸同源的序列的杂交引物(表7)用于天然mgsA基因的缺失。前面构建了质粒pLOI2398,以便于将乳酸片球菌(Pediococcusacidilactici)ldhL整合到E.Coli的染色体ldhA基因中(Zhouetal.,2003a)。发酵预接种物通过将菌落接种到250ml烧瓶(100ml含有2%(w/v)葡萄糖和100mMMOPS的NBS培养基,pH7.4)中进行培养。16h(37℃,120rpm)后,将这种预接种物稀释到含有350mlNBS培养液(含5-12%糖,含或不含1mM甜菜碱)的500ml发酵容器中,以提供33mgdcwL-1。24h(37℃,150rpm,控制pH在7.0)之后,利用这种培养物来提供33mgdcwl-1的初始接种。活性最大的24h期间的体积生产率给予报道。单位生产率的计算是用体积生产率除以24h时的细胞量所得的商数。代谢演化将来自pH控制发酵的细胞以24或48h间隔进行连续转移,以有利于通过竞争性,基于生长的选择的代谢演化。每次转移时,将接种物稀释(1/100到1/350)入预热的新鲜培养基。从这些选择分离出的克隆则赋予新的菌株名称。分析细胞量利用Bausch&LombSpectronic70分光光度计在550nm测量光密度而估计。总有机酸产量(主要是乳酸盐)通过用于保持pH7.0的KOH消耗进行估计。通过高性能液相色谱在发酵结束时分析酸性产物和手性纯度。通过碱消耗估计的有机酸始终低于通过HPLC测量的乳酸盐,可能是由于矿物的代谢。结果与讨论恢复D-㈠-乳酸盐生产的手性纯度结果表明菌株SZ194在无机盐培养基中可以99%的手性纯度从葡萄糖高效产生D-(-)-乳酸盐(Zhouetal.,2006)。高糖浓度下,甜菜碱能够增加乳酸盐生产率(效价和速率)但也将手性纯度降低到95%(表8)。E.Coli中L-(+)-乳酸盐有若干可能的来源(图9A)。L-(+)-乳酸盐可从乳醛(lactaldehyde)产生,乳醛是鼠李糖(rhamnose)或岩藻糖分解代谢过程的中间体(Badiaetal.,1991),而这两种糖均不存在于我们的培养基中,并且作为来自糖酵解的丙酮醛支路的产物(图9A和9B)。丙酮醛支路表示一个短溢出途径(spilloverpathway),由快速糖酵解过程中的二羟基丙酮-磷酸酯(盐)堆积以及ATP合成时的磷酸酯(盐)限制引起。二羟基丙酮-磷酸酯堆积和磷酸酯限制均可因高速的糖酵解代谢流(flux)而加剧。为了检验这种假设,将编码第一个承诺步骤(committedstep)(丙酮醛合成酶)的mgsA基因缺失。所得的菌株TG112产生手性纯的D-(-)-乳酸盐(表8和9;图10A和10B)。与SZ194相比,这种缺失提高了了生长和初始生产率。基于HPLC分析的产率类似(表8)。对TG112的进一步改进通过81天生长过程中的代谢演化进行选择(图11)。发酵培养物在含有10%(w/v)葡萄糖、12%(w/v)葡萄糖、以及12%(w/v)葡萄糖和1mM甜菜碱的无机盐培养基中连续转移。在第28天分离到一个克隆(TG113)并且在富集结束时分离到另一个克隆(TG114)。D-(-)-乳酸盐生产率(速度、效价以及产率)和细胞产率在这个过程中得到改进(图10A和10B;表8和表9)。利用SZ114的发酵在大多数实验中48h内基本上完成,终产率为0.98gD-(-)-乳酸盐g-1葡萄糖。高接种物为单位生产率和体积生产率提供了适度的进一步改进。利用这些缺失mgsA的菌株的副产物和手性杂质低于检测水平(<0.1%)。通过TG114生产D-(-)-乳酸盐不亚于其他生物催化剂,并且可以利用简单的培养基和发酵条件提供较高生产率、效价、产率和手性纯度(表10)。手性纯L-㈩-乳酸盐的生产为了主要生产L-(+)-乳酸盐,用来自乳酸片球菌的编码L-(+)-乳酸脱氢酶的ldhL基因替换编码D-(-)-乳酸脱氢酶的天然ldhA基因,重新改造D-(-)-乳酸盐菌株SZ194(Zhouetal.,2003a)。所得菌株TG102主要生产L-(+)-乳酸盐,并在含有5%(w/v)葡萄糖、10%(w/v)葡萄糖以及含有1mM甜菜碱的10%(w/v)葡萄糖的基本培养基中进行连续转移,以选择生长和生产率改善的菌株。24天之后,选出一个克隆并命名为TG103。尽管由于糖酵解代谢流增加而改善了乳酸盐生产率(图10C和10D;表9),但是L-(+)-乳酸盐被5%的D-(-)-乳酸盐污染(表8)。由于丙酮醛支路可以生产两种手性形式的乳酸盐,所以mgsA被确定为手性杂质的最可能的来源。缺失mgsA之后就恢复了绝对手性纯度。所得菌株TG105只生产L-(+)-乳酸盐(图10和10D;表8)。进一步的改进则通过在含有10%-12%(w/v)葡萄糖和1mM甜菜碱的无机盐培养基的连续转移(接种稀释度为1∶100到1:350)过程中共选择(co-select)生长和生产率而实现。在10次和22次转移后分别分离TG106和TG107作为中间菌株(图10E和10F)。在99天之后分离出菌株TG108。所有菌株只以高产率生产L-(+)-乳酸盐而副产物达到最低水平(表8)。虽然单位生产率基本保持不变,但在菌株选择过程中12%(w/v)葡萄糖中的体积生产率和细胞产率均增加(表9)。利用较高接种物时,可以观察到最大体积生产率(活性最大的24h期间)稍有增加。菌株TG108用于L-(+)-乳酸盐生产可以比拟其他生物催化剂(表10)。这种菌株的效价和产率高于其他微生物报道的效价和产率。其他的优点包括简单的发酵条件和培养基以及手性纯度。结论高效价(48h内>100gl-1)手性纯的L-(+)和D-(-)-乳酸盐(>99.9%的手性纯度)可通过重组E.ColiB在补充了1mM甜菜碱的无机盐培养基中容易地进行生产。消除丙酮醛支路对于去除乳酸盐的D-(+)及L-(-)对映异构体中的杂质是必要的。实施例4通过去除来自TG114的所有外源DNA构建TG128为了促进用于乳酸盐生产的重组E.Coli菌株的商业应用,进一步构建了TG114的衍生物,其中去除了前面基因改造过程中在染色体中留下的所有外源DNA。去除的DNA包括含有用来去除抗生素标记的翻转酶(flippase)(重组酶)的FRT识别位点的scar区、来自运动发酵单胞菌的DNA小片段、产酸克雷伯氏菌casAB的部分、以及菊欧文氏菌celY的部分。这些外源DNA片段被彻底去除,只留下天然染色体DNA,其中所选基因的中心区域已被去除用以提高乳酸盐的生产。菌株、质粒、培养基以及培养条件本研究中使用的E.Coli菌株、质粒和引物列于表11中。菌株TG114之前从E.ColiB(ATCC11303)的衍生物构建并用作构建的起始点(Grabaretal.,2006)。菌株构建过程中,有氧条件下培养物在含有2%(w/v)葡萄糖和阿拉伯糖或10%蔗糖的Luria培养液(每升:10gDifco胰胨,5gDifco酵母提取物以及5g氯化钠)(Miller1992)中、于30℃、37℃、或39℃进行培养。如果需要,则加入氨苄青霉素(50mg/L)、金霉素(10mg/L)或氯霉素(40mg/L)。将培养物保持在含有补充了2%(w/v)葡萄糖的NBS无机盐培养基(Causeyetal.,2004)的平皿上。在缺少pH控制的条件(平皿、试管、摇瓶(shakenflasks))下将MOPS缓冲液(100mM,pH7.4)加入固体和液体培养基。为了进行发酵试验,将菌株在37℃没有抗生素的情况下在AM1无机盐培养基(每升:2.63g(NH4)2HPO4,0.87gNH4H2PO4,0.37gMgSO4·7H2O,2.4mgFeCl3,0.3mgCoCl2·6H2O,0.15mgCuCl2,0.3mgZnCl2·4H2O,0.3mgNaMoO4,0.075mgH3BO3,0.075mgMnCl2·4H2O2,1mM甜菜碱以及120g/L葡萄糖)中进行培育。遗传学方法基因克隆(Invitrogen)、DNA纯化(Qiagen)、限制性内切核酸酶消化(NewEnglandBiolabs)、DNA扩增(StratageneandInvitrogen)以及转化,依照厂商方案和标准方法。用于染色体缺失和整合的方法之前已经描述过(Causeyetal.,2004,Zhouetal.,2003,DatsenkoandWanner2000,Martinez-Moralesetal.,1999)。所使用质粒和引物的说明列于表11中。质粒构建通过设计在E.ColiBgDNA的frdABCD操纵子‘frdAfrdBfrdC和frdD’内部扩增的引物、随后连接到Invitrogen克隆载体pCR2.1-TOPO中而产生质粒pLOI4411(表11)。质粒pLOI4412、pLOI4413、pLOI4415以及pLOI4416用类似的方法进行构建,以便分别克隆感兴趣的天然基因ackA、adhE、focA-plfB以及mgsA。利用反义引物产生编码无缝基因敲除(knockout)的质粒。设计具有5’磷酸酯基团的反义引物以扩增除要去除的基因区以外的整个质粒(图12)。例如,pLOI4417通过在pLOI4411上进行PCR而生成,一引物在frdA的3’端退火(向上游延伸)、另一引物在frdC内退火(向下游延伸)。所得4263bpPCR产物用限制性内切酶DpnI进行处理,以消化天然pLOI4411质粒。消化的PCR产物随后进行自身连接,以产生新的质粒pLOI4417。利用列于表11中的引物组,以类似的方式产生质粒pLOI4418、pLOI4419、pLOI4421以及pLOI4422。这些质粒作为PCR模板,用来扩增含有预期缺失并缺少所有异源序列的线性DNA。将这种线性DNA用来通过同源重组(双向跨接过程,doublecross-overevent)替换FRTscar区、异源DNA、以及sacB。用这种方式就在染色体上形成了所选基因的无缝去除。从TG114去除FRTScar和异源DNA由DatsenkoandWanner(2000)的一步去除法留下的~85bpFLP识别靶标(FRT)scar可通过两步法去除。首先,FRT位点单独地通过环形FRT-cat-sacB盒进行靶向,导致这种盒的单跨接整合(singlecross-overintergration)到染色体上的FRTscar(FRT-cat-sacB-FRT)。含有cat-sacB-FRT盒的环形DNA构建如下:1)利用质粒pLOI4151作为模板进行FRT-cat-sacB盒的PCR扩增。被扩增的盒包括侧翼的PstI位点;2)用PstI消化,随后通过自身连接以产生不能自动复制的闭合环。这种环形DNA用于整合。cat基因的表达赋予氯霉素抗性,并允许直接选择含有该整合的细胞。第二步中,sacB基因允许直接选择细胞,其中含有sacB的DNA区已通过同源重组(双向跨接过程)而去除。在有10%蔗糖的情况(Ausubeletal.,2005;Leeetal.,2001)下表达sacB的细胞通过多糖的胞内生产而被杀灭和溶解。利用这种方法,那些DNA已通过插入而替换sacB的重组体(recombinant)可从大的种群中进行筛选。在本实施例中,通过双向跨接过程的线性DNA片段的整合可得到完全敲除(cleanknockout)的菌株,只含有天然DNA或伴有缺失的天然DNA,不含任何异源DNA。第二步中使用天然DNA序列或含有预期基因缺失的天然序列,以彻底去除FRTscar区和来自TG114的所有异源DNA。线性DNA片段经电穿孔之后,将细胞在1mLSOC培养基中于37℃振摇孵育4小时。经过4小时的扩大培养(outgrown)后,将该细胞在50mL补充了10%(w/v)蔗糖的无盐的LB(每升:10gDifco胰胨,5gDifco酵母提取物)中孵育16-20小时。然后将细胞刮涂(struckonto)到补充了5%蔗糖的无盐的LB琼脂平板上。保留FRT-cat-sacB的多数细胞在有蔗糖的情况下延长周期之后不能存活。存活且形成菌落的细胞则缺少sacB和scar区。连续使用这种方法,将存在于TG114中的全部5个FRTscar消除并恢复lac操纵子。在这个过程中使用的引物、质粒以及菌株总结在表11中。利用两步法的修改将整合到lacA中的casAB基因去除并用这个区域中的野生型序列加以替换。整合到frdA中的菊欧文氏菌celY片段(和运动发酵单胞菌DNA)在构建无缝frdBC缺失体的过程中被去除。所得菌株TG128缺少FRTscar并且只含有天然DNA序列,这些序列含有在表11所示的基因中的缺失区。示出了对于TG114和TG128的基因缺失区中的初始序列的对比(表12)。天然lac操纵子的恢复TG114含有来自以前构建体的在lacYlacA区中的异源DNA片段(来自运动发酵单胞菌、产酸克雷伯氏菌casAB、菊欧文氏菌celY的部分的DNA片段)(Grabaretal.,2006)。从TG120去除这些而产生TG122,并且在TG122随后的衍生物中不存在。用于这种构建的引物列在表11中。为了实现这个目的,将来自E.ColiB从天然lacZ’到cynX的区域加以扩增并克隆到pCR2.1-TOPO中,以产生pLOI3956。为了有利于重组体的选择,如下将pLOI3956的中心区(lacZ’-lacA’)用含有cat和sacB基因的盒加以替换。以pLOI3956作为模板,使用含有NheI位点的反义引物来扩增lacZ’、pCR2.1以及cynX(省去lacZ’的部分、lacY、lacA和cynX’的部分)。使用第二组含有NheI位点的引物扩增来自pEL04(最初通称pK04,Ausubeletal.,2005andLeeetal.,2001)的cat-sacB盒。在利用NheI的消化之后,将这些PCR产物加以连接而产生pLOI3957,这是含有lacZ’-sacB-cat-cynX’的pCR2.1的衍生物。以质粒pLOI3957作为模板,将lacZ’-sacB-cat-cynX’经PCR扩增并整合到具有用于氯霉素抗性的选择性的TG120中,以产生TG121。利用pLOI3956作为模板,将野生型lacZ’-lacY-lacA-cynX’区经PCR扩增并整合到TG121中。通过在有蔗糖的生长过程中sacB功能缺乏而选择含有lacZYA操纵子的天然序列的整合体。将这些整合体中的一种命名为TG122。菌株TG122是TG128构建过程中的中间菌株。发酵预接种物通过将菌落接种到250mL烧瓶(盛有100mL含有2%(w/v)葡萄糖和100mMMOPS(pH7.4)的NBS培养基(Causeyetal.,2004)进行培养。16h(37℃,120rpm)后,将这种预接种物稀释到盛有350mLAF1无机盐培养基(12%糖)的500mL发酵容器中,以提供33mgdcw/L。24h(37℃,150rpm,pH控制在7.0)后得到的培养物用来提供33mgdcw/L的初始接种物,并在相同条件下孵育96h。移出样品用于分析。分析利用Bausch&LombSpectronic70分光光度计在550nm检测光密度而估计细胞量。总有机酸产量(主要是乳酸盐)通过用于保持pH7.0的KOH消耗进行估计。发酵结束时通过高性能液相色谱分析酸性产物和手性纯度。通过碱消耗估计的有机酸始终低于通过HPLC测量的乳酸盐,可能是由于矿物的代谢。利用TG128的葡萄糖发酵菌株TG114经过深入的(intensive)遗传学操作而去除FRTscar和其他外源DNA。所得微生物菌株TG128只含有天然的、野生型DNA序列和基因的无缝缺失,从而消除不需要的发酵产物。这种菌株的发酵性能基本上与TG114相当,最大理论产率的96%-98%的乳酸盐产率(表13)。通过代谢演化选择用于乳酸盐生产的耐热突变体利用代谢演化来选择能够在高温下进行有效乳酸盐生产的菌株。pH控制发酵的细胞以24h的间隔进行连续转移,以利于通过竞争性、基于生长选择的代谢演化。每次转移均将接种物稀释(1/100到1/350)到预热的新鲜培养基中。孵育温度在生长允许的情况下,每次增加一度的一小部分(afractionofadegree),以有利于分离出耐热菌株。菌株TG128的发酵优选在37°C。在连续转移的过程中,突变体通过孵育温度递增而获得。用这种方法,从TG128筛选出新的菌株,其能够在39℃(菌株TG129)和43℃(菌株TG130)进行有效发酵。这两种耐热菌株在高温下的产率相当于TG114在37℃的产率(表13)。利用TG128的葡萄糖发酵菌株TG114经过深入的遗传学操作而去除FRTscar和其他外源DNA。所得微生物菌株TG128只含有天然的、野生型DNA序列和基因的无缝缺失,从而消除不需要的发酵产物。这种菌株的发酵性能基本上与TG114相当,最大理论产率的96%-98%的乳酸盐产率(表13)。选择用于乳酸盐生产的耐热突变体利用代谢演化来选择能够在高温下进行有效乳酸盐生产的菌株。连续转移到发酵罐中(其中温度以一度的一小部分增加)允许TG129(其发酵优选在39℃)和TG130(其发酵优选在43℃)的分离。这两种在高温下的产率相当于TG114在37℃的产率(表13)。a除非另有说明,NBS无机盐培养基含有12%(w/v)葡萄糖(pH7.0)。当有指明时,还需加入1mM甜菜碱。粗体字菌株生产D-(-)-乳酸盐。下划线的菌株生产L-(+)-乳酸盐。b产率基于代谢的糖,假定每摩尔己糖有2摩尔乳酸盐的最大理论产率(等重量转化)。a无机盐(培养基)NBS含有12%(w/v)葡萄糖、1mM甜菜碱、控制pH在7.0(除非另有说明)。粗体字菌株生产D-(-)-乳酸盐。下划线的菌株生产L-(+)-乳酸盐。bpH控制在7.5。c5%接种物。d10%接种物。e10%葡萄糖。f产率最高的24h期间的平均值。a用粗体字示出的是基因的5’和3’区的部分序列,斜体是FRTscar以及下划线的是菌株TG128中缺失的区域。b这个区还含有来自运动发酵单胞菌启动子的部分序列以及来自菊欧文氏菌celY基因的部分序列(以下划线的正常体示出)。c破折号表示缺失的区域a含有12%(w/v)葡萄糖、1mM甜菜碱、控制pH在7.0的无机盐(培养基)(NBS)。b产率是基于代谢的糖,假定每摩尔己糖有2摩尔乳酸盐的最大理论产率(等重量转化)。将本文所参照或引用的所有专利、专利申请以及出版物的全部内容结合于此作为参考,包括所有图形和表格,到它们与本说明书显而易见的内容相一致的程度。应当理解本文所描述的实施例和具体实施方式只是用于说明的目的,并且对本领域技术人员来说,建议根据其作出各种更改和变化,且这些修改和变化都包括在本申请的精神和范围内。参考文献U.S.PatentNo.4,963,486U.S.PublishedPatentApplicationNo.20040005677U.S.PublishedPatentApplicationNo.20030003553U.S.PublishedPatentApplicationNo.20050112737U.S.PublishedPatentApplicationNo.20040029256WO99/14335Adachietal.(1998)“ModificationofmetabolicpathwaysofSaccharomycescerevisiaebytheexpressionoflactatedehydrogenaseanddeletionofpyruvatedecarboxylasegenesforthelacticacidfermentationatlowpHvalue”J.Ferment.Bioeng.86:284-289.Agrawal,A.K.(2003)“Advancesintheproductionofpoly(lacticacid)fibers.Areview.”J.Macromolec.Science-PolymerRev.,4:479-503.Arntzen,C.E.,Dale,B.E.(1999)Biobasedindustrialproducts,prioritiesforresearchandcommercialization,NationalAcademyPress,Washington,D.C.Ausubel,F.M.etal.(1995)CurrentProtocolsinMolecularBiology,JohnWiley&Sons,Inc.Ausubel,F.M.,R.Brent,R.E.Klingston,D.D.Moore,J.G.Deidman,J.A.SmithandK.Struhl(eds.)(2005)Currentprotocolsinmolecularbiology.JohnWiley&Sons,Inc.NewYork,N.Y.Axe,D.D.andJ.E.Bailey(1995)“TransportoflactateandacetatethroughtheenergizedcytoplasmicmembraneofEscherichiacoli”Biotechnol.Bioeng.47:8-19.Badia,J.,Gimenez,R.,Baldoma,L.,Barnes,E.,Fessner,W.D.,Aguilar,J.(1991)“L-lyxosemetabolismemploystheL-rhamlnosepathwayinmutant-cellsofEscherichiacoli-adaptedtogrowonxylose”J.Bacteriol.173:5144-5150.Bianchi,M.M.,Brambilla,L.,Protani,F.,Liu,C.,Lievense,Porro,D.(2001)“EfficienthomolacticfermentationbyKluveromyceslactisstrainsdefectiveinpyruvateutilizationandtransformedwithheterologousLDHgene”Appl.Environ.Microbiol.67:5621-5625.Causey,T.B.etal.(2004)“EngineeringEscherichiacoliforefficientconversionofglucosetopyruvate”Proc.Natl.Acad.Sci.USA101:2235-2240.Chang,D.E.etal.(1999)“HomofermentativeproductionofD-(-)orL-(+)lactateinmetabolicallyengineeredEcherichiacoliRR1”ApplEnvironMicrobiol65:1384-1389.Chotani,G.etal.(2000)“Thecommercialproductionofchemicalsusingpathwayengineering”Biochim.Biophys.Acta1543:434-455.Datsenko,K.A.,Wanner,B.L.(2000)“One-stepinactivationofchromosomalgenesinEscherichiacoliK-12usingPCRproducts”Proc.Natl.Acad.Sci.USA97:6640-6645.Datta,R.etal.(1995)“Technologicalandeconomicpotentialofpoly(lacticacid)andlacticacidderivatives”FEMSMicrobiol.Rev.16:221-231.Demirci,A.andA.L.Pometto(1992)“EnhancedproductionofD-(-)-lacticacidbymutantsofLactobacillusdelbrueckiiATCC9649”J.Ind.Microbiol.Biotechnol.11:23-28.Dien,B.S.etal.(2001)“RecombinantEscherichiacoliengineeredforproductionofL-lacticacidfromhexoseandpentosesugars”JInd.MicrobiolBiotechnol.27:259-264.Grabar,T.B.etal.(2006)“MethylglyoxalbypassidentifiedassourceofchiralcontaminationinL(+)andD(-)lactatefermentationsbyrecombinantEscherichiacoli”BiotechnologyLetters,inpublication.Hofvendahl,K.,andHahn-Hagerdal,B.(2000)“Factorsaffectingfermentativelacticacidproductionfromrenewableresources”Enzy.Microbiol.Technol26:87-107.Konings,W.N.(2002)“Thecellmembraneandthestruggleforlifeoflacticacidbacteria”AntonievanLeeuwenhoek82:3-27.Kyla-Nikkila,K.etal.(2000)“MetabolicengineeringofLactobacillushelveticusCNR32forproductionofpureL-(+)-lacticacid”Appl.Environ.Microbiol.66:3835-3841.Lee,E-C.,Yu,K.,MartinezdeVelasco,J.,Tessarollo,L.,Swing,D.A.,Court,D.L.,Jenkins,N.A.,andCopeland,N.G.(2001)“AhighlyefficientEscherichiacoli-basedchromosomeengineeringsystemadaptedforrecombinogenictargetingandsubcloningofBACDNA”Genomics73:56-65.Maniatis,T.etal.(1989)MolecularCloning:ALaboratoryManual,ColdSpringsHarborLaboratory.Martinez-Morales,F.etal.(1999)“ChromosomalintegrationofheterologousDNAinEscherichiacoliwithpreciseremovalofmarkersandrepliconsduringconstruction”J.Bacteriol.,181:7143-7148.Michels,P.A.M.etal.(1979)“Generationofanelectrochemicalprotongradientinbacteriabytheexcretionofmetabolicendproducts”FEMSMicrobial.Lett5:357-364.Miller,J.H.(1992)Ashortcourseinbacterialgenetics:AlaboratorymanualandhandbookforEscherichiacoliandrelatedbacteria,ColdSpringHarborPress.Moniruzzaman,M.etal.(1997)“Isolationandmolecularcharacterizationofhigh-performancecellobiose-fermentingspontaneousmutantsofethanologenicEscherichiacoliKO11containingtheKlebsiellaoxytocacasABoperon”Appl.Environ.Microbiol.63:4633-4637.Narayananetal.(2004)“L-(+)lacticacidfermentationanditsproductpolymerization,”Elect.J.Beiotechnol.7:167-179.Ohara,H.etal.(2001)“Developmentofindustrialproductionofhighmolecularweightpoly-L-lactatefromrenewableresources”NipponKagakuKaishi6:323-331.Ohta,K.etal.(1991)“GeneticimprovementofEscherichiacoliforethanolproductionbychromosomalintegrationofZymomonasmobilisgenesencodingpyruvatedecarboxylaseandalcoholdehydrogenaseII”ApplEnvironMicrobiol.57:893-900.Poolman,B.(2002)“TransportersandtheirrolesinLABcellphysiology”AntonievanLeeuwenhoek82:147-164.Porro,D.etal.(1999)“Replacementofametabolicpathwayforlarge-scaleproductionoflacticacidfromengineeredyeasts”Appl.Environ.Microbiol.65:4211-4215.Posfai,G.,Koob,M.D.,Kirkpatrick,H.A.,Blattner,F.C.(1997)“Versatileinsertionplasmidsfortargetedgenomemanipulationsinbacteria:Isolation,deletion,andrescueofthepathogenicityislandLEEoftheEscherichiacoliO157:H7genome”J.Bacteriol.179:4426-4428.Purvis,J.E.etal.(2005)“EnhancedtrehaloseproductionimprovesgrowthofEscherichiacoliunderosmoticstress”Appl.Environ.Microbiol.71:3761-3769.Saitoh,S.etal.(2005)“GeneticallyengineeredwineyeastproducesahighconcentrationofL-lacticacidofextremelyhighopticalpurity”Appl.Environ.Microbiol.71:2789-2792.Sambrook,J.etal(1989)MolecularCloning:ALaboratoryM4nual,SecondEdition,ColdSpringHarborLaboratoryPress.Sambrook,J.andRussell,D.W.(2001)Molecularcloning:Alaboratorymanual,ColdSpringHarborPress.Shukla,V.B.etal.(2004)“ProductionofD-(-)-lactatefromsucroseandmolasses,”BiotechnolLett.,26:689-693.vanMaris,A.J.A.etal.(2004a)“Microbialexportoflacticand3-hydroxypropanoicacid:implicationsforindustrialfermentationprocesses”MetabolicEngineering6:245-255.vanMaris,A.J.A.etal.(2004b)“HomofermentativelactateproductioncannotsustainanaerobicgrowthofengineeredSaccharomycescerevisiae:possibleconsequenceofenergy-dependentlactateexport”Appl.Environ.Microbiol.70(5):2898-2905.Warnecke,T.andR.T.Gill(2005)“Organicacidtoxicity,tolerance,andproductionofEscherichiacolibiorefiningapplications”MicrobialCellFactories4:25.ZhouS.,IngramL.O.(1999)“Engineeringendoglucanase-secretingstrainsofethanologenicKlebsiellaoxytocaP2”J.Ind.Microbiol.Biotechnol.22:600-607.Zhou,S.etal.(2003)“ProductionofopticallypureD-lacticacidinmineralsaltmediumbymetabolicallyengineeredEscherichiacoliW3110”ApplEnvironMicrobiol.69:399-407.Zhou,S.etal.(2003a)“ProductionsofopticallypureD-lacticacidinmineralsaltmediumbymetabolicallyengineeredEscherichiacoliD-(-)-lactatedehydrogenasegene(ldhA)withtheL-(+)-lactatedehydrogenasegene(ldhL)fromPediococcusacidilactici”Appl.Environ.Microbiol.69:2237-2244.Zhou,S.,Yomano,L.P.,Shanmugam,K.T.,Ingram,L.O.(2005)“Fermentationof10%(w/v)sugartoD-(-)-lactatebyengineeredEscherichiacoliB”Biotechnol.Lett.27:1891-1896.Zhou,S.,Shanmugam,K.T.,Yomano,L.P.,Grabar,T.B.,Ingram,L.O.(2006)“Fermentationof12%(w/v)glucoseto1.2MlactatebyEscherichiacoliBstrainSZ194usingmineralsaltsmedium”Biotechnol.Lett.InPress.Zhu,J.andShimizu,K.(2004)“TheeffectofpflgeneknockoutonthemetabolismforopticallypureD-lactateproductionbyEscherichiacoli”ApplMicrobiolandBiotechnol.64:367-375.
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1