复杂基因集合中的独特标记的重排适应性免疫受体基因的制作方法
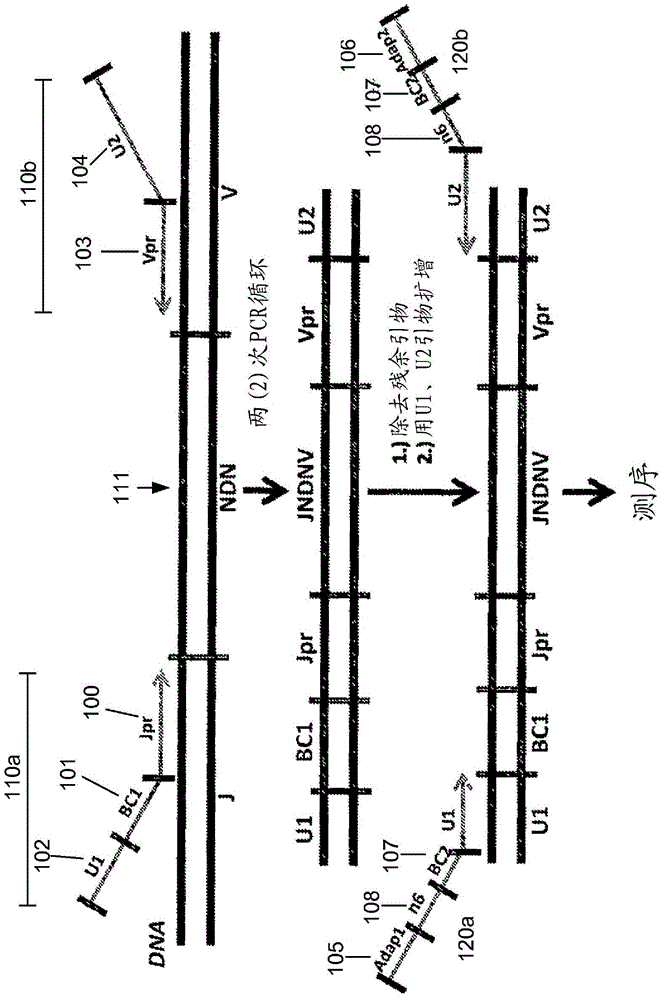
本申请要求提交于2013年3月15日的美国临时申请No.61/789,408和提交于2013年6月14日的国际申请No.PCT/US2013/045994的权益,这两份专利申请全文以引用方式并入。序列表本申请包含序列表,该序列表通过EFS-Web提交并且据此全文以引用方式并入。所述ASCII副本创建日期为2014年3月13日,名称为26371PCT_CRF_sequencelisting.txt,文件大小为4,382,702字节。
背景技术:
:
技术领域:
本公开整体涉及在多重核酸扩增反应中对适应性免疫受体编码DNA或RNA(例如,编码T细胞受体和免疫球蛋白的DNA或RNA)进行定量高通量测序。具体地讲,本文所述的组合物和方法允许对DNA序列进行定量测序,所述DNA序列编码单个细胞中的适应性免疫受体异源二聚体的两条链。本文还公开了克服在适应性免疫受体编码序列的定量中的不期望失真的实施例,所述失真可由多重DNA扩增中的特异性寡核苷酸引物的偏倚的过度利用和/或利用不足所造成。相关领域的说明适应性免疫系统采用若干策略来生成T细胞和B细胞抗原受体即适应性免疫受体的组库,该组库具有足够的多样性以识别众多的潜在病原体。T细胞识别与各种癌或传染性生物体相关的众多抗原的能力由其T细胞抗原受体(TCR)赋予,该T细胞抗原受体是来自TCRA基因座的α(alpha)链与来自TCRB基因座的β(beta)链的异源二聚体,或来自TCRG基因座的γ(gamma)链与来自TCRD基因座的δ(delta)链的异源二聚体。构成这些链的蛋白质由DNA编码,该DNA在淋巴样细胞中采用独特重排机制以便生成极其多样的TCR。该多亚基免疫识别受体与CD3复合物相关,并且结合于由主要组织相容性复合物(MHC)类别I和II蛋白质递呈在抗原递呈细胞(APC)表面上的肽。TCR结合于APC上的抗原肽是T细胞活化过程的中心事件,T细胞活化在免疫突触处T细胞与APC之间的接触点处发生。每个TCR肽包含可变互补性决定区(CDR)以及骨架区(FR)和恒定区。αβT细胞的序列多样性主要由α和β链可变结构域的第三互补性决定区(CDR3)环的氨基酸序列决定,所述多样性分别是β链基因座中的可变(Vβ)、多样性(Dβ)和连接(Jβ)基因区段之间重组,及α链基因座中的类似Vα和Jα基因区段之间重组的结果。TCRα和β链基因座中的多个此类基因区段的存在允许大量不同CDR3序列被编码。TCR基因重排过程期间Vβ-Dβ、Dβ-Jβ和Vα-Jα接合部处核苷酸的独立添加和缺失进一步增加了CDR3序列多样性。在这方面,免疫活性反映于TCR的多样性中。γδTCR与αβTCR不同之处在于其编码的受体与先天免疫系统紧密相互作用,并且以非HLA依赖性方式识别抗原。TCRγδ在发育早期表达,并具有特化的解剖分布、独特的病原体和小分子特异性,以及广谱的先天和适应性细胞相互作用。TCRγV和J区段表达的偏倚模式在个体发生早期建立。因此,成体组织中的多样TCRγ组库是在环境暴露于病原体和有毒分子造成的刺激之后大量外周扩增的结果。由B细胞表达的免疫球蛋白(Ig或IG),本文也称为B细胞受体(BCR),是由四条多肽链组成的蛋白质,所述四条多肽链即来自IGH基因座的两条重链(H链)以及来自IGK(κ)或IGL(λ)基因座的两条轻链(L链),从而形成H2L2结构。H链和L链均包含涉及抗原识别的互补性决定区(CDR),以及恒定结构域。IG的H链最初使用IgM或IgD恒定区同工型表达为膜结合同工型,但在抗原识别后H链恒定区可类别转换为若干另外的同种型,包括IgG、IgE和IgA。与TCR一样,个体内的初始Ig的多样性主要由高变互补性决定区(CDR)决定。与TCR类似,IGH链的CDR3结构域由VH、DH和JH基因区段的组合连接所形成。Ig基因重排过程期间VH-DH、DH-JH和VH-JH接合部处核苷酸的独立添加和缺失进一步增加了高变结构域序列多样性。与TCR不同,在初始B细胞最初识别抗原后整个重排IG基因的体细胞高频突变(SHM)进一步提高了Ig序列多样性。SHM的过程不受限于CDR3,因此可将变化引入生殖系序列中的骨架区、CDR1和CDR2、以及体细胞重排的CDR3。由于适应性免疫系统部分地通过表达独特TCR或BCR的细胞的克隆性扩增来起作用,准确测量每个克隆的总丰度的变化对于理解适应性免疫应答的动态是重要的。例如,健康人体具有数百万独特的TCRβ链,每条链在健康个体的大致万亿T细胞中的数百至数千克隆T细胞中携带。利用高通量测序方面的进步,最近出现了分子免疫学的新领域以对巨大TCR和BCR组库进行谱图分析。用于对重排的适应性免疫受体基因序列测序并用于适应性免疫受体克隆型确定的组合物和方法在如下文献中有所描述,例如Robinsetal.,2009Blood114,4099(Robins等人,2009年,《血液》,第114卷,第4099页);Robinsetalh.,2010Sci.Translat.Med.2:47ra64(Robins等人,2010年,《科学转化医学》,第2卷,第47ra64页);Robinsetal.,2011J.Immunol.Meth.doi:10.1016/j.jim.2011.09.001(Robins等人,2011年,《免疫法杂志》,doi:10.1016/j.jim.2011.09.001);Sherwoodetal.2011Sci.Translat.Med.3:90ra61(Sherwood等人,2011年,《科学转化医学》,第3卷,第90ra61页);U.S.A.N.13/217,126(美国公布No.2012/0058902)、U.S.A.N.12/794,507(美国公布No.2010/0330571)、WO/2010/151416、WO/2011/106738(PCT/US2011/026373)、W02012/027503(PCT/US2011/049012)、U.S.A.N.61/550,311以及U.S.A.N.61/569,118,所有这些文献以引用的方式并入本文。到目前为止,已采用了若干不同策略来对编码适应性免疫受体的核酸以高通量方式定量地测序,并且这些策略的区别可在于例如用于扩增CDR3编码区的方法以及对基因组DNA(gDNA)或信使RNA(mRNA)测序的选择。对mRNA测序是相比于对gDNA测序可能更简便的方法,因为mRNA剪接事件去除了J区段与C区段之间的内含子。这允许在C区使用通用3’聚合酶链反应(PCR)扩增引物扩增具有不同V区和J区的适应性免疫受体(例如,TCR或Ig)。对于每种TCRβ而言,例如十三个J区段均短于60个碱基对(bp)。因此,剪接事件在小于100bp重排VDJ接合部内引入编码TCRh恒定区的相同多核苷酸序列(不论使用的是哪些V和J序列)。然后可使用与mRNA的多聚A尾互补的多聚dT引物、随机小引物(通常为六聚物或九聚物)或C区段特异性寡核苷酸,将剪接的mRNA逆转录为互补DNA(cDNA)。该逆转录应产生TCRcDNA的无偏文库(因为所有cDNA均用相同寡核苷酸引发,不论是多聚dT、随机六聚物,还是C区段特异性寡核苷酸),然后可对所述无偏文库测序以获得有关每种重排中使用的V和J区段以及CDR3特异性序列的信息。此类测序可使用单种、跨CDR3的长读段(“长读段”)技术,或者可替代地涉及分级分离较长序列的多个拷贝并使用较高通量较短序列读段。然而,基于mRNA测序来对表达特定重排TCR(或Ig)的样品中的细胞数量定量的尝试难以说明,因为每个细胞潜在地表达不同数量的TCRmRNA。例如,体外活化的T细胞的每个细胞拥有的mRNA数量是静止期T细胞的10-100倍。到目前为止,有关不同功能状态的T细胞中的TCRmRNA的相对量的信息非常有限,因此大量mRNA的定量不一定能准确测量携带每种克隆TCR的细胞的数量。另一方面,大多数T细胞具有一个有效重排的TCRα和一个有效重排的TCRβ基因(或两个重排的TCRγ和TCRδ),并且大多数B细胞具有一个有效重排的Ig重链基因和一个有效重排的Ig轻链基因(IGK或IGL),因此样品中的编码TCR或BCR的基因组DNA的定量应分别与样品中的T细胞或B细胞的数量直接关联。编码适应性免疫受体链中的任何一种或多种(例如,使用人类TCRβ链作为代表性例子)的多核苷酸的基因组测序有利地需要以等同效率扩增取自受试者淋巴样细胞的含DNA样品中存在的许多可能重排TCRβ编码序列的所有序列,然后进行定量测序,使得可获得每个克隆型的相对丰度的定量量度。然而,采用此类方法遇到了困难,因为可能不易例如对每个重排TCRβ编码克隆实现等同的扩增和测序效率,其中每个克隆采用54个可能的生殖系V区编码基因中的一个和13个可能的J区编码基因中的一个。TCRβ基因组基因座中的高度多样V区段和J区段的特定序列在大量可能的重排间广泛变化,这些重排通过使用不同V基因或J基因引起,原因在于多样性生成机制,诸如以上概述的那些。该序列多样性会产生复杂DNA样品,其中使用多重扩增和高通量测序同时定量多种分子物质的能力受到技术限制,而阻碍了所述样品中所含的多种不同序列的准确测定。另外,现有方法难以对编码TCR或IG异源二聚体的两条链的DNA或RNA定量地测序,该测序方式允许确定这两条链都源自相同淋巴样细胞。一种或多种因素可引起使测序数据输出出现偏态的伪差,从而削弱了从基于TCR或IG基因模板的高度多样集合的多重扩增的测序策略获得可靠定量数据的能力。这些伪差通常是多重扩增步骤过程中多样引物的不等同使用所引起。在使用多样扩增模板的多重反应中可出现随如下的一者或多者而变的一种或多种寡核苷酸引物的此类偏倚利用:模板和/或寡核苷酸引物的核苷酸碱基组成的差异、模板和/或引物长度的差异、所使用的具体聚合酶、扩增反应温度(例如,退火、延伸和/或变性温度)和/或其他因素(例如,Kanagawa,2003J.Biosci.Bioeng.96:317(Kanagawa,2003年,《生物学与生物工程杂志》,第96卷,第317页);Dayetal.,1996Hum.Mol.Genet.5:2039(Day等人,1996年,《人分子遗传学》,第5卷,第2039页);Oginoetal.,2002J.Mol.Diagnost.4:185(Ogino等人,2002年,《分子诊断学杂志》,第4卷,第185页);Barnardetal.,1998Biotechniques25:684(Barnard等人,1998年,《生物技术》,第25卷,第684页);Airdetal.,2011GenomeBiol.12:R18(Aird等人,2011年,《基因组生物学》,第12卷,第R18页))。很明显,仍然需要改善的组合物和方法,其将允许以避免偏态结果的方式并且以允许确定TCR或IG异源二聚体的两条链的编码序列源自相同淋巴样细胞的方式,准确定量复杂样品中的适应性免疫受体编码DNA和RNA的序列多样性,所述偏态结果诸如单独序列失实的过高占比或过低占比的原因在于在用于复杂模板DNA群体的多重扩增的寡核苷酸引物组中一种或多种寡核苷酸引物的利用偏倚。本发明所述的实施例解决了该需求并提供了其他相关优点。技术实现要素:本发明提供了一种识别多个同源对的方法,所述多个同源对包含第一多肽和第二多肽而形成适应性免疫受体异源二聚体,所述适应性免疫受体异源二聚体包含来自样品中的单个克隆的T细胞受体(TCR)或免疫球蛋白(IG),所述样品包含来自哺乳动物受试者的多个淋巴样细胞,所述方法包括:将多个淋巴样细胞分布于多个容器间,每个容器包含多个淋巴样细胞;通过对已由得自所述多个淋巴样细胞的mRNA分子逆转录的cDNA分子进行多重PCR,而在所述多个容器中生成扩增子的文库,所述扩增子的文库包含:i)编码所述第一多肽的多个第一适应性免疫受体扩增子,每个扩增子包含独特可变(V)区编码序列、独特J区编码序列或者独特J区编码序列与独特C区编码序列两者、至少一个条形码序列、至少一个通用接头序列以及测序平台标签序列,和ii)编码所述第二多肽的多个第二适应性免疫受体扩增子,每个扩增子包含独特V区编码序列、独特J区编码序列或者独特J区编码序列与独特C区编码序列两者、至少一个条形码序列、至少一个通用接头序列以及测序平台标签序列;对所述扩增子的文库进行高通量测序以获得多个第一和第二适应性免疫受体扩增子序列的数据集。所述方法包括通过将每个独特第一适应性免疫受体扩增子序列分配至一个或多个容器来确定每个独特第一适应性免疫受体扩增子序列的容器占用模式,以及通过将每个独特第二适应性免疫受体扩增子序列分配至一个或多个容器来确定每个独特第二适应性免疫受体扩增子序列的容器占用模式,其中所述独特第一或第二适应性免疫受体扩增子序列中的每个条形码序列与具体容器相关。对于独特第一和第二适应性免疫受体扩增子序列形成推定同源对的每种可能配对来说,所述方法还包括计算观察到所述容器占用模式、或偶然观察到比预期高的任意共用容器比例的统计概率,前提是所述第一和第二适应性免疫受体扩增子序列不源自淋巴样细胞的相同克隆群体;随后基于分数比预定可能性截断值低的所述统计概率来识别多个推定的同源对。所述方法包括对于每个识别出的推定同源对而言,确定所述独特第一适应性免疫受体扩增子序列和所述独特第二适应性免疫受体扩增子序列的可能错误配对的假发现率估值;以及基于所述统计概率和所述假发现率估值,将独特第一适应性免疫受体序列和第二适应性免疫受体序列的多个同源对识别为编码所述样品中的所述适应性免疫受体的真实同源对。在一些实施例中,统计分数包含计算得到的将独特第一适应性免疫受体扩增子序列和独特第二适应性免疫受体扩增子序列配成每个推定同源对的p值。在一个实施例中,计算所述独特第一和第二适应性免疫受体扩增子序列应当共同占用与观察到的它们实际共同占用的容器数相比一样多或更多的容器数的概率,计算假定没有真实同源配对,并给定了由所述独特第一适应性免疫受体扩增子序列占用的容器数和由所述独特第二适应性免疫受体扩增子序列占用的容器数。在另一个实施例中,基于所述统计概率来识别具有高配对可能性的多个推定的同源对包括对于每个独特第一适应性免疫受体扩增子序列而言,识别具有匹配的最低p值分数的独特第二适应性免疫受体扩增子序列,或者对于每个独特第二适应性免疫受体扩增子序列而言,寻找具有匹配的最低p值分数的独特第一适应性免疫受体扩增子序列。在其他实施例中,确定假发现率估值包括:计算所述样品中鉴定的所述多个推定的同源对每个的p值;将所述多个推定的同源对所有的p值与预期p值分布进行比较,其中计算得到的预期p值分布用来代表不存在真实同源对的实验;然后针对每个推定的同源对,确定假阳性结果的预期比例,由此确定等于或低于所述推定同源对的p值的所有p值,用来代表真实同源配对。在某些实施例中,计算所述预期p值分布包括:置换容器,其中已在无真实同源对的其他方面相同的实验中观察到每个第一和第二适应性免疫受体序列,以及计算与每个推定同源对相关的p值的分布。在一些实施例中,所述方法包括通过选择具有低于基于所述假发现率估值计算的阈值的p值的多个推定同源对,将独特第一适应性免疫受体序列和第二适应性免疫受体序列的多个同源对识别为真实同源对。在一个实施例中,识别出的独特第一和第二适应性免疫受体扩增子序列的同源对具有小于1%的假发现率估值。在另一个实施例中,在足以促进对得自所述多个淋巴样细胞的mRNA分子进行逆转录的条件及持续时间下,使所述多个容器中的每一个与第一逆转录引物组接触。在某些实施例中,(A)第一寡核苷酸逆转录引物组包括能够逆转录编码所述多个第一和第二适应性免疫受体多肽的多个mRNA序列以便生成多个第一和第二逆转录的适应性免疫受体cDNA扩增子的引物,其中编码所述第一适应性免疫受体多肽的所述多个第一逆转录的适应性免疫受体cDNA扩增子包含1)独特V区编码基因序列和2)独特J区编码基因序列,或者独特J区编码基因序列和独特C区编码基因序列两者,并且其中编码所述第二适应性免疫受体多肽的所述多个第二逆转录的适应性免疫受体cDNA扩增子包含1)独特V区编码基因序列和2)独特J区编码基因序列,或者独特J区编码基因序列和独特C区编码基因序列两者。在某些方面,所述方法包括在足以促进对所述第一和第二逆转录的适应性免疫受体cDNA扩增子进行多重PCR扩增的条件和持续时间下,使所述多个容器中的每一个与第二(B)和第三(C)寡核苷酸引物组接触。在一些方面,(B)第二寡核苷酸引物组包括能够扩增所述多个第一逆转录的适应性免疫受体cDNA扩增子的正向引物和反向引物,其中所述正向引物和反向引物各自能够杂交于所述第一逆转录的适应性免疫受体cDNA扩增子。在一个方面,所述第二寡核苷酸引物组中的每对正向引物和反向引物能够扩增所述第一逆转录的适应性免疫受体cDNA扩增子。在另一个方面,所述第二寡核苷酸引物组中的正向引物包含第一通用接头序列和与所述V区编码基因序列互补的区域。在其他方面,所述第二寡核苷酸引物组中的反向引物包含第二通用接头序列和与所述J区编码基因序列或所述C区编码基因序列互补的区域。在另一个方面,(C)第三寡核苷酸引物组包括能够扩增所述多个逆转录的第二适应性免疫受体cDNA扩增子的正向引物和反向引物。在一个实施例中,所述第三寡核苷酸引物组中的每对正向引物和反向引物能够扩增所述第二逆转录的适应性免疫受体cDNA扩增子。在一个方面,所述第三寡核苷酸引物组中的正向引物包含第一通用接头序列和与所述V区编码基因序列互补的区域。在一些方面,所述第三寡核苷酸引物组中的反向引物包含第二通用接头序列和与所述J区编码基因序列互补或与所述C区编码基因序列互补的区域。所述方法还包括生成i)多个第三适应性免疫受体扩增子,每个这种扩增子包含独特V区编码基因序列或其互补序列、独特J区编码基因序列或其互补序列,或者独特J区编码基因序列和独特C区编码基因序列两者或这两者的互补序列,以及所述第一和第二通用接头序列,和ii)多个第四适应性免疫受体扩增子,每个这种扩增子包含独特V区编码基因序列或其互补序列、独特J区编码基因序列或其互补序列,或者独特J区编码基因序列和独特C区编码基因序列两者或这两者的互补序列,以及所述第一和第二通用接头序列。在另一个实施例中,所述方法包括在足以促进对所述多个第三和第四适应性免疫受体扩增子进行第二多重PCR扩增的条件和持续时间下,使所述多个容器中的每一个与第四(D)寡核苷酸引物组和第五(E)寡核苷酸引物组接触。在一个实施例中,(D)第四寡核苷酸引物组包括能够扩增所述多个第三适应性免疫受体扩增子的正向引物和反向引物,其中所述正向引物和反向引物各自能够杂交于所述第三适应性免疫受体扩增子。在一个方面,所述第四寡核苷酸引物组中的每对正向引物和反向引物能够扩增所述第三适应性免疫受体扩增子。在另一个方面,所述第四寡核苷酸引物组中的正向引物包含测序平台标签序列和与所述多个第三适应性免疫受体扩增子中的所述第一通用接头序列互补的区域,并且所述反向引物包含测序平台标签序列和与所述多个第三适应性免疫受体扩增子中的所述第二通用接头序列互补的区域。在另一个实施例中,所述第四寡核苷酸引物组中的所述正向引物和反向引物中的一者或两者包含独特条形码序列,该条形码序列与其中引入了所述第四寡核苷酸引物组的所述容器相关联。在一个实施例中,(E)第五寡核苷酸引物组包括能够扩增所述多个第四适应性免疫受体扩增子的正向引物和反向引物,其中所述正向引物和反向引物各自能够杂交于所述第四适应性免疫受体扩增子。在一个实施例中,所述第四寡核苷酸引物组中的每对正向引物和反向引物能够扩增所述多个第四适应性免疫受体扩增子。在另一个实施例中,所述第五寡核苷酸引物组中的正向引物包含测序平台标签序列和与所述多个第四适应性免疫受体扩增子中的所述第一通用接头序列互补的区域,并且所述第五寡核苷酸引物组中的所述反向引物包含测序平台标签序列和与所述多个第四适应性免疫受体扩增子中的所述第二通用接头序列互补的区域。在其他实施例中,所述第四寡核苷酸引物组的所述正向引物和反向引物中的一者或两者包含独特条形码序列,该条形码序列与其中引入了所述第四寡核苷酸引物组的所述容器相关联,从而生成包含所述多个第一适应性免疫受体扩增子和所述多个第二适应性免疫受体扩增子的所述扩增子的文库。在一个方面,所述方法包括将来自所述多个容器的所述扩增子的文库合并成用于测序的混合物。在另一个方面,所述多个第一适应性免疫受体扩增子包含C区编码序列。在一些方面,所述多个第二适应性免疫受体扩增子包含C区编码序列。在一个实施例中,所述样品包括血液样品。在另一个实施例中,所述样品包括组织样品。在某些实施例中,所述样品包括样品纯化或培养的人类淋巴样细胞。在其他实施例中,所述容器包含至少104个淋巴样细胞。在另一个实施例中,所述样品包含至少104个细胞。在某些方面,所述适应性免疫受体异源二聚体的第一多肽为TCRα(TCRA)链,并且所述适应性免疫受体异源二聚体的第二多肽为TCRβ(TCRB)链。在一个方面,适应性免疫受体异源二聚体的第一多肽为TCRγ(TCRG)链,并且所述适应性免疫受体异源二聚体的所述第二多肽为TCRδ(TCRD)链。在另一个方面,所述适应性免疫受体异源二聚体的第一多肽为免疫球蛋白重(IGH)链,并且适应性免疫受体异源二聚体的所述第二多肽选自免疫球蛋白轻链IGL或IGK。在又一个方面,如果适应性免疫受体异源二聚体的第一多肽为IGH链,并且适应性免疫受体异源二聚体的第二多肽为IGL和IGK两者,则使用三种不同的扩增引物组,包括:针对IGH的第一寡核苷酸扩增引物组、针对IGK的第二寡核苷酸扩增引物组以及针对IGL的第三寡核苷酸扩增引物组。本发明提供了一种识别多个同源对的方法,所述多个同源对包含第一多肽和第二多肽而形成适应性免疫受体异源二聚体,所述适应性免疫受体异源二聚体包含来自样品中的单个克隆的T细胞受体(TCR)或免疫球蛋白(IG),所述样品包含来自哺乳动物受试者的多个淋巴样细胞。所述方法包括将多个淋巴样细胞分布于多个容器间,每个容器包含多个淋巴样细胞,以及通过对得自所述多个淋巴样细胞的基因组分子进行多重PCR,而在所述多个容器中生成扩增子的文库。所述扩增子的文库包含i)编码所述第一多肽的多个第一适应性免疫受体扩增子,每个扩增子包含独特可变(V)区编码序列、独特J区编码序列、至少一个条形码序列、至少一个通用接头序列以及测序平台标签序列,和ii)编码所述第二多肽的多个第二适应性免疫受体扩增子,每个扩增子包含独特V区编码序列、独特J区编码序列、至少一个条形码序列、至少一个通用接头序列以及测序平台标签序列。所述方法包括对所述扩增子的文库进行高通量测序以获得多个第一和第二适应性免疫受体扩增子序列的数据集,以及通过将每个独特第一适应性免疫受体扩增子序列分配至一个或多个容器来确定每个独特第一适应性免疫受体扩增子序列的容器占用模式,及通过将每个独特第二适应性免疫受体扩增子序列分配至一个或多个容器来确定每个独特第二适应性免疫受体扩增子序列的容器占用模式,其中所述独特第一或第二适应性免疫受体扩增子序列中的每个条形码序列与具体容器相关。在一些实施例中,对于独特第一和第二适应性免疫受体扩增子序列形成推定同源对的每种可能配对来说,所述方法包括计算观察到所述容器占用模式、或偶然观察到比预期高的任意共用容器比例的统计概率,前提是所述第一和第二适应性免疫受体扩增子序列不源自淋巴样细胞的相同克隆群体。在一些实施例中,所述方法包括基于分数比预定可能性截断值更低的所述统计概率来识别多个推定的同源对。在其他实施例中,所述方法包括对于每个识别出的推定同源对而言,确定所述独特第一适应性免疫受体扩增子序列和所述独特第二适应性免疫受体扩增子序列的可能错误配对的假发现率估值。在一个实施例中,所述方法包括基于所述统计概率和所述假发现率估值,将独特第一适应性免疫受体序列和第二适应性免疫受体序列的多个同源对识别为编码所述样品中的所述适应性免疫受体的真实同源对。在其他实施例中,统计分数包含计算得到的将独特第一适应性免疫受体扩增子序列和独特第二适应性免疫受体扩增子序列配成每个推定同源对的p值。在其他实施例中,计算所述统计分数的步骤包括计算所述独特第一和第二适应性免疫受体扩增子序列应当共同占用与观察到的它们实际共同占用的容器数相比一样多或更多的容器数的概率,计算假定没有真实同源配对,并给定了由所述独特第一适应性免疫受体扩增子序列占用的容器数和由所述独特第二适应性免疫受体扩增子序列占用的容器数。在另一个实施例中,所述方法包括基于所述统计概率来识别具有高配对可能性的多个推定的同源对,其包括对于每个独特第一适应性免疫受体扩增子序列而言,识别具有匹配的最低p值分数的独特第二适应性免疫受体扩增子序列,或者对于每个独特第二适应性免疫受体扩增子序列而言,寻找具有匹配的最低p值分数的独特第一适应性免疫受体扩增子序列。在其他实施例中,确定假发现率估值的步骤包括:计算所述样品中鉴定的所述多个推定的同源对每个的p值;将所述多个推定的同源对所有的p值与预期p值分布进行比较,其中计算得到的预期p值分布用来代表不存在真实同源对的实验;然后针对每个推定的同源对,确定假阳性结果的预期比例,由此确定等于或低于所述推定同源对的p值的所有p值,用来代表真实同源配对。在一个实施例中,计算所述预期p值分布的步骤包括:置换容器,其中已在无真实同源对的其他方面相同的实验中观察到每个第一和第二适应性免疫受体序列,以及计算与每个推定同源对相关的p值的分布。在另一个方面,所述方法包括通过选择具有低于阈值的p值的多个推定同源对,将独特第一适应性免疫受体序列和第二适应性免疫受体序列的多个同源对识别为真实同源对,所述阈值是基于所述假发现率估值计算的。在一个实施例中,识别出的独特第一和第二适应性免疫受体扩增子序列的同源对具有小于1%的假发现率估值。在另一个实施例中,所述方法包括在足以促进对所述第一和第二适应性免疫受体cDNA扩增子进行多重PCR扩增的条件和持续时间下,使所述多个容器中的每一个与第一(A)和第二(B)寡核苷酸引物组接触。在一个实施例中,(A)第一寡核苷酸引物组包括能够扩增所述多个第一适应性免疫受体扩增子的正向引物和反向引物,其中所述正向引物和反向引物各自能够杂交于所述第一适应性免疫受体扩增子。在一个方面,所述第一寡核苷酸引物组中的每对正向引物和反向引物能够扩增所述第一适应性免疫受体扩增子。在另一个方面,所述第一寡核苷酸引物组中的正向引物包含第一通用接头序列和与所述V区编码基因序列互补的区域。在一个方面,所述第二寡核苷酸引物组中的反向引物包含第二通用接头序列和与所述J区编码基因序列互补的区域。在另一个方面,(B)第二寡核苷酸引物组包括能够扩增所述多个第二适应性免疫受体扩增子的正向引物和反向引物。在一个实施例中,所述第二寡核苷酸引物组中的每对正向引物和反向引物能够扩增所述第二适应性免疫受体扩增子。在另一个实施例中,所述第二寡核苷酸引物组中的正向引物包含第一通用接头序列和与所述V区编码基因序列互补的区域。在另一个方面,所述第二寡核苷酸引物组中的反向引物包含第二通用接头序列和与所述J区编码基因序列互补的区域。在一些实施例中,所述方法还包括生成i)多个第三适应性免疫受体扩增子,每个这种扩增子包含独特V区编码基因序列或其互补序列、独特J区编码基因序列或其互补序列,以及所述第一和第二通用接头序列,和ii)多个第四适应性免疫受体扩增子,每个这种扩增子包含独特V区编码基因序列或其互补序列、独特J区编码基因序列或其互补序列,以及所述第一和第二通用接头序列。所述方法还包括在足以促进对所述多个第三和第四适应性免疫受体扩增子进行第二多重PCR扩增的条件和持续时间下,使所述多个容器中的每一个与第三(C)寡核苷酸引物组和第四(D)寡核苷酸引物组接触的步骤。在一个实施例中,(C)第三寡核苷酸引物组包括能够扩增所述多个第三适应性免疫受体扩增子的正向引物和反向引物,其中所述正向引物和反向引物各自能够杂交于所述第三适应性免疫受体扩增子。在另一个实施例中,所述第三寡核苷酸引物组中的每对正向引物和反向引物能够扩增所述第二适应性免疫受体扩增子。在又一个实施例中,所述第三寡核苷酸引物组中的正向引物包含测序平台标签序列和与所述多个第三适应性免疫受体扩增子中的所述第一通用接头序列互补的区域,并且所述第三寡核苷酸引物组中的所述反向引物包含测序平台标签序列和与所述多个第二适应性免疫受体扩增子中的所述第二通用接头序列互补的区域。在一个方面,所述第四寡核苷酸引物组中的所述正向引物和反向引物中的一者或两者包含独特条形码序列,该条形码序列与其中引入了所述第三寡核苷酸引物组的所述容器相关联。在另一个方面,(D)第四寡核苷酸引物组包括能够扩增所述多个第四适应性免疫受体扩增子的正向引物和反向引物,其中所述正向引物和反向引物各自能够杂交于所述第四适应性免疫受体扩增子。在一个实施例中,所述第四寡核苷酸引物组中的每对正向引物和反向引物能够扩增所述多个第四适应性免疫受体扩增子。在一些实施例中,所述第四寡核苷酸引物组中的正向引物包含测序平台标签序列和与所述多个第四适应性免疫受体扩增子中的所述第一通用接头序列互补的区域,并且所述第四寡核苷酸引物组中的所述反向引物包含测序平台标签序列和与所述多个第四适应性免疫受体扩增子中的所述第二通用接头序列互补的区域。在又一个实施例中,所述第四寡核苷酸引物组的所述正向引物和反向引物中的一者或两者包含独特条形码序列,该条形码序列与其中引入了所述第四寡核苷酸引物组的所述容器相关联,从而生成包含所述多个第一适应性免疫受体扩增子和所述多个第二适应性免疫受体扩增子的所述扩增子的文库。在其他实施例中,所述方法包括将来自所述多个容器的所述扩增子的文库合并成用于测序的混合物。在一些实施例中,所述样品包括血液样品。在一个实施例中,所述样品包括组织样品。在另一个实施例中,所述样品包括样品纯化或培养的人类淋巴样细胞。在一些实施例中,每个容器包含至少104个淋巴样细胞。在其他实施例中,所述样品包含至少104个细胞。在其他实施例中,所述适应性免疫受体异源二聚体的第一多肽为TCRα(TCRA)链,并且所述适应性免疫受体异源二聚体的第二多肽为TCRβ(TCRB)链。在一些实施例中,适应性免疫受体异源二聚体的第一多肽为TCRγ(TCRG)链,并且所述适应性免疫受体异源二聚体的所述第二多肽为TCRδ(TCRD)链。在另一个实施例中,所述适应性免疫受体异源二聚体的第一多肽为免疫球蛋白重(IGH)链,并且适应性免疫受体异源二聚体的所述第二多肽选自免疫球蛋白轻链IGL或IGK。在其他实施例中,如果适应性免疫受体异源二聚体的第一多肽为IGH链,并且适应性免疫受体异源二聚体的第二多肽为IGL和IGK两者,则使用三种不同的扩增引物组,包括:针对IGH的第一寡核苷酸扩增引物组、针对IGK的第二寡核苷酸扩增引物组以及针对IGL的第三寡核苷酸扩增引物组。附图说明参照下列描述和附图,本发明的这些和其他特征、方面和优点将变得更好理解,其中:图1描绘了某些本文所述的组合物和方法的示意图。U1和U2代表通用接头寡核苷酸。BC1和BC2代表条形码寡核苷酸。J代表适应性免疫受体连接(J)区基因,并且Jpr代表J特异性寡核苷酸引物特异性地退火至其上的这种基因的区域。V代表适应性免疫受体可变(V)区基因,并且Vpr代表V特异性寡核苷酸引物特异性地退火至其上的这种基因的区域。NDN代表存在于一些适应性免疫受体编码基因中的多样性(D)区域,其在任一侧上侧接接合核苷酸(N),所述接合核苷酸可包括非模板核苷酸。Adap1和Adap2代表测序平台特异性接头。示出为“n6”的区段代表任何核苷酸序列的间隔区核苷酸区段,在这种情况下为六个随机选择的核苷酸的间隔区。图2描绘了某些本文所述的组合物和方法的示意图,其中单独的第一和第二微滴接触而允许单个第一和第二微滴之间的融合事件,通过所述融合事件,将来自单独淋巴样细胞(例如,T细胞或B细胞)的DNA在融合的微滴内引入到第一和第二寡核苷酸扩增引物组,所述第一和第二寡核苷酸扩增引物组能够分别扩增来自相同细胞的第一和第二适应性免疫受体多肽编码基因的DNA编码序列(例如,CDR3编码DNA)。因此,如本文所述设想了来自相同细胞的至少两个重排DNA基因座的扩增和寡核苷酸条形码标记,例如[IGH+IGL]、[IGH+IGK]、[IGH+IGK+IGL]、[TCRA+TCRB]、[TCRG+TCRG]等等。图3描绘了某些本文所述的组合物和方法的示例性示意图,根据所述示意图,例如,将来自单独淋巴样细胞(例如,T细胞或B细胞)的DNA或已由单个淋巴样细胞的mRNA逆转录的cDNA在融合的微滴内引入到第一和第二寡核苷酸扩增引物组,所述第一和第二寡核苷酸扩增引物组能够分别扩增来自相同细胞的第一和第二适应性免疫受体多肽编码基因的DNA编码序列(例如,CDR3编码DNA),之后破坏单独的微滴(例如,通过化学、物理和/或机械溶解、解离、破裂等等),并且用通用寡核苷酸引物和测序平台特异性接头扩增释放的带条形码的双链DNA以允许大规模多重定量测序。缩写参见图1的简述。图4描绘了通过使用引导寡核苷酸条形码和通用接头寡核苷酸序列掺入cDNA中的寡核苷酸逆转录引物,在逆转录期间标记适应性免疫受体多肽编码cDNA的示意图。图5描绘了通过使用引导寡核苷酸条形码和通用接头寡核苷酸序列掺入cDNA中的寡核苷酸逆转录引物,在逆转录期间标记适应性免疫受体多肽编码cDNA的示意图。图6给出了适于在用Illumina测序接头修饰扩增的适应性免疫受体多肽编码cDNA后进行测序的DNA产物的示意图,所述扩增的适应性免疫受体多肽编码cDNA已通过使用引导寡核苷酸条形码和通用接头寡核苷酸序列掺入的寡核苷酸逆转录引物,在逆转录期间进行标记。图7示出了占用T细胞亚群的数量与共用亚群的概率的关系。对于使用本发明方法的模拟实验,其中T细胞被分为各包含70,000个T细胞的96个亚群,该曲线图给出了存在于给定数量亚群(x轴)中的任何克隆将出现于完全相同的亚群中的概率(y轴)。图8示出了随机分配至96孔板上的每个孔的固定数量的T细胞(例如,70,000个T细胞),在每个孔中将mRNA提取、转化为cDNA,并通过TCR特异性引物进行扩增。附接孔特异性条形码,并且合并TCR分子以便测序,然后进行计算逆多重化以将每个TCR序列定位回其所来源的孔。免疫组库是高度多样的,并且两个克隆将共用孔模式的概率是极小的,因此可推测共用孔模式的任何TCRA/TCRB对来自于相同克隆。图9A和图9B示出了根据本发明实施例的高通量配对实验的结果。图9A示出了采用96孔和70,000个T细胞/孔的高通量配对实验的假发现率曲线。在FDR<1%(红色虚线)情况下,判定了总计34,763对TCR序列。图9B示出了随孔占用数而变的经测序(浅灰色)和配对(黑色)的TCRB克隆的数量。该插图提供了>20孔中所见的克隆的放大视图。图10示出了根据本发明实施例的高通量配对实验的示例性工作流程。图11示出了根据本发明实施例的孔占用数与配对的TCRB序列的分率的关系。来自采用96孔和70,000个T细胞/孔的高通量配对实验的TCRB克隆的配对收率(配对的序列的分率)。按照其中观察到TCRB克隆的孔的数量(x轴)对TCRB克隆进行分组,并且计算在FDR<1%时配对的这些序列的分率(y轴)。图12示出了根据本发明实施例的双受试者验证实验的示意图。从两名受试者X和Y采集外周血。使用深度免疫测序来表征每名受试者的TCRA和TCRB组库。然后将来自这两名受试者的PBMC混合,并且使用所得的混合物进行本发明的高通量配对实验。真阳性对必须包括来自相同受试者的TCRA和TCRB,而大约一半的假阳性结果将为跨受试者的TCRA/TCRB对。图13A和图13B示出了使用两个供体的高通量配对方法的验证。在图13A中,示出了实验的假发现率曲线,其中将来自两名受试者(‘X’和‘Y’)的PBMC混合并且将25,000个细胞分配到96个孔中的每一个。将各对分为名称为‘X/X’(对的两个成员仅见于受试者X)、‘Y/Y’(对的两个成员仅见于受试者Y)和‘X/Y’(对的一个成员仅见于受试者X并且一个成员仅见于受试者Y)的组。垂直虚线示出了估计FDR<1%的截断值;低于该阈值时,有1,115个X/X对、706个Y/Y对和7个X/Y对。在图13B中,示出了来自样品混合实验的预测的log10FDR与经验log10FDR的关系。对于每个可能的p值截断值,通过本发明的统计方法提供预测的FDR值,并且在假设X/A对代表错误总数的一半的情况下,将经验FDR值计算为X/Y对的数量除以所判定的对的数量的商的两倍。垂直虚线对应于图13A中所用的截断值。图14A至图14D示出了mRNA子样品中及各对内的孔丢失率。图14A示出了由70,000个T细胞/孔的本发明高通量配对方法得出的mRNA的第一子样品中的孔丢失率。中值丢失率(垂直虚线)对于TCRA为20%,而对于TCRB为14%。图14B示出了由本发明高通量配对方法得出的mRNA的第二子样品中的孔丢失率。中值丢失率对于TCRA为24%,而对于TCRB为21%。图14C示出了由mRNA的第一子样品中确信配对的序列(FDR<1%)估计的孔丢失率(与图A中相同的数据)。中值丢失率对于TCRA为14%,而对于TCRB为10%,这些中值丢失率从图A中的估值向下偏倚。图14D示出了由mRNA的全部合并样品中确信配对的序列(FDR<1%)估计的孔丢失率,即,用于我们发现的约35,000对的数据。中值丢失率对于TCRA为7%,而对于TCRB为3%;在考虑偏倚之后,我们预计真实中值丢失率分别为10%和5%。图15示出了随克隆频率和输入T细胞的数量而变的模拟孔占用数。本发明高通量配对方法的关键设计参数是分配至每个孔的T细胞的数量。图15示出了对于96孔板及在100-100,000个细胞/孔范围内的T细胞输入(x轴)而言,本发明的模拟实验的结果。不同颜色描绘了具有不同组库频率的克隆,并且该曲线图示出了这些克隆在我们模拟中占用的孔的数量。为了从不同频带捕获克隆,可以简单地改变实验中输入T细胞的数量。也可以通过改变板上各孔的输入T细胞的数量,而在单个实验中捕获多个频带。具体实施方式在某些实施例中及如本文所述,本发明提供了这样的组合物和方法,所述组合物和方法可用于可靠地定量并确定编码适应性免疫受体诸如免疫球蛋白(IG)和/或T细胞受体(TCR)的重排基因的大而结构多样的群体的序列。这些重排基因可存在于包含来自受试者或生物来源(包括人类受试者)的淋巴样细胞的DNA的生物样品中,和/或这些重排基因的mRNA转录物可存在于这种样品中并且用作通过逆转录进行cDNA合成的模板。本文公开了用于独特且明确地标记单独的、序列不同的IG和TCR编码基因区段或其mRNA转录物或已由此类mRNA转录物逆转录的cDNA的出乎意料有利的方法,这种标记是在通过既有的核酸扩增技术扩增此类基因区段或其转录物(包括逆转录物)的群体的常规步骤之前进行的。不希望受理论的束缚,在用于生成足够数量的DNA拷贝以便测序的通常实行的扩增步骤之前,通过标记如本文所述单独的TCR和IG编码基因区段或其转录物(包括通过逆转录生成的互补DNA),本发明实施例提供了多样TCR和IG编码序列的检测和定量方面前所未有的灵敏度,与此同时避免了在使用扩增引物的序列多样性组由初始样品进行多轮核酸扩增期间可能由于寡核苷酸引物利用偏倚而出现的失实、不准确或不完整结果。另外本文在某些实施例中描述了前所未有的组合物和方法,所述组合物和方法允许定量确定编码来自单个细胞的适应性免疫受体异源二聚体的两个多肽诸如来自T细胞的TCRA和TCRB两者或来自B细胞的IgH和IgL两者的序列。通过提供从复杂样品诸如包含来自受试者的T细胞和/或B细胞的异质混合物的样品获得此类信息的能力,这些和相关实施例允许比以前更准确地确定样品中具体T细胞和/或B细胞克隆群体的相对占比。某些实施例设想了对寡核苷酸引物组进行如本文所述的修饰,所述寡核苷酸引物组用于多重核酸扩增反应,以在此类扩增产物的定量高通量测序之前,由包含编码适应性免疫受体的重排基因的生物样品生成经扩增的重排DNA分子的群体。重排的TCR和BCR编码DNA序列的多重扩增和高通量测序例如在如下文献中有所描述:Robinsetal.,2009Blood114:4099(Robins等人,2009年,《血液》,第114卷,第4099页);Robinsetal.,2010Sci.Translat.Med.2:47ra64(Robins等人,2010年,《科学转化医学》,第2卷,第47ra64页);Robinsetal.,2011J.Immunol.Meth.doi:10.1016/j.jim.2011.09.001(Robins等人,2011年,《免疫法杂志》,doi:10.1016/j.jim.2011.09.001);Sherwoodetal.2011Sei.Translat.Med.3:90ra61(Sherwood等人,2011年,《科学转化医学》,第3卷,第90ra61页);U.S.A.N.13/217,126(美国公布No.2012/0058902)、U.S.A.N.12/794,507(美国公布No.2010/0330571)、WO/2010/151416、WO/2011/106738(PCT/US2011/026373)、WO2012/027503(PCT/US2011/049012)、U.S.A.N.61/550,311以及U.S.A.N.61/569,118;因此,这些公开内容以引用方式并入并且适于根据本文所述的实施例使用。根据某些实施例,在包含多个序列多样性TCR或IG编码基因区段的样品诸如包含来自淋巴样细胞的DNA(或由其转录的mRNA或由这种mRNA逆转录的cDNA)的样品中,其中发生了DNA重排以编码功能性TCR和/或IG异源二聚体(或其中非功能性TCR或IG假基因涉及DNA重排),多个单独的TCR或IG编码序列可各自通过单轮核酸扩增(例如,聚合酶链反应PCR),用如本文所述的特异性寡核苷酸条形码序列进行独特标记。然后可扩增标记的多核苷酸的群体以获得标记的分子的文库,接着可通过现有程序诸如如下文献所述的那些对其进行定量测序:U.S.A.N.13/217,126(美国公布No.2012/0058902)、U.S.A.N.12/794,507(美国公布No.2010/0330571)、WO/2010/151416、WO/2011/106738(PCT/US2011/026373)、WO2012/027503(PCT/US2011/049012)、U.S.A.N.61/550,311以及U.S.A.N.61/569,118。在这些序列读段的过程中,对掺入的条形码标签序列进行测序,并且在收集和分析由此获得的序列数据的过程中,所述序列可用作标识符。在某些实施例中,设想了对于每个条形码标签序列而言,可确定相关TCR或IG序列的共有序列。然后可应用聚类算法以识别由相同初始克隆细胞群体生成的分子。通过这种方法,可以以克服与测序假象相关的不准确性的方式获得高质量的序列数据。图1中描绘了示例性实施例,根据该图,由来自包含淋巴样细胞的群体的基因组DNA或cDNA的起始模板群体,使用寡核苷酸引物组合物进行PCR的两个或更多个循环,所述寡核苷酸引物组合物包含具有如本文所述的通式UI-BIn-X的引物。如图1中所示,J特异性引物110a包含与J区段的一部分互补的J引物序列100、图1中的条形码标签(BCl)101或通式中的B1n,并且还包含第一外部通用接头序列(Ul)102,而V特异性引物110b包含与V区段的一部分互补的V引物序列103和第二外部通用接头序列(U2)104。然而,本发明不必如此所限,并且还设想了相关实施例,诸如这样的实施例,其中条形码可替代地或可除此之外还作为V特异性引物的部分存在,并且位于V序列与第二通用接头之间。应当理解,基于本公开,本领域技术人员可设计其他合适的引物,通过所述引物引入本文所述的条形码标签以独特地标记单独的TCR和/或IG编码基因区段。如本文所述,较大数量(最多至4n,其中n为条形码序列的长度)的不同条形码序列存在于包含具有如本文所述的通式U1-B1n-X的引物的寡核苷酸引物组合物中,使得在适当V特异性引物和J特异性引物的特异性退火后,大量不同扩增事件的PCR产物被有差别地标记。在一些实施例中,条形码序列的数量最多至或少于4n。在一个实施例中,基于长度n=8的条形码,使用一组192个不同条形码序列。条形码“n”的长度决定条形码的可能数量(如本文所述的4n),但在一些实施例中,使用较小亚组,以避开紧密相关的条形码或具有不同退火温度的条形码。在其他实施例中,如本文所述,m和n条形码序列的组用于后续扩增步骤(例如,以单独地标记每个重排TCR或IG序列,然后均匀地标记(“加尾”)一组得自相同来源或样品的序列。在优选实施例中,V引物100和J引物103能够促进TCR或Ig编码序列的扩增,所述TCR或Ig编码序列包括CDR3编码序列,其在图1中包括NDN区111。正如在图1中也指出过的,在不超过两个扩增循环后,第一扩增引物组110a、110b与双链DNA产物分离。通过这个步骤,根据非限制性理论,据信避免了产物制备物被后续几轮扩增所污染,否则污染物可通过用扩增引物扩增新形成的双链DNA分子而产生,所述扩增引物存在于复杂反应体系中,但该引物不同于在最初的一个或两个扩增循环中用于生成双链DNA的引物。用于将双链DNA与寡核苷酸扩增引物分离的多种化学和生物化学技术是本领域已知的。一旦移除第一扩增引物组110a、110b(已通过该引物组引入独特条形码标签序列),就可使用如本文所述和图1中描绘的第二扩增引物组120a、120b扩增标记的双链DNA(dsDNA)产物,以获得适用于测序的DNA文库。第二扩增引物组有利地利用了在前述步骤期间将通用接头序列102、104(例如,图1中的U1和U2)引入dsDNA产物中。因此,由于这些通用接头序列位于图1中的独特条形码标签(BCl)101外部,包含待测序的DNA文库的扩增产物保留与每个具体重排V-J基因区段组合相连的独特条形码标识符序列,同时适于经由通用接头进行扩增。这种第二引物组(也称为“加尾”引物)的示例性组示于表7中。在优选实施例中并且如在图1中也描绘的,第二扩增引物组120a、120b可引入测序平台特异性寡核苷酸序列(图1中的Adap1105和Adap2106),然而这些序列在某些其他相关实施例中不是必要的。第二扩增引物组120a、120b也可任选地引入第二寡核苷酸条形码标识符标签(图1中的BC2107),诸如单个条形码序列,其可有利地识别由具体样品扩增的所有产物(例如,作为来源受试者标识码)并且易于多重分析多个样品而允许更高通量。条形码(BC2;图1中的107)是提高测定法的通量(例如,允许在测序仪上对样品进行多重分析)的修饰,但并非必需。或者,可使用无接头的通用引物来扩增标记的分子。在扩增后,可另外用平台特异性寡核苷酸序列来标记分子。第二样品标识条形码的这种添加可在来自若干不同受试者的样品混合时有利地帮助识别样品起源,或帮助识别一种样品制备物被来自另一种样品制备物的材料意外污染。如图1所示,第二扩增引物组也可任选地包含间隔区核苷酸(“n6”;图1中的108),其可有利于测序平台特异性序列的操作。所述间隔区改善了测序数据的质量,但其在某些实施例中并不需要或并不存在。在所述方法的测序步骤的最初12个循环期间特异性地添加所述间隔区从而增加随机碱基对的数量。通过增加最初12个循环的多样性,改善了聚类定义和碱基判定。间隔区核苷酸108可为任何序列(通常为随机生成的序列)的0、1、2、3、4、5、6、7、8、9、10、11-20、21-30个或更多个核苷酸。在可能值得关注的是此类随机序列的存在将导致包含此类序列的寡核苷酸引物间不均匀的退火速率的情况下,可能优选的是进行相对较少数量的扩增循环,通常三个、四个或五个循环或任选地1-6个或不超过八个循环,以降低可使下游结果出现偏态的扩增不均匀的可能性。然后可根据标准方法并使用如本文所提供且本领域已知的可用仪器,对所得DNA文库进行测序。在存在第二样品标识条形码(图1中的BC2107)的情况下,进行包括读取两个这种条形码的测序,其中也会在样品标识条形码107的两次出现之间读取序列信息(包含CDR3编码序列的V-J接合部,以及独特地标记每个不同序列的第一寡核苷酸条形码BC1101)。例如并且参照图1,测序引物可包含第一读段的NDN111的J侧上的通用引物102,然后是条形码序列BC1101、J引物序列100以及CDR3序列。第二组扩增引物包括正向引物,所述正向引物包含J侧上的平台特异性引物(Adap1105)、含随机核苷酸的间隔区序列(标记为“n6”;图1中的108)以及BC2样品标识条形码107。第二组扩增引物中的反向引物包含NDN111的V侧上的通用引物104、含随机核苷酸的间隔区序列108以及BC2样品标识条形码序列107,以及任选的采用反向第二测序平台特异性引物(Adap2106)的末端配对读段。第二测序平台特异性引物(Adap2106)用于测序并“读取”间隔区序列108、样品标识条形码序列BC2107、通用接头序列104、V序列103以及NDN111。为了捕获CDR3序列,可以使用J扩增引物、C扩增引物或V扩增引物。可使用BC2样品标识条形码107分选序列数据,然后根据包含通用第一条形码BCl101的序列进一步分选。在此类分选的序列内,可使用用于聚类分析的多种已知算法中的任何一种(例如,BLASTClust、UCLUST、CD-HIT或其他,或如Robinsetal.,2009Blood114:4099(Robins等人,2009年,《血液》,第114卷,第4099页)中所述)对CDR3序列进行聚类分析,以确定是否存在不止一种序列聚类。除此之外或作为另外一种选择,可基于那些被发现至少两次的序列来分选和选择序列数据。然后可通过序列比较(例如以修正测序错误)来确定共有序列。在其他方面共用通用共有序列的序列间检测到多个独特标识符条形码标签(BC1101)的情况下,识别出的此类条形码标签的数量可被视为反映了来自相同T细胞或B细胞克隆的样品中的分子数量。识别来自单个适应性免疫细胞的TCR或IG异源二聚体的两条链如同在上文也指出的,在某些其他实施例中,本文提供了一种用于确定编码单个淋巴样细胞中适应性免疫受体异源二聚体的第一和第二多肽序列的重排DNA序列(或由其转录的mRNA序列或已由这种mRNA逆转录的cDNA)的方法。所述方法包括用独特条形码序列独特地标记每个重排DNA序列以便识别具体细胞和/或样品。简而言之,并且以举例说明而非限制的方式,这些和相关实施例包括一种方法,所述方法包括如下步骤:(1)在多个平行反应中的每一个中,使第一和第二微滴接触,并且允许它们在容许核酸扩增的条件下融合,以生成双链DNA产物(或单链cDNA产物),这些产物全都包含相同条形码寡核苷酸序列并且对应于适应性免疫受体异源二聚体的两条链;(2)破坏融合的微滴以获得双链(或单链)DNA产物的异质混合物;(3)扩增双链DNA(或单链)产物的异质混合物以获得用于测序的DNA文库;以及(4)对文库进行测序以获得编码异源二聚体的第一和第二多肽的DNA序列的数据集。所述方法包括使如下两者接触并允许以成对方式融合:(A)单独第一微滴,所述单独第一微滴各自(或每第n小滴中m个)包含单个淋巴样细胞或从其分离的基因组DNA,或已由mRNA逆转录为cDNA,与(B)来自多个第二液体微滴的单独第二微滴,所述单独第二微滴各自包含两个寡核苷酸扩增引物组,第一组用于扩增编码适应性免疫受体异源二聚体的第一链(例如,IGH链或TCRA链)的任何重排DNA,并且第二组用于扩增编码异源二聚体的第二链(例如,IGL链或TCRB链)的任何重排DNA。明显地,在一个给定的第二微滴中,所有寡核苷酸扩增引物将包含相同的条形码寡核苷酸,但在不同的第二微滴内,引物组将包含不同的条形码序列。所述接触的步骤受到控制,使得在多个事件的每一个中,单个第一微滴与单个第二微滴融合而获得融合的微滴。第一微滴和第二微滴每者的内容物在融合的微滴中彼此接触。能够扩增编码给定TCR或IG多肽的任何重排DNA的寡核苷酸扩增引物组描述于本文别处及就此类公开内容并入的参考文献中。熟悉本领域的技术人员将会知道可借助其来制备具有限定内容物和性质(诸如能可控制地发生融合的能力)的微滴组合物的多种微流体设备和装置中的任何一种,诸如RainDanceTM微滴数字PCR系统(马萨诸塞州列克星敦的雷恩丹斯技术公司(RainDanceTechnologies,Lexington,MA))或例如如下文献中所述的系统中的任何一种:Pekinetal.,2011LabChip11:2156(Pekin等人,2011年,《芯片实验室》,第11卷,第2156页);Milleretal.,2012Proc.Nat.Acad.Sci.USA109:378(Miller等人,2012年,《美国国家科学院院刊》,第109卷,第378页);Brouzesetal.,2009Proc.Nat.Acad.Sci.USA106:14195(Brouzes等人,2009年,《美国国家科学院院刊》,第106卷,第14195页);Joenssonetal.,2009Angew.Chem.Int.Ed.81:4813(Joensson等人,2009年,《应用化学国际版》,第81卷,第4813页);Baretetal.,2009LabChip9:1850(Baret等人,2009年,《芯片实验室》,第9卷,第1850页);Frenzetal.,2009LabChip9:1344(Frenz等人,2009年,《芯片实验室》,第9卷,第1344页);Kissetal.,2008Anal.Chem.80:8975(Kiss等人,2008年,《分析化学》,第80卷,第8975页);Leamonetal.,2006Nat.Meths.3:541(Leamon等人,2006年,《自然方法》,第3卷,第541页);这些文献可通过从本公开看来是常规的修改而适于具体方法,诸如本文所述的那些方法。作为非限制性例子,某些实施例可利用使用微流体通道分散在油相中的水相微滴的性质。微滴可为油包水乳液、水包油乳液或者类似的水性和非水性乳液组合物。微滴也可称为微滴或胶束微滴。常规油包水(WO)乳液在生物学方面存在许多应用,包括下一代测序(Marguliesetal.,Nature2005,437,376-380(Margulies等人,《自然》,2005年,第437卷,第376-380页))、稀有突变检测(Diehl,F.etal.Proc.Natl.Acad.Sei.U.S.A.2005,102,16368-16373(Diehl,F.等人,《美国国家科学院院刊》,2005年,第102卷,第16368-16373页);Li,M.etal.,Nat.Methods2006,3,95-97(Li,M.等人,《自然方法》,2006年,第3卷,第95-97页);Diehl,F.etal.,Nat.Med.2008,14,985-990(Diehl,F.等人,《自然医学》,2008年,第14卷,第985-990页))和DNA甲基化的定量检测(Li,M.etal.,NatBiotechnol2009,27,858-U118(Li,M.等人,《自然生物技术》,2009年,第27卷,第858-U118页)),但在用于形成乳液的机械搅拌期间这些乳液会遭受可使细胞破坏的小滴多分散性和剪切应力。微流体的使用克服了这些限制并且得到了基于生物化学和细胞的测定法的改善性能(Zeng,Y.etal,Anal.Chem.2010,82,3183-3190(Zeng,Y.等人,《分析化学》,2010年,第82卷,第3183-3190页))。具有10-100μm的通道直径的微流体芯片通常使用标准软光刻技术由石英、硅、玻璃或聚二甲基硅氧烷(PDMS)制成(A.Manz,N.GraberandH.M.Widmer:MiniaturizedtotalChemicalAnalysissystems:ANovelConceptforChemicalSensing,SensorsandActuators,BChemical(1990)244-248(A.Manz、N.Graber和H.M.Widmer,微型全化学分析系统:一种化学传感的新概念,《传感器与致动器,B化学》,1990年,第244-248页))。通常通过使一个通道中的水溶液流入油流中而以约1-10Hz的频率生成小滴。流动聚焦喷嘴的使用使得能够生成水相的受控尺寸小滴。对于给定喷嘴几何形状而言,小滴尺寸和小滴生成速率受到油相流速与水相流速的比率的控制。芯片通道表面通常被改性为疏水性的,例如通过许多已公布的硅烷化化学反应之一进行改性(Zeng,Y.etal.,Anal.Chem.2010,82,3183-3190(Zeng,Y.等人,《分析化学》,2010年,第82卷,第3183-3190页))。对于作为完全功能性的微容器的小滴,疏水和疏脂的油的使用可能是有利的,因为小滴之间的分子扩散被最小化,油具有对于水相中所含的生物试剂的低溶解度,并且具有良好气体溶解度,这确保了在某些应用中包封的细胞的活力。另外,根据某些实施例,表面活性剂可有利地混合于油相中,因为小滴趋于聚结。表面活性剂也可抑制生物分子吸附在微滴界面处。已描述了一种新型类别的嵌段共聚物表面活性剂,包括偶联到聚乙二醇(PEG)的全氟聚醚(PFPE),以便与氟碳油一起使用,所述氟碳油例如为氟化油FC-40(西格玛公司(Sigma)),即全氟三正丁胺与二(全氟(正丁基))全氟甲胺的混合物(Holtze,C.etal.,LabChip,2008(Holtze,C.等人,《芯片实验室》,2008年),DOI:10.1039/b806706f)。这些组合物已得到了非常稳定的生物相容性乳液(Brouzes,E.,etal.,PNAS2009,106(34),14195-14200(Brouzes,E.等人,《美国国家科学院院刊》,2009年,第106卷,第34期,第14195-14200页))。在微流体通道中行进的小滴可借助其表面张力保持为离散微滴。也已提出了各种方法以在需要时克服表面张力并允许小滴合并,从而允许试剂混合,例如,通过如下方式实现:在通道中微加工被动阻流元件(Niu,X.etal.,LabChip2008,8,1837-1841(Niu,X.等人,《芯片实验室》,2008年,第8卷,第1837-1841页))、使用静电荷(电聚结)(Zagnoni,M.etal.,Langmuir,2010,26(18),14443-14449(Zagnoni,M.等人,《朗缪尔》,2010年,第26卷,第18期,第14443-14449页))或操纵微通道几何形状(DolomiteMerger芯片;另参见WO/2012/083225)。经由皮升注射器(通过电场操作的垂直于小滴通道的加压试剂填充通道)将试剂加入微流体通道中的小滴的方法最近已有所公布(Abate,A.R.etal.,PNAS2010,107(45),19163-19166(Abate,A.R.等人,《美国国家科学院院刊》,2010年,第107卷,第45期,第19163-19166页))并且也可根据如本文所述的某些本发明设想的实施例进行修改。微滴内容物及接触的步骤被选择为容许基因组DNA与扩增引物之间的核酸扩增相互作用。核酸扩增(例如,PCR)试剂和条件是熟知的。这种扩增被允许至少进行到获得第一和第二双链DNA产物,所述产物包含如本文提供的第一和第二寡核苷酸扩增引物的核苷酸序列及其互补序列。因此,例如,任何单个融合的微滴可包含(i)第一双链DNA产物,所述第一双链DNA产物至少包含第一通用接头序列、条形码序列、编码异源二聚体的第一适应性免疫受体多肽的一部分的V区和J区或C区序列,以及第二通用接头序列,和(ii)第二双链DNA产物,所述第二双链DNA产物至少包含第三通用接头序列、与(i)中相同的条形码序列、编码异源二聚体的第二适应性免疫受体多肽的一部分的V区和J区或C区序列,以及第四通用接头序列。在下一步骤之前停止在融合的微滴中用于扩增步骤的条件。这可通过如下实现:改变其中含有微滴(例如,包含于容器或孔中)的环境的温度以停止扩增过程。在一些实施例中,所述方法包括破坏所述多个融合的微滴以获得第一和第二双链产物的异质混合物。破坏可基于微滴的化学性质和组成来选择,并且可例如通过化学、生物化学和/或物理操纵来实现,诸如引入稀释剂、清洁剂、离液剂、表面活性剂、渗透剂或其他化学试剂,或者使用超声处理、压力、电场或其他破坏条件。应当理解,优选的条件将涉及将水性溶剂用于微滴内所含的体积和/或用于通过破坏步骤获得的异质混合物。通过使用微滴而非单独细胞作为测定法格式,可以分析有关样品中的输入细胞的数量的数据。可以修正PCR和测序错误,并且就IG分子而言,可以区分因体细胞高频突变(SHM)而致的非生殖系序列与因PCR错误引入的非生殖系序列。在一些实施例中,所述方法包括用于使第一和第二双链DNA产物的混合物与本文所述的第三和第四扩增引物组接触的后续步骤。用于该步骤的条件可以类似地使用用于DNA扩增以获得测序用的DNA文库的公认方法来实现,其也可根据多种既有DNA测序技术中的任何一种来实现。在某些相关实施例中,与使用各自包含单个淋巴样细胞或从其分离的基因组DNA的第一液体微滴相反,第一液体微滴每者包含已由单个淋巴样细胞的mRNA逆转录的互补DNA(cDNA),诸如编码适应性免疫受体异源二聚体的第一链的第一cDNA以及编码异源二聚体的第二链的第二cDNA。在某些相关实施例中,单独第二微滴可各自包含第三寡核苷酸引物组,所述第三寡核苷酸引物组能够扩增编码一个或多个淋巴细胞状态指示分子的另外cDNA序列,用存在于处于微滴中的第一和第二引物组中的相同条形码序列标记第三引物组。在此类实施例中,对于从其识别给定TCR或IG异源二聚体序列的单个来源细胞而言,可确定生物状态。生物状态可为活化与静止、成熟状态,初始与记忆,调节与效应子等等。示例性淋巴细胞状态指示分子包括例如lck、fyn、FoxP3、CD4、CD8、CDlla、CD18、CD25、CD28、CD29、CD44、CD45、CD49d、CD62、CD69、CD71、CD103、CD137(4-1BB)、HLA-DR等等。某些实施例包括第三寡核苷酸引物组,所述第三寡核苷酸引物组能够扩增编码淋巴细胞状态指示分子的第三cDNA序列,其中用存在于第一和第二引物组中的相同条形码序列标记第三寡核苷酸引物组,并且其中淋巴细胞状态指示分子包括如下的一者或多者:FoxP3、CD4、CD8、CDlla、CD18、CD21、CD25、CD29、CCD30、CD38、CD44、CD45、CD45RA、CD45RO、CD49d、CD62、CD62L、CD69、CD71、CD103、CD137(4-1BB)、CD138、CD161、CD294、CCR5、CXCR4、IgGl-4H链恒定区、IgAH链恒定区、IgEH链恒定区、IgDH链恒定区、IgMH链恒定区、HLA-DR、IL-2、IL-5、IL-6、IL-9、IL-10、IL-12、IL-13、IL-15、IL-21、TGF-β、TLRl、TLR2、TLR3、TLR4、TLR5、TLR6、TLR7、TLR8、TLR9以及TLR10。表1:示例性淋巴细胞状态指示分子然而,这些和相关实施例不必如此所限,使得还有这样的设想实施例,根据所述实施例,除此之外或作为另外一种选择,可包括第三寡核苷酸引物组,所述第三寡核苷酸引物组能够扩增编码淋巴细胞状态指示分子的第三cDNA序列,其中用存在于第一和第二引物组中的相同条形码序列标记第三引物组,并且其中淋巴细胞状态指示分子包含细胞表面受体。细胞表面受体的例子包括下列受体等等:CD2(例如,GenBank登录号Y00023、SEG_HUMCD2、M16336、M16445、SEG_MUSCD2、M14362)、4-1BB(CDw137,Kwonetal,1989Proc.Nat.Acad.Sci.USA86:1963(Kwon等人,1989年,《美国国家科学院院刊》,第86卷,第1963页),4-1BB配体(Goodwinetal,1993Eur.J.Immunol.23:2361(Goodwin等人,1993年,《欧洲免疫学杂志》,第23卷,第2361页);Meleroetal,1998Eur.J.Immunol.3:116(Melero等人,1998年,《欧洲免疫学杂志》,第3卷,第116页))、CD5(例如,GenBank登录号X78985、X89405)、CD10(例如,GenBank登录号M81591、X76732)、CD27(例如,GenBank登录号M63928、L24495、L08096)、CD28(Juneetal,1990Immunol.Today11:211(June等人,1990年,《今日免疫学》,第11卷,第211页);另参见例如,GenBank登录号J02988、SEG_HUMCD28、M34563)、CD152/CTLA-4(例如,GenBank登录号L15006、X05719、SEG-HUMIGCTL)、CD40(例如,GenBank登录号M83312、SEGMUSC040A0、Y10507、X67878、X96710、U15637、L07414)、干扰素-γ(IFN-γ;参见例如,Farraretal.1993Ann.Rev.Immunol.11:571(Farrar等人,1993年,《免疫学年鉴》,第11卷,第571页)及其中引用的文献,Grayetal.1982Nature295:503(Gray等人,1982年,《自然》,第295卷,第503页),Rinderknechtetal.1984J.Biol.Chem.259:6790(Rinderknecht等人,1984年,《生物化学杂志》,第259卷,第6790页),DeGradoetal.1982Nature300:379(DeGrado等人,1982年,《自然》,第300卷,第379页))、白介素-4(IL-4;参见例如,53rdForuminImmunology,1993ResearchinImmunol.144:553-643(第53届免疫学论坛,1993年,《免疫学研究》,第144卷,第553-643页);Banchereauetal.,1994inTheCytokineHandbook,2nded.,A.Thomson,ed.,AcademiePress,NY,p.99(Banchereau等人,1994年,载于《细胞因子手册》,第2版,A.Thomson编辑,纽约学术出版社,第99页);Keeganetal.,1994JLeukocyt.Biol..55:272(Keegan等人,1994年,《白细胞生物学杂志》,第55卷,第272页)及其中引用的文献)、白介素-17(IL-17)(例如,GenBank登录号U32659、U43088)以及白介素-17受体(IL-17R)(例如,GenBank登录号U31993、U58917)。另外的细胞表面受体包括下列受体等等:CD59(例如,GenBank登录号SEG_HUMCD590、M95708、M34671)、CD48(例如,GenBank登录号M59904)、CD58/LFA-3(例如,GenBank登录号A25933、Y00636、E12817;另参见JP1997075090-A)、CD72(例如,GenBank登录号AA311036、S40777、L35772)、CD70(例如,GenBank登录号Y13636、S69339)、CD80/B7.1(Freemanetal,1989J.Immunol.43:2714(Freeman等人,1989年7月,《免疫学杂志》,第43卷,第2714页);Freemanetal,1991J.Exp.Med.174:625(Freeman等人,1991年,《实验医学杂志》,第174卷,第625页);另参见例如,GenBank登录号U33208、1683379)、CD86/B7.2(Freemanetal,1993J.Exp.Med.178:2185(Freeman等人,1993年,《实验医学杂志》,第178卷,第2185页),Borielloetal,1995J.Immunol.155:5490(Boriello等人,1995年,《免疫学杂志》,第155卷,第5490页);另参见例如,GenBank登录号AF099105、SEG_MMB72G、U39466、U04343、SEG_HSB725、L25606、L25259)、B7-H1/B7-DC(例如,Genbank登录号NM014143、AF177937、AF317088;Dongetal,2002Nat.Med.Jun24(Dong等人,2002年,《自然医学》,6月24日)[刊载之前的电子版],PMID12091876;Tsengetal,2001J.Exp.Med.193:839(Tseng等人,2001年,《实验医学杂志》,第193卷,第839页);Tamuraetal,2001Blood97:1809(Tamura等人,2001年,《血液》,第97卷,第1809页);Dongetal,1999Nat.Med.5:1365(Dong等人,1999年,《自然医学》,第5卷,第1365页))、CD40配体(例如,GenBank登录号SEGHUMCD40L、X67878、X65453、L07414)、IL-17(例如,GenBank登录号U32659、U43088)、CD43(例如,GenBank登录号X52075、J04536)、ICOS(例如,Genbank登录号AH011568)、CD3(例如,Genbank登录号NM_000073(γ亚基)、NM_000733(ε亚基)、X73617(δ亚基))、CD4(例如,Genbank登录号NM000616)、CD25(例如,Genbank登录号NM_000417)、CD8(例如,Genbank登录号M12828)、CD11b(例如,Genbank登录号J03925)、CD14(例如,Genbank登录号XM039364)、CD56(例如,Genbank登录号U63041)、CD69(例如,Genbank登录号NM001781)以及VLA-4(α4β7)(例如,GenBank登录号L12002、X16983、L20788、U97031、L24913、M68892、M95632)。以下细胞表面受体通常与B细胞相关:CD19(例如,GenBank登录号SEG_HUMCD19W0、M84371、SEG_MUSCD19W、M62542)、CD20(例如,GenBank登录号SEGHUMCD20、M62541)、CD22(例如,GenBank登录号1680629、Y10210、X59350、U62631、X52782、L16928)、CD30(例如,Genbank登录号M83554、D86042)、CD153(CD30配体,例如,GenBank登录号L09753、M83554)、CD37(例如,GenBank登录号SEG_MMCD37X、X14046、X53517)、CD50(ICAM-3,例如,GenBank登录号NM002162)、CD106(VCAM-1)(例如,GenBank登录号X53051、X67783、SEG_MMVCAM1C,另参见美国专利No.5,596,090)、CD54(ICAM-1)(例如,GenBank登录号X84737、S82847、X06990、J03132、SEG_MUSICAM0)、白介素-12(参见例如Reiteretal,1993Crit.Rev.Immunol.13:1(Reiter等人,1993年,《免疫学评论》,第13卷,第1页)及其中引用的文献)、CD134(0X40,例如,GenBank登录号AJ277151)、CD137(41BB,例如,GenBank登录号L12964、NM_001561)、CD83(例如,GenBank登录号AF001036、AL021918)、DEC-205(例如,GenBank登录号AF011333、U19271)。其他细胞表面受体的例子包括下列受体等等:HER1(例如,GenBank登录号U48722、SEG_HEGFREXS、KO3193)、HER2(Yoshinoetal.,1994J.Immunol.152:2393(Yoshino等人,1994年,《免疫学杂志》,第152卷,第2393页);Disisetal.,1994Canc.Res.54:16(Disis等人,1994年,《癌症研究》,第54卷,第16页);另参见例如,GenBank登录号X03363、M17730、SEGHUMHER20)、HER3(例如,GenBank登录号U29339、M34309)、HER4(Plowmanetal.,1993Nature366:473(Plowman等人,1993年,《自然》,第366卷,第473页);另参见例如GenBank登录号L07868、T64105)、表皮生长因子受体(EGFR)(例如,GenBank登录号U48722、SEGHEGFREXS、KO3193)、血管内皮细胞生长因子(例如,GenBank号M32977)、血管内皮细胞生长因子受体(例如,GenBank登录号AF022375、1680143、U48801、X62568)、胰岛素样生长因子-I(例如,GenBank登录号X00173、X56774、X56773、X06043,另参见欧洲专利No.GB2241703)、胰岛素样生长因子-II(例如,GenBank登录号X03562、X00910、SEG_HUMGFIA、SEG_HUMGFI2、M17863、M17862)、转铁蛋白受体(TrowbridgeandOmary,1981Proc.Nat.Acad.USA78:3039(Trowbridge和Omary,1981年,《美国国家科学院院刊》,第78卷,第3039页);另参见例如,GenBank登录号X01060、Ml1507)、雌激素受体(例如,GenBank登录号M38651、X03635、X99101、U47678、M12674)、孕酮受体(例如,GenBank登录号X51730、X69068、M15716)、卵泡刺激素受体(FSH-R)(例如,GenBank登录号Z34260、M65085)、视黄酸受体(例如,GenBank登录号L12060、M60909、X77664、X57280、X07282、X06538)、MUC-1(Barnesetal.,1989Proc.Nat.Acad.Sci.USA86:7159(Barnes等人,1989年,《美国国家科学院院刊》,第86卷,第7159页);另参见例如,GenBank登录号SEGMUSMUCIO、M65132、M64928)、NY-ESO-1(例如,GenBank登录号AJ003149、U87459)、NA17-A(例如,欧洲专利No.WO96/40039)、Melan-A/MART-1(Kawakamietal.,1994Proc.Nat.Acad.Sci.USA91:3515(Kawakami等人,1994年,《美国国家科学院院刊》,第91卷,第3515页);另参见例如,GenBank登录号U06654、U06452)、酪氨酸酶(Topalianetal.,1994Proc.Nat.Acad.Sci.USA91:9461(Topalian等人,1994年,《美国国家科学院院刊》,第91卷,第9461页);另参见例如,GenBank登录号M26729、SEG_HUMTYRO,另参见Weberetal.,J.Clin.Invest(1998)102:1258(Weber等人,《临床研究杂志》,1998年,第102卷,第1258页))、Gp-100(Kawakamietal.,1994Proc.Nat.Acad.Sci.USA91:3515(Kawakami等人,1994年,《美国国家科学院院刊》,第91卷,第3515页);另参见例如,GenBank登录号S73003,另参见欧洲专利No.EP668350;Ademaetal.,1994J.Biol.Chem.269:20126(Adema等人,1994年,《生物化学杂志》,第269卷,第20126页))、MAGE(vandenBruggenetal.,1991Science254:1643(vandenBruggen等人,1991年,《科学》,第254卷,第1643页);另参见例如,GenBank登录号U93163、AF064589、U66083、D32077、D32076、D32075、U10694、U10693、U10691、U10690、U10689、U10688、U10687、U10686、U10685、L18877、U10340、U10339、L18920、U03735、M77481)、BAGE(例如,GenBank登录号U19180,另参见美国专利No.5,683,886和5,571,711)、GAGE(例如,GenBank登录号AF055475、AF055474、AF055473、U19147、U19146、U19145、U19144、U19143、Ul9142)、CTA类别的受体中的任何一种,尤其包括由SSX2基因编码的HOM-MEL-40抗原(例如,GenBank登录号X86175、U90842、U90841、X86174)、癌胚抗原(CEA,GoldandFreedman,1985J.Exp.Med.121:439(Gold和Freedman,1985年,《实验医学杂志》,第121卷,第439页);另参见例如,GenBank登录号SEGHUMCEA、M59710、M59255、M29540)以及PyLT(例如,GenBank登录号J02289、J02038)。淋巴细胞状态指示分子还可包括一种或多种细胞凋亡信号传导多肽,所述多肽的序列是本领域已知的,如例如以下文献中所综述的:WhenCellsDie:AComprehensiveEvaluationofApoptosisandProgrammedCellDeath(R.A.Lockshinetal,Eds.,1998JohnWiley&Sons,NewYork(当细胞死亡时:细胞凋亡与程序性细胞死亡的综合评价,R.A.Lockshin等人编辑,1998年,约翰·威立父子出版公司,纽约);另参见例如,Greenetal,1998Science281:1309(Green等人,1998年,《科学》,第281卷,第1309页)及其中引用的文献;Ferreiraetal,2002Clin.Cane.Res.8:2024(Ferreira等人,2002年,《临床癌症研究》,第8卷,第2024页);Gurumurthyetal,2001CancerMetastas.Rev.20:225(Gurumurthy等人,2001年,《癌与转移评论》,第20卷,第225页);Kanducetal,2002Int.J.Oncol.21:165(Kanduc等人,2002年,《国际肿瘤学杂志》,第21卷,第165页))。通常,细胞凋亡信号传导多肽序列包含受体死亡结构域多肽的全部或一部分或衍生自受体死亡结构域多肽,所述受体死亡结构域多肽例如为,FADD(例如,Genbank登录号U24231、U43184、AF009616、AF009617、NM_012115)、TRADD(例如,Genbank登录号NM003789)、RAIDD(例如,Genbank登录号U87229)、CD95(FAS/Apo-1;例如,Genbank登录号X89101、NM003824、AF344850、AF344856)、TNF-α-受体-l(TNFR1,例如,Genbank登录号S63368、AF040257)、DR5(例如,Genbank登录号AF020501、AF016268、AF012535)、ITIM结构域(例如,Genbank登录号AF081675、BC0l5731、NM006840、NM006844、NM006847、XM017977;参见例如,Billadeauetal.,2002J.Clin.Invest.109:161(Billadeau等人,2002年,《临床研究杂志》,第109卷,第161页))、ITAM结构域(例如,Genbank登录号NM005843、NM003473、BC030586;参见例如,Billadeau等人,2002年)或本领域已知的其他细胞凋亡相关的受体死亡结构域多肽,例如,TNFR2(例如,Genbank登录号L49431、L49432)、半胱天冬酶/半胱天冬酶原-3(例如,Genbank登录号XM_54686)、半胱天冬酶/半胱天冬酶原-8(例如,AF380342、NM_004208、NM_001228、NM033355、NM033356、NM033357、NM033358)、半胱天冬酶/半胱天冬酶原-2(例如,Genbank登录号AF314174、AF314175)等等。可检查生物样品中疑似发生细胞凋亡的细胞的形态学变化、渗透性变化、生物化学变化、分子遗传学变化或其他变化,这些变化对于熟悉本领域的技术人员而言是显而易见的。这些和相关方法首次允许快速确定编码来自单个细胞的TCR或IG异源二聚体的两条链的重排DNA序列。此类实施例由于允许对来自多个单一细胞每者的适应性免疫受体编码序列进行高通量测序而将可用于诊断和预后目的,并且通常也将为TCR或IG异源二聚体配对及其内在分子机制的免疫学研究提供信息。大量TCR和/或IG异源二聚体的两个亚基的DNA序列信息的快速且大规模的可用性将加速合成抗体技术及相关技术的开发,例如,其中通常可将抗体或者完全或部分TCR或IG抗原结合区工程改造为诊断组合物、治疗组合物、仿生组合物、酶或催化组合物(例如,抗体酶)或其他工业上有用的组合物。借助于本公开所提供的高通量TCR和/或IG测序的定量性质,存在于样品中的TCR和/或IG异源二聚体序列的定量表征的高精度将有利地改善确定属于特异性T细胞或B细胞克隆的细胞的数量的能力。如上文所指出,根据用于识别来自单个适应性免疫细胞的TCR或IG异源二聚体的两条链的这些实施例,在任何给定的第二微滴中,所有寡核苷酸扩增引物将包含相同的条形码寡核苷酸,但在不同的第二微滴内,引物组将包含不同的条形码序列。因此,在对如上文所述获得的DNA文库进行测序以获得序列的数据集之后,可将数据集中的序列分选为具有相同条形码序列的序列的组,并且可将此类条形码组进一步分选为具有X1序列或X2序列(其包含V区和J区或C区的部分)的那些组,所述X1序列或X2序列将指示给定序列反映的是第一TCR或IG编码链(例如,TCRA或IGH链)还是第二TCR或IG编码链(例如,TCRB或IGL链)的扩增产物。已通过条形码或通过TCR或IG链分选的序列可进一步使用用于聚类分析的多种已知算法中的任何一种(例如,BLASTClust、UCLUST、CD-HIT,另参见IEEERevBiomedEng.2010;3:120-54.doi:10.1109/RBME.2010.2083647(《IEEE生物医学工程评论》,2010年,第3卷,第120-154页,doi:10.1109/RBME.2010.2083647);Clusteringalgorithmsinbiomedicalresearch:areview,XuR,WunschDC2nd;MolBiotechnol.2005Sep;31(1):55-80(生物医学研究中的聚类算法:综述,XuR、WunschDC(第二作者);《分子生物技术》,2005年9月,第31卷,第1期,第55-80页);Dataclusteringinlifesciences.ZhaoY,KarypisG;MethodsMolBiol.2010;593:81-107.doi:10.1007/978-1-60327-194-3_5(生命科学中的数据聚类分析,ZhaoY、KarypisG;《分子生物学方法》,2010年,第593卷,第81-107页,doi:10.1007/978-1-60327-194-3_5);Overviewontechniquesinclusteranalysis.FradesI,MatthiesenR(有关聚类分析的技术的概述,FradesI、MatthiesenR)以及就如下序列而言的错误修正来进行聚类分析,所述序列无法与具有共用条形码序列的其他序列聚类,而相反地将与具有相差单个核苷酸的条形码的序列聚类。参见例如ProcNatlAcadSciUSA.2012Jan24;109(4):1347-52.doi:10.1073/pnas.1118018109.Epub2012Jan9(《美国国家科学院院刊》,2012年1月24日,第109卷,第4期,第1347-1352页,doi:10.1073/pnas.1118018109,电子版2012年1月9日)。数字RNA测序使序列依赖性偏倚和扩增噪声最小化,且具有优化的单分子条形码。ShiroguchiK,JiaTZ,SimsPA,XieXS;ProcNatlAcadSciUSA.2012Sep4;109(36):14508-13.doi:10.1073/pnas.1208715109.Epub2012Aug1(ShiroguchiK、JiaTZ、SimsPA、XieXS;《美国国家科学院院刊》,2012年9月4日;第109卷,第36期,第14508-14513页,doi:10.1073/pnas.1208715109,电子版2012年8月1日)。Detectionofultra-raremutationsbynext-generationsequencing.SchmittMW,KennedySR,SalkJJ,FoxEJ,HiattJB,LoebLA(通过下一代测序来检测超罕见突变,SchmittMW、KennedySR、SalkJJ、FoxEJ、HiattJB、LoebLA)。因此,某些实施例包括一种方法,所述方法包括如下步骤:(a)根据其中识别出的寡核苷酸条形码序列来分选序列(如上文所述获得)的数据集,以获得多个条形码序列集合,每个条形码序列集合具有独特条形码;(b)将(a)的每个条形码序列集合分选为含X1序列的子集和含X2序列的子集;(c)根据X1序列和X2序列对含X1序列和含X2序列的子集每者的成员进行聚类分析,以分别获得一个或多个X1序列聚类集合和一个或多个X2序列聚类集合,及对所述X1和X2序列聚类集合中的任何一个或多个内的单个核苷酸条形码序列错配进行错误修正;以及(d)识别为源自相同细胞序列,所述相同细胞序列为属于相同一个或多个条形码序列集合的X1和X2序列聚类集合的成员。应当理解,根据非限制性理论,与条形码序列相同集合一同出现的第一和第二适应性免疫受体链编码序列具有极其高的概率源自相同的融合微滴,从而来自相同来源细胞。例如,在104个不同条形码用于构建第一和第二寡核苷酸扩增引物的情况下,将获得具有相同条形码序列的两个独立的(即,源自不同细胞)双链第一和第二产物的概率为108分之一。因此,如果根据本文所述的方法,第一和第二适应性免疫受体多肽编码序列(例如,X1和X2)的给定集合的三个或更多个拷贝共用通用条形码序列(例如,属于相同条形码序列集合),则序列具有独立细胞起源的概率接近零。相似地,应当理解,根据本发明方法获得的序列的数据集的分析也可用于表征基因组DNA的淋巴样细胞来源的生物状态。例如,由于在B细胞中已知IGH基因重排先于IGL基因重排,如本文所述的条形码序列分析可揭示多个单一淋巴样细胞基因组具有相同重排IGH序列但具有不同IGL序列,指示了这些序列在免疫初始细胞中的起源。或者,所述分析可利用这样的观察结果:T细胞表达对于其功能具有特异性的蛋白质,诸如如本文所述的淋巴细胞状态指示分子。例如,调节性T细胞表达蛋白质FOXP3。如果对已由T细胞mRNA逆转录的cDNA进行后续扩增,则共扩增产物可包括反映编码表型特异性蛋白质诸如FOXP3的其他mRNA的cDNA物质,以及编码TCRB和TCRA分子的cDNA。该方法可允许识别由具有特异性表型的T细胞诸如T调节细胞或效应T细胞表达的适应性免疫受体。因此,本文提供了一种用于确定编码单个淋巴样细胞中适应性免疫受体异源二聚体的第一和第二多肽序列的重排DNA序列的方法,所述方法包括(1)使如下两者接触:(A)单独第一微滴,所述单独第一微滴各自包含单个淋巴样细胞或从其分离的基因组DNA,与(B)来自多个第二液体微滴的单独第二微滴,所述单独第二微滴各自包含(i)第一寡核苷酸扩增引物组,所述第一寡核苷酸扩增引物组能够扩增编码适应性免疫受体异源二聚体的第一多肽的第一互补性决定区-3(CDR3)的重排DNA序列,和(ii)第二寡核苷酸扩增引物组,所述第二寡核苷酸扩增引物组能够扩增编码适应性免疫受体异源二聚体的第二多肽的第二互补性决定区-3(CDR3)的重排DNA序列。第一寡核苷酸扩增引物组包括包含具有如下通式的多个寡核苷酸序列的多个寡核苷酸的组合物:U1/2-B1-X1,其中U1/2包含这样的寡核苷酸,当B1存在时其包含第一通用接头寡核苷酸序列,或者当没有B1时其包含第二通用接头寡核苷酸序列。在一些实施例中,B1包含没有序列或具有6、7、8、9、10、11、12、13、14、15、16、17、18、19或20个连续核苷酸的第一寡核苷酸条形码序列的寡核苷酸,并且Xl包含如下之一的寡核苷酸:(a)多核苷酸,其包含适应性免疫受体异源二聚体的所述第一多肽的适应性免疫受体可变(V)区编码基因序列或者其互补序列的至少20、30、40或50个并且不超过100、90、80、70或60个连续核苷酸,并且在通式U1/2-B1-X1的所述多个寡核苷酸序列每者中,X1包含独特寡核苷酸序列,以及(b)多核苷酸,其包含如下任一者的至少15-30或31-50个并且不超过80、70、60或55个连续核苷酸:(i)适应性免疫受体异源二聚体的所述第一多肽的适应性免疫受体连接(J)区编码基因序列或者其互补序列,或者(ii)适应性免疫受体异源二聚体的所述第一多肽的适应性免疫受体恒定(C)区编码基因序列或者其互补序列,并且在通式U1/2-B1-X1的所述多个寡核苷酸序列每者中,X1包含独特寡核苷酸序列。第二寡核苷酸扩增引物组可包括包含具有如下通式的多个寡核苷酸序列的多个寡核苷酸的组合物:U3/4-B2-X2,其中U3/4包含这样的寡核苷酸,当B2存在时其包含第三通用接头寡核苷酸序列,或者当没有B2时其包含第四通用接头寡核苷酸序列,B2包含没有序列或具有来源与B1相同的6、7、8、9、10、11、12、13、14、15、16、17、18、19或20个连续核苷酸的第二寡核苷酸条形码序列的寡核苷酸,并且X2包含如下之一的寡核苷酸:(a)多核苷酸,其包含适应性免疫受体异源二聚体的所述第二多肽的适应性免疫受体可变(V)区编码基因序列或者其互补序列的至少20、30、40或50个并且不超过100、90、80、70或60个连续核苷酸,并且在通式U3/4-B2-X2的所述多个寡核苷酸序列每者中,X2包含独特寡核苷酸序列,以及(b)多核苷酸,其包含如下任一者的至少15-30或31-50个并且不超过80、70、60或55个连续核苷酸:(i)适应性免疫受体异源二聚体的所述第二多肽的适应性免疫受体连接(J)区编码基因序列或者其互补序列,或者(ii)适应性免疫受体异源二聚体的所述第二多肽的适应性免疫受体恒定(C)区编码基因序列或者其互补序列,并且在通式U3/4-B2-X2的所述多个寡核苷酸序列每者中,X2包含独特寡核苷酸序列。接触步骤可在足以实现第一微滴之一与第二微滴之一之间的多个融合事件以产生多个融合微滴的条件和持续时间下发生,其中基因组DNA与第一和第二寡核苷酸扩增引物组之间发生核酸扩增相互作用,以在所述多个融合的微滴一者或多者的每一个中获得:第一双链DNA产物,所述第一双链DNA产物包含至少一个第一通用接头寡核苷酸序列、至少一个第一寡核苷酸条形码序列、适应性免疫受体异源二聚体的所述第一多肽的至少一个X1寡核苷酸V区编码基因序列、至少一个第二通用接头寡核苷酸序列、以及适应性免疫受体异源二聚体的所述第一多肽的至少一个X1寡核苷酸J区或C区编码基因序列。所述条件还允许在所述多个融合微滴一者或多者的每一个中获得:第二双链DNA产物,所述第二双链DNA产物包含至少一个第三通用接头寡核苷酸序列、至少一个第二寡核苷酸条形码序列、适应性免疫受体异源二聚体的所述第二多肽的至少一个X2寡核苷酸V区编码基因序列、至少一个第四通用接头寡核苷酸序列、以及适应性免疫受体异源二聚体的所述第二多肽的至少一个X2寡核苷酸J区或C区编码基因序列。所述方法还包括破坏所述多个融合的微滴以获得所述第一和第二双链DNA产物的异质混合物,以及使第一和第二双链DNA产物的混合物与第三扩增引物组和第四扩增引物组接触。在一些实施例中,第三扩增引物组包含(i)多个第一含测序平台标签的寡核苷酸,其各自包含能够特异性地杂交于第一通用接头寡核苷酸的寡核苷酸序列及连接到第一通用接头寡核苷酸序列并位于第一通用接头寡核苷酸序列的5’的第一测序平台特异性寡核苷酸序列,以及(ii)多个第二含测序平台标签的寡核苷酸,其各自包含能够特异性地杂交于第二通用接头寡核苷酸序列的寡核苷酸序列及连接到第二通用接头寡核苷酸序列并位于第二通用接头寡核苷酸序列的5’的第二测序平台特异性寡核苷酸序列。在其他实施例中,第四扩增引物组包含(i)多个第三含测序平台标签的寡核苷酸,其各自包含能够特异性地杂交于第三通用接头寡核苷酸的寡核苷酸序列及连接到第三通用接头寡核苷酸序列并位于第三通用接头寡核苷酸序列的5’的第三测序平台特异性寡核苷酸序列,以及(ii)多个第四含测序平台标签的寡核苷酸,其各自包含能够特异性地杂交于第四通用接头寡核苷酸序列的寡核苷酸序列及连接到第四通用接头寡核苷酸序列并位于第四通用接头寡核苷酸序列的5’的第四测序平台特异性寡核苷酸序列。接触步骤可在一些条件下发生并持续足够的时间以扩增(2)的第一和第二双链DNA产物的两条链,从而获得用于测序的DNA文库。所述方法还包括对(3)中获得的DNA文库进行测序,以获得编码适应性免疫受体异源二聚体的第一多肽序列和第二多肽序列的序列的数据集。图2示出了一种方法,通过所述方法将包含单个淋巴样细胞或基因组DNA的多个第一微滴210与多个单独第二微滴220融合,以形成多个融合的微滴230。第二多个小滴可包含如本文所述的扩增引物组,并且可将融合的小滴置于一定条件下,在所述条件下,扩增引物可扩增存在于微滴内的单个淋巴样细胞或基因组DNA(或cDNA)中的DNA。这些和相关实施例允许对编码适应性免疫受体异源二聚体的来自相同细胞的两条链诸如IGH加IGL、或IGH加IGK、或TCRA加TCRB、或TCRG加TCRD的重排基因进行高通量测序。有利的是,该方法还允许对具有给定TCR或IG的细胞的数量进行定量。示例性实施例的示意图示于图3,根据该图,执行与上述步骤高度相似的步骤,然而,明显的是,其中来自单个淋巴样细胞的DNA与如本文所述的第一和第二扩增引物组接触以实现第一扩增反应并由此掺入独特分子标记条形码的步骤,是在单个微滴内发生的,所述微滴诸如为由乳液形成的微滴,其用于RainDanceTM微滴数字PCR系统(RainDanceTMmicrodropletdigitalPCRsystem)(美国马萨诸塞州列克星敦的雷恩丹斯技术公司(RainDanceTechnologies,Lexington,MA))(例如,Pekinetal,2011Lab.Chip11(13):2156(Pekin等人,2011年,《芯片实验室》,第11卷,第13期,第2156页);Zhongetal,2011Lab.Chip11(13):2167(Zhong等人,2011年,《芯片实验室》,第11卷,第13期,第2167页);Tewheyetal,2009NatureBiotechnol.27:1025;2010NatureBiotechnol.28:178(Tewhey等人,2009年,《自然生物技术》,第27卷,第1025页;2010年,《自然生物技术》,第28卷,第178页))或其他相当的系统,技术人员可对任何一种系统进行修改以便与本文所述的组合物和方法一起使用。在将所述多个独特分子标记条形码掺入多个不同dsDNA产物中之后,可破坏微滴,并且可如本文所述及图3所示执行包括扩增和引入测序平台特异性寡核苷酸的后续步骤。在这些和相关实施例中,单个标记条形码(BC1)可被所有J引物共用(或在某些实施例中被所有V引物共用),并且可能有利的是产生具有特异性且预先识别出的条形码序列的有限集合的此类引物。然而,在第一步骤期间,仅单个标记条形码序列(BC1)将存在于任何给定微滴内。因此,在以包含如本文提供的多个异质淋巴样细胞的样品开始进行操作时,即使在测序步骤后获得序列信息的大而多样的集合之后,此类信息的分析可包括确定包含相同标记条形码(BC1)的第一和第二TCR或Ig异源二聚体多肽链编码序列,由此概率依据将指示两条链都是相同细胞的产物的可能性极高。因此,本公开首次提供了用于确定并定量在相同细胞中表达的TCR或Ig异源二聚体的两条链在样品中的相对占比的组合物和方法。在无微滴的情况下确定克隆异源二聚体序列根据某些其他实施例,可在不首先制备第一和第二微滴的单独群体的情况下,实现编码单个细胞中的第一和第二适应性免疫受体异源二聚体多肽序列的重排DNA序列的确定,所述第一和第二微滴分别包含单个淋巴样细胞基因组DNA(或已由来自其的mRNA逆转录的cDNA)和寡核苷酸扩增引物组。相反地,这些可供选择的实施例设想了通过将细胞分布到多个容器诸如多孔细胞培养板或测定板(例如,96、384或1536孔格式)的多个孔,将包含淋巴样细胞的细胞悬液(例如,来自受试者的血细胞制备物或其细胞亚群)的细胞分成亚群。熟悉本领域的人员将知道用于将细胞悬液分布到此类多个容器中的多种装置和方法,例如使用荧光活化细胞分选(FACS)或用自动化低容量分配设备或采取有限稀释,以使每孔、每个容器、每管、每个隔室等获得所需数量的细胞。在某些实施例中,可能优选的是,将基本上相同数量的细胞分布到每个容器,但某些其他设想的实施例不必如此限制。简而言之,根据这些和相关实施例,分离的淋巴样细胞亚群可提供用作逆转录产生cDNA分子的模板的mRNA分子,所述cDNA分子同时在逆转录(RT)步骤期间被标记(参见图4和图5)。图4描绘了通过使用引导寡核苷酸条形码和通用接头寡核苷酸序列掺入cDNA中的寡核苷酸逆转录引物,在逆转录期间标记适应性免疫受体多肽编码cDNA的示意图。用包含pGEX-Rev序列、条形码BC和N6间隔区序列(BC-N6)及“Cn-RC”序列的引物扩增cDNA链。经扩增的cDNA链的3’端包含pGEX-FRC序列、条形码BC-N6间隔区序列及“SmarterUAII”序列。将经扩增的cDNA的孔或容器合并,并且对第一cDNA链库进行SPRI微珠纯化。使用加尾-pGEXF/R序列进行PCR扩增。基于尺寸来纯化和选择扩增子。所得的cDNA扩增子示于图4中。图5描绘了通过使用引导寡核苷酸条形码和通用接头寡核苷酸序列掺入cDNA中的寡核苷酸逆转录引物,在逆转录期间标记适应性免疫受体多肽编码cDNA的示意图。图6给出了适于在用Illumina测序接头修饰扩增的适应性免疫受体多肽编码cDNA后进行测序的DNA产物的示意图,所述扩增的适应性免疫受体多肽编码cDNA已通过使用引导寡核苷酸条形码和通用接头寡核苷酸序列掺入的寡核苷酸逆转录引物,在逆转录期间进行标记。如本文所提供,此类实施例中的寡核苷酸RT引物包含特异性杂交于靶适应性免疫受体编码区诸如V区、J区或C区序列的寡核苷酸序列,并且还包含作为分子标签的寡核苷酸条形码序列,以及如本文所述的通用接头寡核苷酸序列。由适应性免疫受体编码mRNA逆转录的过程可因此伴随在cDNA产物中掺入(i)作为来源标识符的寡核苷酸条形码序列,和(ii)如本文所述有利于自动化高通量测序的通用接头。以举例说明而非限制的方式,在这些实施例的某些中,与单个具体容器(例如,多孔板的一个孔)的内容物接触的寡核苷酸RT引物组中的所有RT引物共用通用条形码寡核苷酸序列(B),并且不同的条形码寡核苷酸序列(B)存在于每个单独容器(诸如多孔板的每个孔)中。例如,细胞悬液(例如,血细胞或其部分,诸如有核细胞、淋巴样细胞等)可通过随机分布而在多孔板的不同孔间分配,以将细胞物理分离为亚群。然后可根据多种常规程序中的任何一种来裂解或以其他方式处理每孔中的细胞亚群,以释放存在于细胞内的mRNA,其可包括编码由细胞表达的TCR(例如,TCRA和TCRB,或TCRG和TCRG)或IG(例如,IGH和IGL)异源二聚体的两条链的mRNA,并且其也可包括编码一个或多个淋巴细胞状态指示分子的mRNA。然后可将mRNA用作模板,以便通过修改既有的逆转录(RT)方案,使用如本文所述的寡核苷酸逆转录引物组进行cDNA合成,所述寡核苷酸逆转录引物组能够在每个单独孔中向cDNA产物引入独特寡核苷酸条形码序列,所述独特寡核苷酸条形码序列连接到TCR或IG编码序列或其互补序列(参见例如图4至图5)。在条形码的外部(例如,相对于条形码而言,远离TCR或IG编码序列),寡核苷酸逆转录引物组也可被设计为引入如本文所述的通用接头寡核苷酸序列和/或其他已知的寡核苷酸序列特征,诸如可有利于下游扩增、处理和/或其他操纵步骤(诸如将与自动化高通量定量测序相容的步骤)的寡核苷酸序列特征。在逆转录cDNA产物的DNA扩增后,多孔板的给定孔内每个经扩增的DNA分子将具有相同的寡核苷酸条形码序列,而每个不同孔中的扩增产物的条形码序列将彼此不同。这样,在每个孔内,编码适应性免疫受体异源二聚体(例如,IGH和IGL、TCRA和TCRB、TCRG和TCRD)的任一条链的所有DNA分子将具有相同的寡核苷酸条形码序列。可将扩增产物合并,并使用如本文别处所述的自动化高通量DNA测序进行定量测序,以获得序列的数据集,其包括TCR和/或IG序列以及相关的寡核苷酸条形码序列。如本文所公开,在某些优选实施例中,可通过组合学方法分析序列的数据集,所述组合学方法允许使适应性免疫受体异源二聚体亚基编码序列的具体对相匹配以将它们识别为源自相同的淋巴样细胞。作为非限制性示例性例子,可从其中分布有淋巴样细胞悬液的100个孔的集合获得序列的假设数据集。在每个孔中,使用分别对于TCRA和TCRB编码序列的部分具有特异性的第一和第二寡核苷酸逆转录引物组,来逆转录细胞的mRNAcDNA。在每个不同孔中,寡核苷酸逆转录引物组也将不同的寡核苷酸条形码序列引入cDNA产物中。如果假设地说,具有单一通用克隆起源的T细胞(例如,表达相同TCRA/B序列的T细胞)被随机分布于所述100个孔的五个不同孔中,则序列数据集将包括五种不同的情况,其中TCRA和TCRB序列的独特对出现于共用相同条形码序列的DNA扩增产物中。换句话讲,在这五个不同孔的每个孔中,寡核苷酸逆转录引物组促进了具有相同重排TCRA和TCRB序列的cDNA的生成,但每个孔的cDNA产物包含不同的孔特异性条形码序列。根据非限制性理论,依据概率,这样的可能性将极其高:独特TCRA/TCRB序列对源自相同T细胞克隆,其成员应已随机分布于这五个不同的孔中。根据某些实施例,用于确定编码单个淋巴样细胞中适应性免疫受体异源二聚体的第一和第二多肽序列的重排DNA序列的该高通量方法的更详细描述如下:使用密度梯度离心(例如,新泽西州皮斯卡特维的通用电气医疗集团生命科学部(GEHealthcareBio-Sciences,Piscataway,NJ))或通过结合于抗体包被磁珠诸如得自美国加利福尼亚州奥本的美天旎公司(MiltenyiBiotec(Auburn,CA))的CD45微珠,从抗凝全血样品分离淋巴样细胞。或者,可通过结合于CD3+磁珠,从全血样品纯化T淋巴细胞,并且可通过结合于CD19+磁珠,从全血样品纯化B淋巴细胞。然后可检查分离的细胞群体的活力。可用过滤器,例如使用美天旎(MiltenyiBiotec)死细胞移除试剂盒,从样品中移除死细胞。取决于应用,可将分离的活的淋巴样细胞(例如,以可存在于未分选的外周血单核细胞(PBMC)中的形式,或以特异性细胞亚群的制备物的形式)在短期细胞培养中培养,并且在某些实施例中,细胞可通过多种已知活化范例中的任何一种来活化,诸如通过暴露于细胞因子、趋化因子、特异性抗体、有丝分裂原、多克隆活化剂等中的一者或多者来活化。可通过如下方式制备最终细胞样品:将细胞重悬于培养基(例如,含10%胎牛血清的RPMI)或适当等渗缓冲溶液(例如,磷酸盐缓冲盐水,PBS)中,其补充有防止细胞凝集的试剂(例如,0.1%BSA、1%F-68)。或者,可利用未经分选的全血或PBMC。作为最一般的情况,可使用作为水溶液中的悬液存在的包含B细胞或T细胞的任何细胞群。将包含多个淋巴样细胞的细胞制备物分为多个物理分离的亚群,例如通过将细胞的悬液分布于能够容纳细胞的多个容器或隔室间,以获得各自包含淋巴样细胞亚群的多个容器或隔室,其中每个亚群包含一个淋巴样细胞或多个淋巴样细胞,并且其中每个容器或隔室物理地分离,以使得内容物彼此不流体连通。优选地,将细胞分布或分配到多个容器中以使得每个容器包含基本上相等数量的细胞,这可导致每个容器中有相同数量的细胞,或每个容器中的细胞数量处于任何其他容器中的细胞数量的1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、21-30%、31-50%、51-70%、71-80%或81-100%内。示例性容器可为多孔培养板或测定板诸如6、12、24、48、96、384或1536孔的多孔板或任何其他多孔板格式的孔;还设想了管阵列、过滤器、微加工的孔阵列、激光产生的基质或能够容纳细胞的任何其他合适容器。在某些示例性实施例中,可通过荧光活化细胞分选(FACS)将细胞分布于多个容器间:可使用FACS将预定数量的细胞分离、分选并置于多孔(例如,96、384或1536)反应板中。可采用多种方法和仪器中的任何一种,其使用能够在多孔板上对细胞进行预备分选的流式细胞仪(例如,碧迪(BecktonDickinson)III、BeckmanMoFloTMXDP等)。FACS允许特异性细胞亚群通过抗体染色、活力染色或特异性细胞染色试剂的多色组合而分离。可采用细胞分选仪对靶细胞计数并将指定数量的细胞置于收集多孔板的每个孔中(10-20%CV)。或者,可使用能够优选地以非接触方式将均匀细胞悬液分配到高密度微孔板(384、1536、3456孔)上的自动化低容量(每孔nl至μl容量)分配器,诸如贝克曼库尔特(BeckmanCoulter)BioRAPTRFRDTM、LambdaJetTMIIIMT(赛默飞世尔科技公司(ThermoFisherScientific))、CyBiTMDrop(耶拿分析仪器公司(JenaAnalytik))、FurukawaPerflowTM或类似仪器,以高精度和再现性(10-20%CV)将指定数量的细胞置于收集多孔板的每个孔中。然后用附接到所有样品的独特的孔特异性条形码寡核苷酸从每个孔扩增适应性免疫受体编码多核苷酸序列。这样做的一种方法是通过逆转录将细胞mRNA转化为cDNA,并且在逆转录步骤期间向cDNA产物添加寡核苷酸条形码形式的分子标签。可将相同条形码添加至与mRNA互补的cDNA,所述mRNA编码孔内的每个异源二聚体适应性免疫受体分子的两条链,例如,免疫球蛋白重链和轻链、TCRA和TCRB链以及TCRG和TCRD链。在这个和相关实施例中,由cDNA扩增抗原受体编码序列,所述cDNA通过mRNA逆转录而制成;未扩增基因组DNA(gDNA)。为了做到这点,微孔板的每个孔可包括含RNA酶抑制剂的介质,以及经设计以保护细胞中的RNA(诸如QiagenRNAlaterTM,美国加利福尼亚州瓦伦西亚的凯杰公司(Qiagen,Valencia,CA))或裂解细胞并分离RNA(Trizol,异硫氰酸胍-QiagenRNeasyTM等)的介质。然后可使用机器人液体处理器将提取的总细胞RNA转移到另一个多孔板中以便进行逆转录反应。或者,可直接在含RNA酶抑制剂的逆转录反应混合物中裂解所分选的细胞。可通过将细胞RNA暴露于反应混合物来引发逆转录反应(RT),所述反应混合物包含适当缓冲液、dNTP、酶(逆转录酶)和一组寡核苷酸逆转录引物。这些引物一般将包含可退火至IgG、IgM、IgA、IgD、IgE、Igκ、Igλ、TCRα、β、γ和δ恒定区(C区段)基因特异性寡核苷酸序列的多样性引物亚组,以及通用模板转换寡核苷酸(例如,ClontechSmarterTMUAII寡核苷酸,美国加利福尼亚州山景城的Clontech公司(Clontech,MountainView,CA))。例如,C区段基因特异性引物或SmarterTMUAII寡核苷酸或这两者将被DNA条形码独特标记,所述DNA条形码将为6、7、8、9、10、11、12、13、14、15个…等碱基对长的独特序列。RT反应板的每个孔将包含相同多样性的引物,其中混合物中的每种引物将被相同DNA条形码标记,但每个孔中将使用不同条形码。因此,在逆转录反应完成时,将用相同DNA条形码序列为给定孔中的每个第一链cDNA分子编上条形码。用于第1cDNA链合成的BCR/TCRC区段引物的列表:因此,在将细胞分布于多个容器的步骤后,使每个容器在足以促进所述多个容器中的淋巴样细胞中的mRNA逆转录的条件和持续时间下,与第一和第二寡核苷酸逆转录引物组接触,其中(A)第一寡核苷酸逆转录引物组能够逆转录编码适应性免疫受体异源二聚体的多个第一多肽的多个第一mRNA序列,并且(B)第二寡核苷酸逆转录引物组能够逆转录编码适应性免疫受体异源二聚体的多个第二多肽的多个第二mRNA序列,并且其中:(I)第一寡核苷酸逆转录引物组包括包含具有如下通式的多个寡核苷酸序列的多个寡核苷酸的组合物:U1/2-B1-X1其中Ul/2包含这样的寡核苷酸,当B1存在时其包含第一通用接头寡核苷酸序列,或者当没有B1时其包含第二通用接头寡核苷酸序列,B1包含没有序列或具有6、7、8、9、10、11、12、13、14、15、16、17、18、19或20个连续核苷酸的第一寡核苷酸条形码序列的寡核苷酸,并且X1包含如下之一的寡核苷酸:(a)多核苷酸,其包含适应性免疫受体异源二聚体的所述第一多肽的适应性免疫受体可变(V)区编码基因序列或者其互补序列的至少20、30、40或50个并且不超过100、90、80、70或60个连续核苷酸,并且在通式U1/2-B1-X1的所述多个寡核苷酸序列每者中,X1包含独特寡核苷酸序列,以及(b)多核苷酸,其包含如下任一者的至少15-30或31-50个并且不超过80、70、60或55个连续核苷酸:(i)适应性免疫受体异源二聚体的所述第一多肽的适应性免疫受体连接(J)区编码基因序列或者其互补序列,或者(ii)适应性免疫受体异源二聚体的所述第一多肽的适应性免疫受体恒定(C)区编码基因序列或者其互补序列,并且在通式U1/2-B1-X1的所述多个寡核苷酸序列每者中,X1包含独特寡核苷酸序列,并且(II)第二寡核苷酸逆转录引物组包括包含具有如下通式的多个寡核苷酸序列的多个寡核苷酸的组合物:U3/4-B2-X2其中U3/4包含这样的寡核苷酸,当B2存在时其包含第三通用接头寡核苷酸序列,或者当没有B2时其包含第四通用接头寡核苷酸序列,B2包含没有序列或具有6、7、8、9、10、11、12、13、14、15、16、17、18、19或20个连续核苷酸的第二寡核苷酸条形码序列的寡核苷酸,对于与所述多个容器的单个容器接触的第一和第二逆转录引物组每者而言,所述第二寡核苷酸条形码序列与B1相同,并且X2包含如下之一的寡核苷酸:(a)多核苷酸,其包含适应性免疫受体异源二聚体的所述第二多肽的适应性免疫受体可变(V)区编码基因序列或者其互补序列的至少20、30、40或50个并且不超过100、90、80、70或60个连续核苷酸,并且在通式U3/4-B2-X2的所述多个寡核苷酸序列每者中,X2包含独特寡核苷酸序列,以及(b)多核苷酸,其包含如下任一者的至少15-30或31-50个并且不超过80、70、60或55个连续核苷酸:(i)适应性免疫受体异源二聚体的所述第二多肽的适应性免疫受体连接(J)区编码基因序列或者其互补序列,或者(ii)适应性免疫受体异源二聚体的所述第二多肽的适应性免疫受体恒定(C)区编码基因序列或者其互补序列,并且在通式U3/4-B2-X2的所述多个寡核苷酸序列每者中,X2包含独特寡核苷酸序列,所述接触步骤在一定条件和持续时间下发生以足以在所述多个容器一者或多者的每一个中获得:第一逆转录的互补DNA(cDNA)产物,其包含至少一个第一通用接头寡核苷酸序列、至少一个第一寡核苷酸条形码序列、适应性免疫受体异源二聚体的所述第一多肽的至少一个X1寡核苷酸V区编码基因序列、至少一个第二通用接头寡核苷酸序列、以及适应性免疫受体异源二聚体的所述第一多肽的至少一个X1寡核苷酸J区或C区编码基因序列,并且也足以在所述多个容器一者或多者的每一个中获得:第二逆转录的cDNA产物,其包含至少一个第三通用接头寡核苷酸序列、至少一个第二寡核苷酸条形码序列、适应性免疫受体异源二聚体的所述第二多肽的至少一个X2寡核苷酸V区编码基因序列、至少一个第四通用接头寡核苷酸序列、以及适应性免疫受体异源二聚体的所述第二多肽的至少一个X2寡核苷酸J区或C区编码基因序列。在接触步骤之后,执行将来自所述多个容器的第一和第二逆转录的cDNA产物组合的步骤,以获得逆转录的cDNA产物的混合物。组合步骤之后是使第一和第二逆转录的cDNA产物的混合物与第一寡核苷酸扩增引物组和第二寡核苷酸扩增引物组接触,其中第一扩增引物组包含(i)多个第一含测序平台标签的寡核苷酸,其各自包含能够特异性地杂交于第一通用接头寡核苷酸的寡核苷酸序列及连接到第一通用接头寡核苷酸序列并位于第一通用接头寡核苷酸序列的5’的第一测序平台特异性寡核苷酸序列,以及(ii)多个第二含测序平台标签的寡核苷酸,其各自包含能够特异性地杂交于第二通用接头寡核苷酸序列的寡核苷酸序列及连接到第二通用接头寡核苷酸序列并位于第二通用接头寡核苷酸序列的5’的第二测序平台特异性寡核苷酸序列,并且其中第二寡核苷酸扩增引物组包含(i)多个第三含测序平台标签的寡核苷酸,其各自包含能够特异性地杂交于第三通用接头寡核苷酸的寡核苷酸序列及连接到第三通用接头寡核苷酸序列并位于第三通用接头寡核苷酸序列的5’的第三测序平台特异性寡核苷酸序列,以及(ii)多个第四含测序平台标签的寡核苷酸,其各自包含能够特异性地杂交于第四通用接头寡核苷酸序列的寡核苷酸序列及连接到第四通用接头寡核苷酸序列并位于第四通用接头寡核苷酸序列的5’的第四测序平台特异性寡核苷酸序列,所述接触步骤在一些条件下发生并持续足够的时间以扩增第一和第二逆转录的cDNA产物两者,从而获得用于测序的DNA文库。一旦由此获得用于测序的DNA文库,就在接下来的步骤中进行DNA文库的测序,以获得编码适应性免疫受体异源二聚体的第一和第二多肽序列的序列的数据集。然后可基本按照本文别处所述的那样进行序列的数据集的分析,以确定编码源自单个(即,相同)淋巴样细胞的适应性免疫受体异源二聚体的第一和第二多肽的重排DNA序列。简而言之,所述方法还可包括如下步骤:(a)根据本文鉴定的寡核苷酸条形码序列来分选序列的数据集,以获得多个条形码序列集合,每个条形码序列集合具有独特条形码;(b)将(a)的每个条形码序列集合分选为含X1序列的子集和含X2序列的子集;(c)根据X1序列和X2序列对含X1序列和含X2序列的子集每者的成员进行聚类分析,以分别获得一个或多个X1序列聚类集合和一个或多个X2序列聚类集合,及对所述X1和X2序列聚类集合中的任何一个或多个内的单个核苷酸条形码序列错配进行错误修正;以及(d)基于已知的X1序列和X2序列来鉴定每个第一和第二适应性免疫受体异源二聚体多肽编码序列,其中每个X1序列和每个X2序列与一个或多个独特B序列相关,以鉴定源自其的每个B序列相关X1序列和每个B序列相关X2序列的容器;以及(e)基于B序列与通用第一和第二适应性免疫受体异源二聚体多肽编码序列一致的概率,将(d)的B序列相关X1和X2序列在组合学上匹配为具有共同克隆起源,由此确定编码适应性免疫受体异源二聚体的第一和第二多肽序列的重排DNA序列源自单个淋巴样细胞。因此且概括地说,在本文所公开的实施例的某些中,可将测序接头放置在所有经逆转录/扩增的TCR和/或IG编码区段的每端上,例如通过在孔特异性条形码之外的每个cDNA分子的每端上合成通用接头序列。然后,可在加尾PCR反应中在每个分子上合成接头。在此类实施例中,可合成融合RT引物,并将该引物用于第一cDNA链合成。这些引物都将包含相同的独特DNA条形码以及通用(例如,pGEX)引发位点。在通过逆转录进行第一cDNA链合成完成后,将所有板孔的内容物以定量方式回收并合并(例如,通过翻转式离心到槽上)、纯化并且随后分到多个孔中,以便用包含“尾”序列的通用接头引物(pGEX)进行PCR,所述“尾”序列被设计为掺入要用于使用下一代序列分析系统(例如,美国加利福尼亚州圣地亚哥的亿明达公司(Illumina,SanDiego,CA))进行扩增和测序的序列。或者,可将测序平台特异性接头连接到标记的分子的末端(例如,亿明达(Illumina)TrueSeqTM样品制备方法)。将来自所有孔的分子合并,从而生成独特标记的BCR或TCRds-cDNA产物的高复杂性测序文库。所述分子均使用高通量测序进行测序。可有利地使用与测序平台特异性接头互补的通用测序引物。这将允许多个样品的样品索引,其中样品特异性索引将用于源自96、384、1536等初始RT反应孔的独特标记的IGH/TCR产物的每个库。或者,可使用以通用UAII正向引物/多重V、J或C反向引物的混合物进行的多重PCR,来扩增特异性靶片段,同时保留初始细胞转录物条形编码。如果使用Illumina测序平台(MiSeqTM),则2x250bp的末端配对测序将跨越全部BCR/TCR重链和轻链(α/β;γ/δ)序列的大多数,从而允许回收每个受体结构域的全部编码序列。或者,具有延长读段长度的测序平台(Roche454、LifeIonTorrent、OGT等)可用于在一个方向上通读单个测序读段中的所有文库片段。在测序之后,可使来自每个样品的读段逆多重化,前提条件是不止一个样品位于相同测序通道中。可通过将测序读段分配给用作通用测序接头的部分的多个索引之一,来进行逆多重化。对于每个样品逆多重化的序列读段,可通过孔特异性条形码来划分所有读段。具有特异性条形码的读段的每个集合可单独地聚类,以修正PCR和测序错误并且确定每个条形码的独特序列:已通过条形码或通过TCR或IG链分选的序列可进一步使用用于聚类分析的多种已知算法中的任何一种(例如,BLASTClust、UCLUST、CD-HIT)以及就如下序列而言的错误修正来进行聚类分析,所述序列无法与具有共用条形码序列的其他序列聚类,而是相反地将与具有相差单个核苷酸的条形码的序列聚类。可通过对已知受体序列的序列匹配,将独特序列识别为IG重链或轻链(κ或λ)或识别为TCR(α或β;γ或δ)链。每个重链和轻链序列可因此与对应于初始样品孔位置的条形码的列表相关联。然后可按照序列对数据进行重排序。与每个独特序列相关的将是多孔板孔特异性条形码的集合,在所述集合内存在该序列。对于每个B或T细胞克隆,重链和轻链序列可与来自存在克隆的一个或多个拷贝的所有孔的条形码相关联。然后可使用组合学来匹配来自相同克隆的重链和轻链。例如,在96孔板中,如果具体重链和轻链序列两者均与相同12个条形码相关联,则可认为该具体对重链和轻链源自相同克隆,因为两条序列随机具有96个中的完全相同12个条形码的概率极其小。示例性算法应当理解,根据非限制性理论,与条形码序列相同集合一同出现的第一和第二适应性免疫受体链编码序列具有较高的概率源自相同的板孔,从而来自相同来源细胞。例如,在103个不同条形码用于构建第一和第二寡核苷酸逆转录引物组的情况下,将获得具有相同条形码序列的两个独立的(即,源自不同细胞)双链cDNA第一和第二产物的概率为106分之一,前提条件是每个板孔分选一个细胞。因此,如果根据本文所述的方法,第一和第二适应性免疫受体多肽编码序列(例如,X1和X2)的给定集合的三个或更多个拷贝共用通用条形码序列(例如,属于相同条形码序列集合),则序列具有独立细胞起源的概率接近零。在某些实施例中,条形码寡核苷酸B(B1、B2)可任选地包含第一和第二寡核苷酸条形码序列,其中选择第一条形码序列以独特地识别特定的V寡核苷酸序列,并且选择第二条形码序列以独特地识别特定的J寡核苷酸序列。条形码寡核苷酸B1和B2与通用接头(U)的相对定位有利地允许通过短序列读段及自动化DNA测序仪(例如,IlluminaHiSeqTM或Illumina或GeneAnalyzerTM-2,美国加利福尼亚州圣地亚哥的亿明达公司(IlluminaCorp.,SanDiego,CA))上的末端配对测序,对给定独特模板寡核苷酸的扩增产物进行快速识别和定量。具体地讲,这些和相关实施例允许对存在于扩增产物中的V序列和J序列的特异性组合进行快速高通量确定,从而表征可存在于样品(诸如包含重排的TCR或BCR编码DNA的样品)中的V特异性引物和J特异性引物的每种组合的退火靶标的相对占比。可通过较长序列读段来实现扩增产物的种类和/或数量的验证。大量适应性免疫受体可变(V)区和连接(J)区基因序列已知为核苷酸和/或氨基酸序列,包括TCR和Ig基因座的非重排基因组DNA序列,以及此类基因座处的有效重排的DNA序列及其编码的产物。参见例如,U.S.A.N.13/217,126;U.S.A.N.12/794,507;PCT/US2011/026373;PCT/US2011/049012。本领域已知的这些和其他序列可根据本公开用于包含于本发明所提供的组合物和方法中的寡核苷酸的设计和产生。V区特异性寡核苷酸可包含适应性免疫受体(例如,TCR或BCR)可变(V)区基因序列或其互补序列的至少20、30、40、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、210、220、230、240、250、260、270、280、290、300、310、320、330、340、350、360、370、380、390、400或450个并且不超过1000、900、800、700、600或500个连续核苷酸的多核苷酸序列,并且在所述多个寡核苷酸序列每者中,V包含独特寡核苷酸序列。人类及其他物种的TCR和BCRV区基因的基因组序列是已知的并且可从公共数据库诸如Genbank获得;V区基因序列包含编码表达的重排TCR和BCR基因的产物的多核苷酸序列,并且还包含已在V区基因座中识别出的假基因的多核苷酸序列。可掺入到本发明所公开的寡核苷酸中的多样性V多核苷酸序列在长度上、在核苷酸组成(例如,GC含量)上、以及在实际的线性多核苷酸序列上可广泛变化,并且例如已知包括表现出具体序列多样性的“热点”或高变区。多核苷酸V可因此包含TCR或BCR基因的特异性寡核苷酸引物组成员可特异性退火至其上的序列。能够扩增编码多个TCR或BCR的重排DNA的引物组在例如U.S.A.N.13/217,126;U.S.A.N.12/794,507;PCT/US2011/026373;或PCT/US2011/049012等中有所描述;或如本文所述可设计为包含可特异性杂交至具体TCR或BCR基因座(例如,TCRA、TCRB、TCRG、TCRD、IGH、IGK或IGL)中的每种独特V基因和每种J基因的寡核苷酸序列。例如,以举例说明而非限制的方式,能够扩增编码一种或多种TCR或BCR的重排DNA的寡核苷酸引物扩增组的寡核苷酸引物通常可包含15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39或40个连续核苷酸或更多个的核苷酸序列,并且可特异性地退火至如本文所提供的V或J多核苷酸的15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39或40个连续核苷酸的互补序列。在某些实施例中,引物可包含至少19、20、21、22、23、24、25、26、27、28、29或30个核苷酸,并且在某个实施例中,引物可包含不超过15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39或40个连续核苷酸的序列。如本文所公开,还明确设想了其他长度的引物和引物退火位点。V多核苷酸可因此在某些实施例中包含与典型V基因从其起始密码子到其CDR3编码区的长度相比,长度较短、相同或类似的核苷酸序列,并且可以(但不是必需)包含编码CDR3区的核苷酸序列。在某些优选实施例中,V多核苷酸包含CDR3编码核苷酸序列或其互补序列的全部或一部分,并且CDR3序列长度可大幅变化并且已通过若干不同编号方案表征(例如,Leffanc,1999TheImmunologist7:132(Lefranc,1999年,《免疫学家》,第7卷,第132页);Kabatetal.,1991In:SequencesofProteinsofImmunologicalInterest,NIHPublication91-3242(Kabat等人,1991年,载于:《具有免疫学重要性的蛋白质的序列》,NIH出版,第91-3242页);Chothiaetal.,1987J.Mol.Biol.196:901(Chothia等人,1987年,《分子生物学杂志》,第196卷,第901页);Chothiaetal.,1989Nature342:877(Chothia等人,1989年,《自然》,第342卷,第877页);Al-Lazikanietal.,1997J.Mol.Biol.273:927(Al-Lazikani等人,1997年,《分子生物学杂志》,第273卷,第927页);另参见例如,Rocketal.,1994J.Exp.Med.179:323(Rock等人,1994年,《实验医学杂志》,第179卷,第323页);Saadaetal.,2007Immunol.CellBiol.85:323(Saada等人,2007年,《免疫学和细胞生物学》,第85卷,第323页))。简而言之,CDR3区通常跨越从V区段中的高度保守的半胱氨酸残基(由三核苷酸密码子TGY编码;Y=T或C)延伸到TCR的J区段中的高度保守的苯丙氨酸残基(由TTY编码)或延伸到IGH中的高度保守的色氨酸(由TGG编码)的多肽部分。TCRB基因座中的超过90%的天然有效重排具有根据该标准介于24与54个核苷酸之间的CDR3编码长度,对应于介于9与17个编码的氨基酸之间。上述CDR3编码区的编号方案指示保守的半胱氨酸、苯丙氨酸和色氨酸密码子的位置,并且这些编号方案也可应用于假基因,其中编码这些保守的氨基酸的一个或多个密码子可被替换为编码不同氨基酸的密码子。对于不使用这些保守的氨基酸的假基因,可根据以上引用的既有CDR3序列位置编号方案中的一者相对于对应位置限定CDR3长度,在所述对应位置处将观察到保守的残基没有被置换。多核苷酸J可包含这样的多核苷酸,其包含适应性免疫受体连接(J)区编码基因序列或其互补序列的至少15-30、31-50、51-60、61-90、91-120或120-150个且不超过600、500、400、300或200个连续核苷酸,并且在所述多个寡核苷酸序列每者中,J包含独特寡核苷酸序列。多核苷酸J(或其互补序列)包含TCR或BCR基因的特异性寡核苷酸引物组成员可特异性退火至其上的序列。能够扩增编码多个TCR或BCR的重排DNA的引物组在例如U.S.A.N.13/217,126;U.S.A.N.12/794,507;PCT/US2011/026373;或PCT/US2011/049012等中有所描述;或如本文所述可设计为包含可特异性杂交至具体TCR或BCR基因座(例如,TCRα、β、γ或δ,或IgHμ、γ、δ、α或ε,或IgLκ或λ)中的每种独特V基因和每种独特J基因的寡核苷酸序列。在某些实施例中可能优选的是,存在于本文所述的引物组合物中的所述多个J多核苷酸具有模拟已知的天然存在J基因核苷酸序列的全长的长度。本文所述的模板中的J区长度可与天然存在的J基因序列的长度相差不超过1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%或20%。J多核苷酸可因此在某些实施例中包含具有与典型天然存在的J基因的长度相同或类似的长度的核苷酸序列,并且可以(但不是必需)包含编码CDR3区的核苷酸序列,如上文所讨论。人类及其他物种的TCR和BCRJ区基因的基因组序列是已知的并且可从公共数据库诸如Genbank获得;J区基因序列包含编码表达的和未表达的重排TCR和BCR基因的产物的多核苷酸序列。可掺入到本发明所公开的引物中的多样性J多核苷酸序列在长度上、在核苷酸组成(例如,GC含量)上、以及在实际的线性多核苷酸序列上可广泛变化。技术人员可使用本领域中与每个TCR和Ig亚基的基因的V和J编码区的已公布基因序列相关的知识,基于本公开来选择用于构建本文所述的V区段和J区段寡核苷酸引物的本文所述的V序列和J序列的替代物。人类适应性免疫受体序列的参考Genbank条目包括:TCRα:(TCRA/D):NC000014.8(chr14:22090057..23021075);TCRβ:(TCRB):NC000007.13(chr7:141998851..142510972);TCRγ:(TCRG):NC000007.13(chr7:38279625..38407656);免疫球蛋白重链,IgH(IGH):NC_000014.8(chrl4:106032614..107288051);免疫球蛋白轻链-κ,IgLκ(IGK):NC_000002.11(chr2:89156874..90274235);以及免疫球蛋白轻链-λ,IgLλ(IGL):NC000022.10(chr22:22380474..23265085)。小鼠适应性免疫受体基因座序列的参考Genbank条目包括:TCRβ:(TCRB):NC000072.5(chr6:40841295..41508370),以及免疫球蛋白重链,IgH(IGH):NC_000078.5(chrl2:114496979..117248165)。可进行引物设计分析以及靶位点选择考虑,例如使用OLIGO引物分析软件和/或BLASTN2.0.5算法软件(Altschuletal.,NucleicAcidsRes.1997,25(17):3389-402(Altschul等人,《核酸研究》,1997年,第25卷,第17期,第3389-3402页))或本领域可利用的其他类似程序进行。因此,基于本公开及考虑到用于包含在本发明寡核苷酸中的这些已知的适应性免疫受体基因序列和寡核苷酸设计方法,本领域技术人员可设计多个V区特异性和J区特异性的多核苷酸序列,其各自独立地包含给定V基因和J基因分别特有的寡核苷酸序列。相似地,根据本公开及考虑到已知的适应性免疫受体序列,本领域技术人员还可设计引物组,所述引物组包含多个V区特异性和J区特异性的寡核苷酸引物,所述引物各自独立地能够退火至给定V基因和J基因分别特有的特异性序列,由此所述多个引物能够扩增给定适应性免疫受体编码基因座(例如,人类TCR或IGH基因座)中的基本上所有V基因和基本上所有J基因。此类引物组允许在多重(例如,使用多种正向和反向引物对)PCR中生成第一端由重排V区编码基因区段编码并且第二端由J区编码基因区段编码的扩增产物。通常以及在某些实施例中,此类扩增产物可包含CDR3编码序列,但本发明并非旨在如此限制并设想了不包含CDR3编码序列的扩增产物。引物可被优选地设计用于产生扩增产物,所述扩增产物具有足够部分的如本文所述的V序列和J序列及在某些优选实施例中也有条形码(B)序列,使得通过对产物(扩增子)测序,可以基于每个基因区段特有的序列识别(i)具体V基因和(ii)具体J基因,在所述具体J基因附近V基因经历重排而产生重排的适应性免疫受体编码基因。通常以及在优选实施例中,PCR扩增产物大小不会超过600个碱基对,这根据非限制理论将不包括来自非重排适应性免疫受体基因的扩增产物。在某些其他优选实施例中,扩增产物大小不会超过500、400、300、250、200、150、125、100、90、80、70、60、50、40、30或20个碱基对,诸如可有利地通过短序列读段提供序列不同的扩增子的快速、高通量定量。引物根据本公开,在包含多个V区段引物和多个J区段引物的寡核苷酸引物组中提供寡核苷酸引物,其中所述引物组能够扩增生物样品中编码适应性免疫受体的重排DNA,所述生物样品包含淋巴样细胞DNA。合适的引物组是本领域已知的并且在本文公开,例如,US2012/0058902;U.S.A.N.13/217,126;U.S.A.N.12/794,507;PCT/US2011/026373;或PCT/US2011/049012等中的引物组;或表1中所示的那些。在某些实施例中,引物组被设计为包含多个V序列特异性引物,其对于样品中的每个独特V区基因(包括假基因)而言包含可特异性退火至独特V区序列的至少一个引物;以及对于样品中的每个独特J区基因而言可特异性退火至独特J区序列的至少一个引物。根据已知的TCR和BCR基因组序列,可通过常规方法实现引物设计。因此,引物组优选地能够扩增可由TCR或BCR基因座中的DNA重排所产生的每种可能的V-J组合。还如下文所述,某些实施例设想了这样的引物组,其中一个或多个V引物可能能够特异性退火至可由两个或更多个V区共用但并非所有V区共有的“独特”序列,和/或其中一个或多个J引物可能能够特异性退火至可由两个或更多个J区共用但并非所有J区共有的“独特”序列。在具体实施例中,用于本文所述组合物和方法中的寡核苷酸引物可包含至少约15个核苷酸长的核酸或由其组成,所述核酸具有与靶V区段或J区段的15个核苷酸长的连续序列(即,编码V区或J区多肽的基因组多核苷酸的部分)相同的序列或与之互补。较长引物,例如,具有与靶V区或J区编码多核苷酸区段的连续序列相同的序列或与之互补的序列的约16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、45或50个核苷酸长的那些引物,也可用于某些实施例。设想本发明所述的寡核苷酸引物的所有中间长度均可用于本文。如技术人员将认识到的,引物可具有添加的额外序列(例如,可不与靶V区或J区编码多核苷酸区段相同或互补的核苷酸),诸如限制性酶识别位点、用于测序的接头序列、条形码序列等等(参见,例如,本文表格和序列表中提供的引物序列)。因此,引物的长度可更长,诸如长度为约55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、80、85、90、95、100或更多个核苷酸或更长,这取决于特定用途或需要。还设想用于某些实施例中的是与本文示出了其核苷酸序列的寡核苷酸引物(包括序列表中示出的那些)具有高度序列同一性的适应性免疫受体V区段或J区段寡核苷酸引物变体。因此,在这些和相关实施例中,适应性免疫受体V区段或J区段寡核苷酸引物变体可与本文所公开的适应性免疫受体V区段或J区段寡核苷酸引物序列具有实质的同一性,例如,使用本文所述的方法(例如,使用标准参数的BLAST分析)与参考多核苷酸序列诸如本文所公开的寡核苷酸引物序列相比,此类寡核苷酸引物变体可具有至少70%的序列同一性,优选地至少75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%或更高的序列同一性。本领域技术人员将认识到可通过考虑密码子简并性、阅读框定位等来适当地调节这些值以确定寡核苷酸引物变体退火至适应性免疫受体区段编码多核苷酸的相应能力。通常,寡核苷酸引物变体将包含一个或多个置换、添加、缺失和/或插入,优选地使得相对于本文明确示出的适应性免疫受体V区段或J区段寡核苷酸引物序列而言,变体寡核苷酸的退火能力不会实质上减弱。表2以非限制性例子示出了能够扩增生物样品中编码TCRβ链(TCRB)的有效重排DNA的寡核苷酸引物组,所述生物样品包含来自受试者的淋巴样细胞的DNA。在该引物组中,J区段引物具有实质的序列同源性,因此可在不止一种靶J多核苷酸序列间充当交叉引物,但V区段引物被设计为特异性退火至V的CDR2区内的靶序列,因此是每个V区段特有的。然而,就在紧密相关靶基因的家族内序列相同的情况下的若干V引物而言,存在例外(例如,在V区段的整个编码序列中V6-2和V6-3在核苷酸水平上是相同的,因此可具有单种引物TRB2V6-2/3)。表2.示例性寡核苷酸引物组(hsTCRBPCR引物)名称序列SEQIDNO:TRBJl-1TTACCTACAACTGTGAGTCTGGTGCCTTGTCCAAA1631TRBJl-2ACCTACAACGGTTAACCTGGTCCCCGAACCGAA1632TRBJl-3ACCTACAACAGTGAGCCAACTTCCCTCTCCAAA1633TRBJl-4CCAAGACAGAGAGCTGGGTTCCACTGCCAAA1634TRBJl-5ACCTAGGATGGAGAGTCGAGTCCCATCACCAAA1635TRBJl-6CTGTCACAGTGAGCCTGGTCCCGTTCCCAAA1636TRBJ2-1CGGTGAGCCGTGTCCCTGGCCCGAA1637名称序列SEQIDNO:TRBJ2-2CCAGTACGGTCAGCCTAGAGCCTTCTCCAAA1638TRBJ2-3ACTGTCAGCCGGGTGCCTGGGCCAAA1639TRBJ2-4AGAGCCGGGTCCCGGCGCCGAA1640TRBJ2-5GGAGCCGCGTGCCTGGCCCGAA1641TRBJ2-6GTCAGCCTGCTGCCGGCCCCGAA1642TRBJ2-7GTGAGCCTGGTGCCCGGCCCGAA1643TRB2V10-1AACAAAGGAGAAGTCTCAGATGGCTACAG1644TRB2V10-2GATAAAGGAGAAGTCCCCGATGGCTATGT1645TRB2V10-3GACAAAGGAGAAGTCTCAGATGGCTATAG1646TRB2V6-2/3GCCAAAGGAGAGGTCCCTGATGGCTACAA1647TRB2V6-8CTCTAGATTAAACACAGAGGATTTCCCAC1648TRB2V6-9AAGGAGAAGTCCCCGATGGCTACAATGTA1649TRB2V6-5AAGGAGAAGTCCCCAATGGCTACAATGTC1650TRB2V6-6GACAAAGGAGAAGTCCCGAATGGCTACAAC1651TRB2V6-7GTTCCCAATGGCTACAATGTCTCCAGATC1652TRB2V6-1GTCCCCAATGGCTACAATGTCTCCAGATT1653TRB2V6-4GTCCCTGATGGTTATAGTGTCTCCAGAGC1654TRB2V24-1ATCTCTGATGGATACAGTGTCTCTCGACA1655TRB2V25-1TTTCCTCTGAGTCAACAGTCTCCAGAATA1656TRB2V27TCCTGAAGGGTACAAAGTCTctcgaaaag1657TRB2V26CTCTGAGAGGTATCATGTTTCTTGAAATa1658TRB2V28TCCTGAGGGGTACAGTGTCTCTagagaga1659TRB2V19TATAGCTGAAGGGTACAGCGTCTCTCGGG1660TRB2V4-1CTGAATGCCCCAACAGCTCTCTCTTAAAC1661TRB2V4-2/3CTGAATGCCCCAACAGCTCTCACTTATTC1662TRB2V2PCCTGAATGCCCTGACAGCTCTCGCTTATA1663TRB2V3-1CCTAAATCTCCAGACAAAGCTCACTTAAA1664TRB2V3-2CTCACCTGACTCTCCAGACAAAGCTCAT1665TRB2V16TTCAGCTAAGTGCCTCCCAAATTCACCCT1666名称序列SEQIDNO:TRB2V23-1GATTCTCATCTCAATGCCCCAAGAACGC1667TRB2V18ATTTTCTGCTGAATTTCCCAAAGAGGGCC1668TRB2V17ATTCACAGCTGAAAGACCTAACGGAACGT1669TRB2V14TCTTAGCTGAAAGGACTGGAGGGACGTAT1670TRB2V2TTCGATGATCAATTCTCAGTTGAAAGGCC1671TRB2V12-1TTGATTCTCAGCACAGATGCCTGATGT1672TRB2V12-2GCGATTCTCAGCTGAGAGGCCTGATGG1673TRB2V12-3/4TCGATTCTCAGCTAAGATGCCTAATGC1674TRB2V12-5TTCTCAGCAGAGATGCCTGATGCAACTTTA1675TRB2V7-9GGTTCTCTGCAGAGAGGCCTAAGGGATCT1676TRB2V7-8GCTGCCCAGTGATCGCTTCTTTGCAGAAA1677TRB2V7-4GGCGGCCCAGTGGTCGGTTCTCTGCAGAG1678TRB2V7-6/7ATGATCGGTTCTCTGCAGAGAGGCCTGAGG1679TRB2V7-2AGTGATCGCTTCTCTGCAGAGAGGACTGG1680TRB2V7-3GGCTGCCCAACGATCGGTTCTTTGCAGT1681TRB2V7-1TCCCCGTGATCGGTTCTCTGCACAGAGGT1682TRB2V11-123CTAAGGATCGATTTTCTGCAGAGAGGCTC1683TRB2V13CTGATCGATTCTCAGCTCAACAGTTCAGT1684TRB2V5-1TGGTCGATTCTCAGGGCGCCAGTTCTCTA1685TRB2V5-3TAATCGATTCTCAGGGCGCCAGTTCCATG1686TRB2V5-4TCCTAGATTCTCAGGTCTCCAGTTCCCTA1687TRB2V5-8GGAAACTTCCCTCCTAGATTTTCAGGTCG1688TRB2V5-5AAGAGGAAACTTCCCTGATCGATTCTCAGC1689TRB2V5-6GGCAACTTCCCTGATCGATTCTCAGGTCA1690TRB2V9GTTCCCTGACTTGCACTCTGAACTAAAC1691TRB2V15GCCGAACACTTCTTTCTGCTTTCTTGAC1692TRB2V30GACCCCAGGACCGGCAGTTCATCCTGAGT1693TRB2V20-1ATGCAAGCCTGACCTTGTCCACTCTGACA1694名称序列SEQIDNO:TRB2V29-1CATCAGCCGCCCAAACCTAACATTCTCAA1695在某些优选实施例中,如本文所述的V区段和J区段寡核苷酸引物被设计为包含核苷酸序列,使得在重排适应性免疫受体(TCR或Ig)基因的扩增产物的序列内存在足够的信息以独特地识别在重排适应性免疫受体基因座中产生扩增产物的特异性V基因和特异性J基因两者(例如,V基因重组信号序列(RSS)上游序列的至少1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20个碱基对,优选地V基因重组信号序列(RSS)上游序列的至少约22、24、26、28、30、32、34、35、36、37、38、39或40个碱基对,并且在某些优选实施例中V基因重组信号序列(RSS)上游序列的大于40个碱基对,以及J基因RSS下游的至少1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20个碱基对,优选地J基因RSS下游的至少约22、24、26、28或30个碱基对,并且在某些优选实施例中J基因RSS下游的大于30个碱基对)。该特征与本领域所述的用于扩增TCR编码或Ig编码基因序列的寡核苷酸引物截然不同,后者主要依赖于仅用于V区段和J区段的适当大小产物存在与否的检测的扩增反应(例如,PCR反应产物中存在具体大小的扩增子指示V区段或J区段存在,但无法提供所扩增PCR产物的序列,因此无法确认其种类,例如谱型分析的习惯作法)。寡核苷酸(例如,引物)可通过任何合适的方法制备,包括按照诸如以下的方法直接化学合成:Narangetal,1979,Meth.Enzymol.68:90-99(Narang等人,1979年,《酶学方法》,第68卷,第90-99页)的磷酸三酯法;Brownetal.,1979,Meth.EnzymoL68:109-151(Brown等人,1979年,《酶学方法》,第68卷,第109-151页)的磷酸二酯法;Beaucageetal.,1981,TetrahedronLett.22:1859-1862(Beaucage等人,1981年,《四面体快报》,第22卷,第1859-1862页)的二乙基亚磷酰胺法;以及美国专利No.4,458,066的固相载体法,所述文献各自以引用方式并入本文。寡核苷酸和修饰核苷酸的缀合物的合成方法的综述提供于Goodchild,1990,BioconjugateChemistry1(3):165-187(Goodchild,1990年,《生物共轭化学》,第1卷,第3期,第165-187页),其以引用方式并入本文。如本文所用,术语“引物”是指能够在合适条件下充当DNA合成起始点的寡核苷酸。。此类条件包括这样的条件,其中在存在适当缓冲液中及合适温度下的四种不同的三磷酸核苷和延伸试剂(例如,DNA聚合酶或逆转录酶)的情况下引发与核酸链互补的引物延伸产物的合成。引物优选地为单链DNA。引物的适当长度取决于引物的预期用途,但通常在6至50个核苷酸的范围内,或在某些实施例中,在15-35个核苷酸的范围内。短引物分子通常需要更冷的温度以与模板形成足够稳定的杂交复合体。引物不需要反映模板核酸的准确序列,但必须具有充分互补性以与模板杂交。用于扩增给定靶序列的合适引物的设计是本领域熟知的并且在本文引用的文献中有所描述。如本文所述,引物可掺入另外的特征,所述特征允许引物的检测或固定但不改变引物的充当DNA合成起始点的基本性质。例如,引物可在5'端含有另外的核酸序列,所述另外的核酸序列不杂交至靶核酸,但有利于扩增产物的克隆、检测或测序。与要杂交的模板充分互补的引物区在本文是指杂交区。如本文所用,如果当用于足够严格条件下的扩增反应时引物主要杂交至靶核酸,则引物对于靶序列是“特异性的”。通常,如果引物-靶标双链体稳定性大于引物与样品中存在的任何其他序列之间形成的双链体的稳定性,则引物对于靶序列是特异性的。本领域技术人员将认识到各种因素诸如盐条件以及引物的碱基组成和错配的位置会影响引物的特异性,并且在许多情况下将需要引物特异性的常规实验确认。可选择杂交条件,在所述杂交条件下,引物可仅与靶序列形成稳定双链体。因此,在适当严格的扩增条件下使用靶标特异性引物能够选择性扩增包含靶引物结合位点的那些靶序列。在具体实施例中,用于本文所述方法中的引物包含至少约15个核苷酸长的核酸或由其组成,所述核酸具有与靶V区段或J区段的15个核苷酸长的连续序列相同的序列或与之互补。较长引物,例如,具有与靶V区段或J区段的连续序列相同的序列或与之互补的序列的约16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、45或50个核苷酸长的那些引物,也可用于某些实施例。设想前述引物的所有中间长度均可用于本文。如技术人员将认识到的,引物可具有添加的额外序列(例如,可不与靶V区段或J区段相同或互补的核苷酸),诸如限制性酶识别位点、用于测序的接头序列、条形码序列等等(参见,例如,本文和序列表中提供的引物序列)。因此,引物的长度可更长,诸如长度为55、56、57、58、59、60、65、70、75个核苷酸或更长,这取决于特定用途或需要。例如,在一个实施例中,正向引物和反向引物均在5'端用与DNA测序仪相容的通用正向引物序列修饰。还设想用于某些实施例中的是与本文示出了其核苷酸序列的寡核苷酸引物(包括序列表中示出的那些)具有高度序列同一性的适应性免疫受体V区段或J区段寡核苷酸引物变体。因此,在这些和相关实施例中,适应性免疫受体V区段或J区段寡核苷酸引物变体可与本文所公开的适应性免疫受体V区段或J区段寡核苷酸引物序列具有实质的同一性,例如,使用本文所述的方法(例如,使用标准参数的BLAST分析)与参考多核苷酸序列诸如本文所公开的寡核苷酸引物序列相比,此类寡核苷酸引物变体可具有至少70%的序列同一性,优选地至少75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%或更高的序列同一性。本领域技术人员将认识到可通过考虑密码子简并性、阅读框定位等来适当地调节这些值以确定寡核苷酸引物变体退火至适应性免疫受体区段编码多核苷酸的相应能力。通常,寡核苷酸引物变体将包含一个或多个置换、添加、缺失和/或插入,优选地使得相对于本文明确示出的适应性免疫受体V区段或J区段寡核苷酸引物序列而言,变体寡核苷酸的退火能力不会实质上减弱。还如本文别处指出的,在优选实施例中,适应性免疫受体V区段和J区段寡核苷酸引物被设计为能够扩增包含CDR3编码区的重排TCR或IGH序列。根据本文设想的某些实施例,用于本公开的多重PCR方法的引物可被功能性阻断以防止非T或B细胞序列的非特异性引发。例如,引物可通过如美国专利申请公布US2010/0167353中所述的化学修饰来阻断。根据某些本文所公开的实施例,在本发明多重PCR反应中使用此类阻断的引物涉及可具有其中DNA复制(即,引物延伸)被阻断的失活构型和其中DNA复制继续进行的活化构型的引物。当引物为单链时或者当引物特异性杂交至所关注的靶DNA序列但引物延伸仍然被连接于引物3'端或其附近的化学部分阻断时,存在引物的失活构型。当引物杂交至所关注的靶核酸序列并随后被RNA酶H或另一种切割剂作用而移除3’阻断基团从而允许酶(例如,DNA聚合酶)催化扩增反应中的引物延伸时,存在引物的活化构型。不希望受理论的束缚,据信此类引物的杂交的动力学类似于二级反应,因此是混合物中的T细胞或B细胞基因序列浓度的函数。阻断的引物通过如下要求而使非特异性反应最小化:杂交至靶标,然后在引物延伸可继续进行之前切割。如果引物错误杂交至与所需靶序列相关但差别在于具有导致碱基错配的一个或多个非互补性核苷酸的序列,则引物的切割被抑制,特别是当存在位于切割位点处或其附近的错配时。提高扩增保真度的该策略减少了此类位置处错误引发的频率,从而增加了反应的特异性。如技术人员将认识到的,可优化反应条件,特别是允许每个循环中的杂交和延伸的RNA酶H浓度和时间,以使当引物正确杂交至其真正靶序列时引物的高效切割,与当引物和模板序列(引物可对其不完全退火)之间存在错配时引物的不良切割之间的切割效率的差异最大化。如US2010/0167353所述,本领域已知多个可位于寡核苷酸(例如,引物)的3'端处或其附近的阻断基团以防止延伸。可通过如下方式在3'端核苷酸处对引物或其他寡核苷酸进行修饰以防止或抑制DNA合成的起始:例如添加3'脱氧核糖核苷酸残基(例如,虫草素)、2',3'-双脱氧核糖核苷酸残基、非核苷酸键或烷二醇修饰(美国专利No.5,554,516)。Wilk等人(1990NucleicAcidsRes.18(8):2065(1990年,《核酸研究》,第18卷,第8期,第2065页))和Arnold等人(美国专利No.6,031,091)也已描述了可用于抑制或阻断引物延伸的烷二醇修饰。合适阻断基团的另外例子包括3'羟基取代(例如,3'-磷酸、3'-三磷酸或3'-磷酸与醇诸如3-羟丙基的二酯)、2’3’-环磷酸、末端RNA碱基的2'羟基取代(例如,磷酸或空间上大位阻的基团诸如三异丙基甲硅烷基(TIPS)或叔丁基二甲基甲硅烷基(TBDMS))。Laikhter等人在美国专利申请序列号11/686,894中描述了在寡核苷酸的3'端取代的2'-烷基甲硅烷基诸如TIPS和TBDMS,所述专利申请以引用方式并入本文。大位阻的取代基也可掺入到寡核苷酸的3'端残基的碱基上以阻断引物延伸。在某些实施例中,寡核苷酸可包含位于用于抑制引物延伸的阻断基团的上游(例如,5’端)的切割结构域。作为例子,切割结构域可为RNA酶H切割结构域,或切割结构域可为包含单个RNA残基的RNA酶H2切割结构域,或寡核苷酸可包含用一个或多个替代核苷置换RNA碱基。另外的示例性切割结构域在US2010/0167353中有所描述。因此,多重PCR体系可使用40、45、50、55、60、65、70、75、80、85或更多种正向引物,其中每种正向引物与单种功能性TCR或IgV区段或者功能性TCR或IgV区段的小家族,例如TCRVβ区段互补(参见例如,如表2所示的TCRBV引物,SEQIDNO:1644-1695),并且多重PCR体系可使用例如十三种反向引物,其各自对TCR或IgJ区段诸如TCRJβ区段具有特异性(参见例如,表2中的TCRBJ引物,SEQIDNO:1631-1643)。在另一个实施例中,多重PCR反应可使用各自对一种或多种功能性TCRγV区段具有特异性的四种正向引物和各自对一种或多种TCRγJ区段具有特异性的四种反向引物。在另一个实施例中,多重PCR反应可使用各自对一种或多种功能性V区段具有特异性的84种正向引物和各自对一种或多种J区段具有特异性的六种反向引物。热循环条件可遵照本领域技术人员的方法。例如,在使用PCRExpressTM热循环仪(英国阿什福德的Hybaid公司(Hybaid,Ashford,UK))时,可使用下列循环条件:95℃下15分钟循环1次,94℃下30秒、59℃下30秒和72℃下1分钟循环25至40次,然后72℃下10分钟循环一次。如技术人员将认识到的,可例如通过修改退火温度、退火时间、循环次数和延伸时间来优化热循环条件。如技术人员将认识到的,可优化引物和其他PCR试剂的用量以及PCR参数(例如,退火温度、延伸时间和循环次数)以实现所需的PCR扩增效率。或者,在也在本文设想的某些相关实施例中,可使用“数字PCR”方法对样品中的靶基因组数定量,而无需标准曲线。在数字PCR中,在大量大于100个的微孔或小滴中进行单个样品的PCR反应,使得每个小滴扩增(例如,扩增产物的生成提供了微孔或小滴中存在至少一种模板分子的证据)或无法扩增(证明模板不存在于给定的微孔或小滴中)。通过简单地对阳性微孔的数量进行计数,可以直接对存在于输入样品中的靶基因组的数量进行计数。数字PCR方法通常使用端点读数,而非在热循环反应的每次循环之后测量的常规定量PCR信号(参见例如,Pekinetal.,2011Lab.Chip11(13):2156(Pekin等人,2011年,《芯片实验室》,第11卷,第13期,第2156页);Zhongetal,2011Lab.Chip11(13):2167(Zhong等人,2011年,《芯片实验室》,第11卷,第13期,第2167页);Tewheyetal,2009NatureBiotechnol27:1025(Tewhey等人,2009年,《自然生物技术》,第27卷,第1025页);2010NatureBiotechnol28:178(2010年,《自然生物技术》,第28卷,第178页))。因此,本文所述的组合物(例如,适应性免疫受体基因特异性寡核苷酸引物组)和方法中的任一者可适合用于此类数字PCR方法,例如ABIQuantStudioTM12KFlex系统(美国加利福尼亚州卡尔斯巴德的生命技术公司(LifeTechnologies,Carlsbad,CA))、QuantaLifeTM数字PCR系统(美国加利福尼亚州赫拉克勒斯的伯乐公司(BioRad,Hercules,CA))或RainDanceTM微滴数字PCR系统(美国马萨诸塞州列克星敦的雷恩丹斯技术公司(RainDanceTechnologies,Lexington,MA))。接头本文所述的寡核苷酸可在某些实施例中包含第一(U1)和第二(U2)(和任选地第三(U3)和第四(U4))通用接头寡核苷酸序列,或可缺少U1和U2(或U3或U4)中的任一者或两者。通用接头寡核苷酸U因此可要么不存在,要么包含具有选自如下的序列的寡核苷酸:(i)第一通用接头寡核苷酸序列和(ii)连接至第一通用接头寡核苷酸序列并位于其5'端的第一测序平台特异性寡核苷酸序列,并且U2可要么不存在,要么包含具有选自如下的序列的寡核苷酸:(i)第二通用接头寡核苷酸序列和(ii)连接至第二通用接头寡核苷酸序列并位于其5'端的第二测序平台特异性寡核苷酸序列。类似的关系适用于U3和U4。U1和/或U2可例如包含通用接头寡核苷酸序列和/或测序平台特异性寡核苷酸序列,所述序列对所采用的单分子测序技术具有特异性,所述测序技术为例如HiSeqTM或GeneAnalyzerTM-2(GA-2)系统(美国加利福尼亚州圣地亚哥的亿明达公司(Illumina,Inc.,SanDiego,CA))或另一种合适的仪器、试剂和软件的测序套件。包括此类平台特异性接头序列在内使得能使用核苷酸测序方法诸如HiSeqTM或GA2或等价方法,对如本文所述掺入了U的本发明所述dsDNA扩增产物进行直接定量测序。因此该特征有利地允许对dsDNA组合物的定性和定量表征。例如,可生成dsDNA扩增产物,其在两端均具有通用接头序列,使得所述接头序列可用于在每种模板的每一端进一步掺入测序平台特异性寡核苷酸。不希望受理论的束缚,可在变性、退火和延伸的三个循环中使用5’寡核苷酸(5’-平台序列-通用接头1序列-3’)和3’寡核苷酸(5’-平台序列-通用接头2序列-3’),将平台特异性寡核苷酸添加到此类dsDNA的末端,使得组分dsDNA中的每一者在dsDNA组合物中的相对占比在数量上不会改变。独特标识符序列(例如,与单独的V区和/或J区相关并因此对其进行识别的条形码序列B,或者如本文所述的样品标识符条形码)位于邻近接头序列的位置,从而允许在短序列读段中定量测序,以便按照存在的每种独特序列的相对量的标准来表征DNA群体。除了实例中所述和序列表中的示例性模板序列中所包括的接头序列(例如,在SEQIDNO:1-1630的5’端和3’端处)之外,熟悉本领域的技术人员在考虑本公开后,也将知道其他可用作通用接头序列的寡核苷酸序列。另外的接头序列的非限制性例子示于表3中并以SEQIDNO:1710-1731示出。表3.示例性接头序列接头(引物)名称序列SEQIDNO:T7启动子AATACGACTCACTATAGG1710T7终止子GCTAGTTATTGCTCAGCGG1711T3ATTAACCCTCACTAAAGG1712SP6GATTTAGGTGACACTATAG1713MBF(-21)TGTAAAACGACGGCCAGT1714M13F(-40)GTTTTCCCAGTCACGAC1715M13R反向CAGGAAACAGCTATGACC1.716AOX1正向GACTGGTTCCAATTGACAAGC1717AOX1反向GCAAATGGCATTCTGACATCC1718pGEX正向(GST5,pGEX5')GGGCTGGCAAGCCACGTTTGGTG1719pGEX反向(GSrF3,pGEX3')CCGGGAGCTGCATGTGTCAGAGG1720BGFl反向AACTAGAAGGCACAGTCGAGGC1721GFP(C'末端,CFP、YFP或BFP)CACTCTCGGCATGGACGAGC1722GFP反向TGGTGCAGATGAACTTCAGG1723GAGGTTCGACCCCGCCTCGATCC1724GAG反向TGACACACATTCCACAGGGTC1725接头(引物)名称序列SEQIDNO:CYC1反向GCGTGAATGTAAGCGTGAC1726pFastBacF5'-d(GGATTATTCATACCGTCCCA)-3'1777pFastBacR5'-d(CAAATGTGGTATGGCTGATT)-3'1728pBAD正向5'-d(ATGCCATAGCATTTTTATCC)-3'1729pBAD反向5’-d(GATTTAATCTGTATCAGG)-3'1730CMV正向5'-d(CGCAAATGGGCGGTAGGCGTG)-3'1731条形码如本文所述,某些实施例设想了将寡核苷酸序列设计成包含短签名序列(signaturesequence),所述短签名序列允许明确识别其中掺入了所述短签名序列的多核苷酸序列及从而明确识别负责扩增该产物的至少一种引物,而不必对整个扩增产物测序。在本文所述的寡核苷酸中,此类条形码B(例如,B1、B2)各自要么不存在,要么各自包含寡核苷酸B,所述寡核苷酸B包含3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、25、30、35、40、45、50个或更多个连续核苷酸(包括其间的所有整数值)的寡核苷酸条形码序列,其中在所述多个寡核苷酸序列每者中,B包含独特寡核苷酸序列,所述独特寡核苷酸序列独特地识别具体V和/或J寡核苷酸引物序列。示例性条形码可包含独特地识别引物组合物中的每种寡核苷酸引物(例如,V引物或J引物)的5、6、7、8、9、10、11、12、13、14、15或16个核苷酸的第一条形码寡核苷酸,和任选地在某些实施例中独特地鉴定引物组中的每种伴侣引物(例如,J引物或V引物)的5、6、7、8、9、10、11、12、13、14、15或16个核苷酸的第二条形码寡核苷酸,以提供长度分别为5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31或32个核苷酸的条形码,但这些和相关实施例并非旨在如此限制。条形码寡核苷酸可包含任何长度的寡核苷酸序列,只要获得最小条形码长度使具有另外不同序列(例如,V序列和J序列)的两种或更多种产物多核苷酸中不出现给定条形码序列即可。因此,避免用于独特地识别不同V-J序列配对的条形码间的此类冗余的最小条形码长度是X个核苷酸,其中4X大于因具有非同一序列而呈现差异化的不同模板物质的数量。实际上,条形码寡核苷酸序列读段长度可仅受限于要采用的核苷酸测序仪器的序列读段长度极限。对于某些实施例,当进行比对以使在条形码寡核苷酸序列中的具体位置处匹配的核苷酸数最大化时,将区分开模板寡核苷酸的单独物质的不同条形码寡核苷酸应具有至少两个核苷酸错配(例如,最小汉明距离(hammingdistance)为2)。技术人员将熟悉例如至少3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、30、35个或更多个连续核苷酸(包括其间的全部整数值)的寡核苷酸条形码序列的设计、合成及掺入到较大寡核苷酸或多核苷酸构建体中。有关寡核苷酸条形码序列识别策略的设计与实现的非限制性例子,参见例如,deCareeretal,2011Adv.Env.Microbiol.77:6310(deCarcer等人,2011年,《高等环境微生物学》,第77卷,第6310页);Parameswaranetal,2007Nucl.Ac.Res.35(19):330(Parameswaran等人,2007年,《核酸研究》,第35卷,第19期,第330页);Rohetal,2010TrendsBiotechnol.28:291(Roh等人,2010年,《生物技术趋势》,第28卷,第291页)。通常,将条形码放置在寡核苷酸中其非天然存在的位置处,即,条形码包含这样的核苷酸序列,其不同于可在靠近条形码所在位置的序列的附近发现的任何天然存在的寡核苷酸序列(例如,V序列和/或J序列)。根据本文所述的某些实施例,可包括此类条形码序列,作为本发明所公开的寡核苷酸的元件B1和/或B2。因此,本文所述的寡核苷酸组合物中的某些也可在某些实施例中包含一个、两个或更多个条形码,而在某些其他实施例中,这些条形码中的一些或全部可不存在。在某些实施例中,所有条形码序列将具有相同或类似的GC含量(例如,GC含量相差不超过20%、或不超过19%、18%、17%、16%、15%、14%、13%、12%、11%或10%)。测序可使用多种可用的高通量单分子测序仪器和系统中的任何一种进行测序。示例性测序系统包括边合成边测序系统,诸如亿明达基因组分析仪(IlluminaGenomeAnalyzer)和相关仪器(加利福尼亚州圣地亚哥的亿明达公司(Illumina,Inc.,SanDiego,CA))、赫利克斯遗传分析系统(HelicosGeneticAnalysisSystem)(马萨诸塞州剑桥的赫利克斯生物科学公司(HelicosBioSciencesCorp.,Cambridge,MA))、太平洋生物科学PacBioRS(加利福尼亚州门洛帕克的太平洋生物科学公司(PacificBiosciences,MenloPark,CA))或具有类似功能的其他系统。使用杂交至扩增的DNA分子内的限定区域的一组测序寡核苷酸来完成测序。测序寡核苷酸被设计成使得V和J编码基因区段可被基于本公开和根据可公开利用的数据库中出现的已知适应性免疫受体基因序列所生成的序列独特地识别。参见例如,U.S.A.N.13/217,126;U.S.A.N.12/794,507;PCT/US2011/026373;或PCT/US2011/049012。示例性TCRBJ区测序引物示于表4中:表4:TCRBJ测序引物术语“基因”意指DNA中涉及产生多肽链诸如TCR或Ig多肽的全部或一部分(例如,含CDR3的多肽)的区段;其包含编码区之前和之后的区域“前导区和尾随区(trailer)”以及单独的编码区段(外显子)之间的间插序列(内含子),并且也可包含调控元件(例如,启动子、增强子、阻遏蛋白结合位点等),并且也可包含如本文所述的重组信号序列(RSS)。本发明实施例的核酸(本文也称为多核苷酸)可为RNA的形式或DNA的形式,所述DNA包括cDNA、基因组DNA和合成DNA。DNA可为双链的或单链的,并且如果是单链的话,可为编码链或非编码(反义)链。根据本发明实施例使用的编码TCR或免疫球蛋白或其区域(例如,V区、D区段、J区、C区等)的编码序列可与本领域已知的任何给定TCR或免疫球蛋白基因区或多肽结构域(例如,V区结构域、CDR3结构域等)的编码序列相同,或者可为不同编码序列,由于遗传密码的冗余或简并性,所述不同编码序列编码相同TCR或免疫球蛋白区或多肽。在某些实施例中,经扩增的J区编码基因区段可各自具有2、3、4、5、6、7、8、9、10或约15、20或更多个核苷酸的独特序列限定标识符标签,其位于相对于RSS位点的限定位置。例如,可在经扩增的TCRβCDR3编码区的Jβ区编码区段中在RSS位点下游的+11至+14的位置处使用四碱基标签。然而,这些和相关实施例不必如此限制并且还设想了其他相对短的核苷酸序列限定标识符标签,其可在J区编码基因区段中被检测到并且基于其相对于RSS位点的位置被限定。这些在不同适应性免疫受体编码基因座之间可有差别。重组信号序列(RSS)由被12+/-1bp(“12-信号”)或23+/-1bp(“23-信号”)的间隔区隔开的两条保守序列(七聚物5'-CACAGTG-3'和九聚物5'-ACAAAAACC-3')组成。已确定多个核苷酸位置对于重组是重要的,包括七聚物第一位和第二位处的CA二核苷酸,并且七聚物第三位处的C也显示出具有强烈偏好性,九聚物的第5、6、7位处的A核苷酸也是如此。(Ramsden等人,1994;Akamatsu等人,1994;Hesse等人,1989)。其他核苷酸的突变具有极小或不一致的效应。间隔区虽然可变性更强,但也会影响重组,而且单核苷酸置换被证实显著影响重组效率(Fanning等人,1996,Larijani等人,1999;Nadel等人,1998)。已描述了识别具有明显不同重组效率的RSS多核苷酸序列的标准(Ramsden等人,1994;Akamatsu等人,1994;Hesse等人,1989以及Cowell等人,1994)。因此,测序寡核苷酸可杂交至RSS位点下游位置+11至位置+14处的经扩增J编码基因区段内的四碱基标签附近。例如,TCRB的测序寡核苷酸可被设计成退火至刚好在该“标签”下游观察到的共有核苷酸基序,使得序列读段开始的四个碱基将独特地识别J编码基因区段(参见例如,WO/2012/027503)。CDR3编码区(对于TCR,被定义为编码V区段的第二保守半胱氨酸与J区段的保守苯丙氨酸之间的TCR多肽的核苷酸)的平均长度为35+/-3个核苷酸。因此以及在某些实施例中,使用V区段寡核苷酸引物与从具体TCR或IgHJ区(例如,如本文所述的TCRJβ、TCRJγ或IgHJH)的J区段标签开始的J区段寡核苷酸引物进行的PCR扩增将几乎总是捕获50碱基对读段中的完整V-D-J接合部。IgHCDR3区(被定义为V区段中的保守半胱氨酸与J区段中的保守苯丙氨酸之间的核苷酸)的平均长度受约束程度不及TCRβ基因座处,但将通常在约10与约70个核苷酸之间。因此以及在某些实施例中,使用V区段寡核苷酸引物与从IgHJ区段标签开始的J区段寡核苷酸引物进行的PCR扩增将捕获100碱基对读段中的完整V-D-J接合部。退火至错配模板序列并支持在其上的多核苷酸延伸的PCR引物被称为混杂引物(promiscuousprimer)。在某些实施例中,TCR和IgJ区段反向PCR引物可被设计为使之与测序寡核苷酸的重叠最小化,以便使多重PCR情况下的混杂引发最小化。在一个实施例中,TCR和IgJ区段反向引物可通过退火至共有剪接位点基序而锚定在3'端,同时与测序引物的重叠最小化。一般来讲,可使用默认参数下的已知序列/引物设计与分析程序来选择TCR以及IgV和J区段引物,以便在一致的退火温度下在PCR中操作。对于测序反应,示例性的IGHJ测序引物延伸三个核苷酸跨越保守的CAG序列,如WO/2012/027503中所述。样品可从中获得测试生物样品的受试者或生物来源可为人类或非人类动物、或转基因的或克隆的或经组织工程改造(包括通过使用干细胞)的生物体。在本发明的某些优选实施例中,受试者或生物来源可已知患有、或可疑似患有、或面临风险患有循环肿瘤或实体瘤或其他恶性病症、或自身免疫疾病、或炎性病症,并且在本发明的某些优选实施例中,受试者或生物来源可已知没有此类疾病的风险或不存在此类疾病。某些优选实施例设想了这样的受试者或生物来源,其为人类受试者,诸如已根据本领域公认的临床诊断标准诊断为患有癌症或存在形成或罹患癌症的风险的患者,所述临床诊断标准为诸如美国马里兰州贝塞斯达的美国国家癌症研究所(U.S.NationalCancerInstitute(Bethesda,MD,USA))的那些标准或如下文献中所述:DeVita,Hellman,andRosenberg'sCancer:PrinciplesandPracticeofOncology(2008,Lippincott,WilliamsandWilkins,Philadelphia/Ovid,NewYork)(DeVita、Hellman和Rosenberg,《癌症:肿瘤学原理与实践》,2008年,Lippincott、Williams和Wilkins,费城/纽约州奥维德);PizzoandPoplack,PrinciplesandPracticeofPediatricOncology(Fourthedition,2001,Lippincott,WilliamsandWilkins,Philadelphia/Ovid,NewYork)(Pizzo和Poplack,《儿科肿瘤学的原理与实践》,第四版,2001年,Lippincott、Williams和Wilkins,费城/纽约州奥维德);以及VogelsteinandKinzler,TheGeneticBasisofHumanCancer(Secondedition,2002,McGrawHillProfessional,NewYork)(Vogelstein和Kinzler,《人类癌症的遗传基础》,第二版,2002年,麦格劳希尔专业出版,纽约);某些实施例设想了根据此类标准而已知没有患有、形成或罹患癌症的风险的人类受试者。某些其他实施例设想了非人类受试者或生物来源,例如非人类灵长目动物诸如猕猴、黑猩猩、大猩猩、长尾黑颚猴、红毛猩猩、狒狒或其他非人类灵长动物,包括本领域已知可作为临床前模型(包括实体瘤和/或其他癌症的临床前模型)的此类非人类受试者。某些其他实施例设想了作为哺乳动物的非人类受试者,例如,小鼠、大鼠、兔、猪、绵羊、马、牛、山羊、沙鼠、仓鼠、豚鼠或其他哺乳动物;许多此类哺乳动物可为本领域已知作为包括循环肿瘤或实体瘤和/或其他癌症的某些疾病或障碍的临床前模型的受试者(例如,Talmadgeetal,2007Am.J.Pathol.170:793(Talmadge等人,2007年,《美国病理学杂志》,第170卷,第793页);Kerbel,2003Cane.Biol.Therap.2(4Suppl1):S134(Kerbel,2003年,《癌症生物学和疗法》,第2卷,第4期增刊1,第S134页);Manetal,2007Cane.Met.Rev.26:737(Man等人,2007年,《癌症与转移综述》,第26卷,第737页);Cespedesetal,2006Clin.Transi.Oncol.8:318(Cespedes等人,2006年,《临床与转化肿瘤学》,第8卷,第318页))。然而,实施例的范围并非旨在如此限制,使得还存在设想的其他实施例,其中受试者或生物来源可为非哺乳类脊椎动物,例如另一种高等脊椎动物,或鸟类、两栖动物或爬行动物物种,或另一种受试者或生物来源。可通过如下方式提供生物样品:从受试者或生物来源获得血液样品、活检标本、组织外植体、器官培养物、生物流体或者任何其他组织或细胞制备物。优选地,生物样品包含来自受试者或生物来源的淋巴样细胞的DNA,以举例说明而非限制的方式,其可包含一个或多个TCR或BCR基因座处的重排DNA。在某些实施例中,可例如通过手术切除、穿刺活检或获得包含细胞混合物的测试生物样品的其他方式,从实体组织(例如,实体瘤)获得测试生物样品。根据某些实施例,可能有利的是,根据大量既有方法中的任何一种来分离淋巴样细胞(例如,T细胞和/或B细胞),其中分离的淋巴样细胞是从淋巴样细胞天然存在于其中的组织、环境或周围移除或分离的那些淋巴样细胞。因此可从生物样品,诸如从多种组织和生物流体样品(包括骨髓、胸腺、淋巴腺、淋巴结、外周组织和外周血液)获得B细胞和T细胞,但外周血液最容易获得。可对任何外周组织取样,检测其中存在B细胞和T细胞,因而设想将其中的B细胞和T细胞用于本文所述的方法。可从中获得适应性免疫细胞的组织和生物流体包括但不限于皮肤、上皮组织、结肠、脾脏、粘膜分泌物、口腔粘膜、肠粘膜、阴道粘膜或阴道分泌物、宫颈组织、神经节、唾液、脑脊液(CSF)、骨髓、脐带血、血清、浆膜液、血浆、淋巴液、尿液、腹水、胸膜液、心包液、腹膜液、腹腔液、培养液、条件培养液或灌洗液。在某些实施例中,可从血浆分离置换法样品(apheresissample)分离适应性免疫细胞。可通过静脉切开术从受试者获得外周血样品。通过本领域技术人员已知的技术,例如通过密度梯度分离技术来分离外周血单核细胞(PBMC)。在某些实施例中,使用全部PBMC进行分析。对于核酸提取,可使用本领域已知的方法和/或可商购获得的试剂盒,例如通过使用DNA血液小提试剂盒(),从细胞提取总基因组DNA。单个单倍体基因组的近似质量为3pg。优选地,使用至少100,000个至200,000个细胞进行分析,即来自二倍体T细胞或B细胞的约0.6至1.2μgDNA。使用PBMC作为来源,可估计T细胞的数量为总细胞的约30%。也可估计B细胞的数量为PBMC制备物中总细胞的约30%。Ig和TCR基因座含有许多不同可变(V)基因区段、多样性(D)基因区段和连接(J)基因区段,这些基因区段在早期淋巴分化期间经历重排过程。Ig以及TCRV、D和J基因区段序列是本领域已知的,并且可在公共数据库诸如GENBANK中获得。V-D-J重排由重组酶复合体介导,其中RAGl和RAG2蛋白质通过如下方式发挥关键作用:在位于V基因区段下游、D基因区段两侧和J基因区段上游的重组信号序列(RSS)处识别和切割DNA。不适当的RSS可减少、甚至完全避免重排。重组信号序列(RSS)由被12+/-1bp(“12-信号”)或23+/-1bp(“23-信号”)的间隔区隔开的两条保守序列(七聚物5'-CACAGTG-3'和九聚物5'-ACAAAAACC-3')组成。已确定多个核苷酸位置对于重组是重要的,包括七聚物第一位和第二位处的CA二核苷酸,并且七聚物第三位处的C也显示出强烈偏好性,九聚物的第5、6、7位处的A核苷酸也是如此。(Ramsdenetal.1994Nucl.Ac.Res.22:1785(Ramsden等人,1994年,《核酸研究》,第22卷,第1785页);Akamatsuet.al.1994J.Immunol.153:4520(Akamatsu等人,1994年,《免疫学杂志》,第153卷,第4520页);Hesseet.al.1989GenesDev.3:1053(Hesse等人,1989年,《基因与发育》,第3卷,第1053页))。其他核苷酸的突变具有极小或不一致的效应。间隔区虽然可变性更强,但也会影响重组,而且单核苷酸置换被证实显著影响重组效率(Fanninget.al.1996Cell.Immunol.Immunopath.79:1(Fanning等人,1996年,《细胞免疫学与免疫病理学》,第79卷,第1页);Larijanietal.1999Nucl.Ac.Res.27:2304(Larijani等人,1999年,《核酸研究》,第27卷,第2304页);Nadeletal.1998J.Immunol.161:6068(Nadel等人,1998年,《免疫学杂志》,第161卷,第6068页);Nadeletal.,1998J.Exp.Med.187:1495(Nadel等人,1998年,《实验医学杂志》,第187卷,第1495页))。已描述了识别具有明显不同重组效率的RSS多核苷酸序列的标准(Ramsdenetal1994Nucl.Ac.Res.22:1785(Ramsden等人,1994年,《核酸研究》,第22卷,第1785页);Akamatsuet.al.1994J.Immunol.153:4520(Akamatsu等人,1994年,《免疫学杂志》,第153卷,第4520页);Hesseetal.1989GenesDev.3:1053(Hesse等人,1989年,《基因与发育》,第3卷,第1053页);以及Leeetal.,2003PLoS1(1):E1(Lee等人,2003年,公共科学图书馆,第1卷,第1期,第E1页))。就Ig重链(IgH)、TCRβ(TCRB)和TCRδ(TCRD)基因而言,重排过程一般以D到J重排开始,接着是V到D-J重排;而就Igκ(IgK)、Igλ(IgL)、TCRα(TCRA)和TCRγ(TCRG)基因而言,涉及直接V到J重排。重排基因区段之间的序列一般以环形切除产物的形式缺失,所述环形切除产物也称TCR切除环(TREC)或B细胞受体切除环(BREC)。V、D和J基因区段的许多不同组合代表所谓的组合组库,据估计,Ig分子的组合组库大小为约2×l06,TCRαβ分子的组合组库大小为约3×l06,TCRγ分子的组合组库大小为约5×103。重排过程期间,在V、D和J基因区段的接合位点处发生核苷酸的缺失和随机插入,从而产生高度多样的接合区,这些接合区对Ig分子和TCR分子的总组库有显著贡献,估计大于1012。成熟B淋巴细胞在卵泡中心经由体细胞高频突变(即,导致Ig分子亲和力成熟的过程)发生抗原识别,进一步扩充其Ig组库。体细胞高频突变过程集中于IgH和Ig轻链基因的V-(D-)J外显子,并涉及单核苷酸突变,有时还涉及核苷酸插入或缺失。体细胞突变的Ig基因也存在于卵泡或后卵泡起源的成熟B细胞恶性肿瘤中。在本文所述的某些实施例中,可将V区段和J区段引物用于PCR反应,以便扩增测试生物样品中的重排TCR或BCRCDR3编码DNA区,其中每种功能性TCR或IgV编码基因区段包含V基因重组信号序列(RSS),每种功能性TCR或IgJ编码基因区段包含J基因RSS。在这些和相关实施例中,每种经扩增的重排DNA分子可包含(i)TCR或IgV编码基因区段有义链的至少约10、20、30、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900、1000(包括这些数值间的所有整数值)或更多个连续核苷酸,其中至少约10、20、30、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900、1000或更多个连续核苷酸位于V基因RSS的5’端,并且/或者每种经扩增的重排DNA分子可包含(ii)TCR或IgJ编码基因区段有义链的至少约10、20、30、40、50、60、70、80、90、100、200、300、400、500(包括这些数值间的所有整数值)或更多个连续核苷酸,其中至少约10、20、30、40、50、60、70、80、90、100、200、300、400、500或更多个连续核苷酸位于J基因RSS的3’端。除非有明确相反的指示,否则实施本发明的某些实施例将采用微生物学技术、分子生物学技术、生物化学技术、分子遗传学技术、细胞生物学技术、病毒学技术和免疫学技术中的常规方法,这些技术都属于本领域技术,下文出于举例说明目的提及若干这些技术。以下文献和论文充分阐述了此类技术。参见例如,Sambrook,etal.,MolecularCloning:ALaboratoryManual(3rdEdition,2001)(Sambrook等人,《分子克隆:实验指南》,第3版,2001年);Sambrook,etal.,MolecularCloning:ALaboratoryManual(2ndEdition,1989)(Sambrook等人,《分子克隆:实验指南》,第2版,1989年);Maniatisetal.,MolecularCloning:ALaboratoryManual(1982)((Maniatis等人,《分子克隆:实验指南》,1982年);Ausubeletal.,CurrentProtocolsinMolecularBiology(JohnWileyandSons,updatedJuly2008)(Ausubel等人,《现代分子生物学实验技术》,约翰·威利父子出版公司,2008年7月更新);ShortProtocolsinMolecularBiology:ACompendiumofMethodsfromCurrentProtocolsinMolecularBiology,GreenePub.AssociatesandWiley-Interscience(《精编分子生物学实验指南:现代分子生物学实验技术方法汇编》,格林出版公司与威利国际科学公司);Glover,DNACloning:APracticalApproach,vol.I&II(IRLPress,OxfordUniv.PressUSA,1985)(Glover,《DNA克隆:实用方法》,第I和II卷,IRL出版社,美国牛津大学出版社,1985年);CurrentProtocolsinImmunology(Editedby:JohnE.Coligan,AdaM.Kruisbeek,DavidH.Margulies,EthanM.Shevach,WarrenStrober2001JohnWiley&Sons,NY,NY)(《现代免疫学实验技术》,编辑:JohnE.Coligan、AdaM.Kruisbeek、DavidH.Margulies、EthanM.Shevach、WarrenStrober,2001年,约翰·威利父子出版公司,纽约州纽约市);Real-TimePCR:CurrentTechnologyandApplications,EditedbyJulieLogan,KirstinEdwardsandNickSaunders,2009,CaisterAcademicPress,Norfolk,UK(《实时PCR:现代技术与应用》,JulieLogan、KirstinEdwards和NickSaunders编辑,2009年,Caister学术出版社,英国诺福克);Anand,TechniquesfortheAnalysisofComplexGenomes,(AcademicPress,NewYork,1992)(Anand,《分析复杂基因组的技术》,学术出版社,纽约,1992年);GuthrieandFink,GuidetoYeastGeneticsandMolecularBiology(AcademicPress,NewYork,1991)(Guthrie和Fink,《酵母遗传学与分子生物学指南》,学术出版社,纽约,1991年);OligonucleotideSynthesis(N.Gait,Ed.,1984)(《寡核苷酸合成》,N.Gait编辑,1984年);NucleicAcidHybridization(B.Harnes&S.Higgins,Eds.,1985)(《核酸杂交》,B.Harnes和S.Higgins编辑,1985年);TranscriptionandTranslation(B.Harnes&S.Higgins,Eds.,1984)(《转录和翻译》,B.Harnes和S.Higgins编辑,1984年);AnimalCellCulture(R.Freshney,Ed.,1986)(《动物细胞培养》,R.Freshney编辑,1986年);Perbal,APracticalGuidetoMolecularCloning(1984)(Perbal,《分子克隆实用指南》,1984年);Next-GenerationGenomeSequencing(Janitz,2008Wiley-VCH)(《下一代基因组测序》,Janitz,2008年,威利-VCH出版公司);PCRProtocols(MethodsinMolecularBiology)(Park,Ed.,3rdEdition,2010HumanaPress)(《PCR实验技术》(分子生物学方法),Park编辑,第3版,2010年,胡马纳出版社);ImmobilizedCellsAndEnzymes(IRLPress,1986)(《固定化细胞和酶》,IRL出版社,1986年);论文MethodsInEnzymology(AcademicPress,Inc.,N.Y.)(《酶学方法》,学术出版社,纽约);GeneTransferVectorsForMammalianCells(J.H.MillerandM.P.Caloseds.,1987,ColdSpringHarborLaboratory)(《哺乳动物细胞的基因转移载体》,J.H.Miller和M.P.Calos编辑,1987年,冷泉港实验室);HarlowandLane,Antibodies,(ColdSpringHarborLaboratoryPress,ColdSpringHarbor,N.Y.,1998)(Harlow和Lane,《抗体》,冷泉港实验室出版社,纽约冷泉港,1998年);ImmunochemicalMethodsInCellAndMolecularBiology(MayerandWalker,eds.,AcademicPress,London,1987)(《细胞与分子生物学中的免疫化学方法》,Mayer和Walker编辑,学术出版社,伦敦,1987年);HandbookOfExperimentalImmunology,VolumesI-IV(D.M.WeirandCCBlackwell,eds.,1986)(《实验免疫学手册》,第I-IV卷,D.M.Weir和CCBlackwell编辑,1986年);Riott,EssentialImmunology,6thEdition,(BlackwellScientificPublications,Oxford,1988)(Riott,《基础免疫学》,第6版,布莱克威尔科学出版公司,牛津,1988年);EmbryonicStemCells:MethodsandProtocols(MethodsinMolecularBiology)(KurstadTurksen,Ed.,2002)(《胚胎干细胞:方法与实验技术》(分子生物学方法),KurstadTurksen编辑,2002年);EmbryonicStemCellProtocols:Volume1:IsolationandCharacterization(MethodsinMolecularBiology)(KurstadTurksen,Ed.,2006)(《胚胎干细胞实验技术:第1卷:分离与表征》(分子生物学方法),KurstadTurksen编辑,2006年);EmbryonicStemCellProtocols:VolumeII:DifferentiationModels(MethodsinMolecularBiology)(KurstadTurksen,Ed.,2006)(《胚胎干细胞实验技术:第II卷:分化模式》(分子生物学方法),KurstadTurksen编辑,2006年);HumanEmbryonicStemCellProtocols(MethodsinMolecularBiology)(KursadTurksenEd.,2006)(《人胚胎干细胞实验技术》(分子生物学方法),KursadTurksen编辑,2006年);MesenchymalStemCells:MethodsandProtocols(MethodsinMolecularBiology)(DarwinJ.Prockop,DonaldG.Phinney,andBruceA.BunnellEds.,2008)(《间充质干细胞:方法与实验技术》(分子生物学方法),DarwinJ.Prockop、DonaldG.Phinney和BruceA.Bunnell编辑,2008年);HematopoieticStemCellProtocols(MethodsinMolecularMedicine)(ChristopherA.Klug,andCraigT.JordanEds.,2001)(《造血干细胞实验技术》(分子医学方法),ChristopherA.Klug和CraigT.Jordan编辑,2001年);HematopoieticStemCellProtocols(MethodsinMolecularBiology)(KevinD.BuntingEd.,2008)(《造血干细胞实验技术》(分子生物学方法),KevinD.Bunting编辑,2008年);NeuralStemCells:MethodsandProtocols(MethodsinMolecularBiology)(LeslieP.WeinerEd.,2008)(《神经干细胞:方法与实验技术》(分子生物学方法),LeslieP.Weiner编辑,2008年)。除非提供了明确定义,否则结合本文所述的分子生物学、分析化学、合成有机化学和医药化学使用的命名,以及本文所述的分子生物学、分析化学、合成有机化学和医药化学的实验室程序和技术是本领域熟知且常用的那些。标准技术可用于重组技术、分子生物学、微生物学、化学合成、化学分析,药物制备、配制和递送,以及患者治疗。术语“分离的”意指将材料从其初始环境(例如,如果材料是天然存在的,则初始环境为天然环境)移除。举例来说,没有分离天然存在于活体动物体内其初始环境中的那些组织、细胞、核酸或多肽,而是从天然系统内的一部分甚至全部共存材料中分离出相同的组织、细胞、核酸或多肽。这种核酸可以为载体的一部分并且/或者这种核酸或多肽可以为组合物(例如,细胞裂解物)的一部分,但仍然是分离的,因为所述载体或组合物不是这种核酸或多肽的天然环境的一部分。术语“基因”意指DNA中涉及产生多肽链的区段;其包含编码区之前和之后的区域,即“前导区和尾随区”,以及位于多个独立的编码区段(外显子)之间的间插序列(内含子)。除非上下文另外要求,否则在本说明书和权利要求书通篇中,词语“包含”及其变型形式诸如“包括”和“具有”应以开放式包含的意义解释,也就是说,解释为“包括但不限于”。所谓“由…组成”意指包括,并通常限于后接短语“由…组成”的任何事物。所谓“基本上由…组成”意指包括该短语之后所列的任何元素,并限于不妨碍或促成公开内容中规定的所列元素的活性或作用的其他元素。因此,短语“基本上由…组成”表示所列元素是必需的或强制性的,而其他元素不是必需的,可能存在、也可能不存在,具体取决于这些其他元素是否影响所列元素的活性或作用。在本说明书和随附权利要求书中,单数形式“一个”、“一种”和“所述”包括复数指代,除非公开内容明确规定并非如此。如本文所用,在具体实施例中,数值之前的术语“约”或“大约”指示该数值加上或减去5%、6%、7%、8%或9%的范围。在其他实施例中,数值之前的术语“约”或“大约”指示该数值加上或减去10%、11%、12%、13%或14%的范围。在另外其他实施例中,数值之前的术语“约”或“大约”指示该数值加上或减去15%、16%、17%、18%、19%或20%的范围。本说明书通篇提及的“一个实施例”或“实施例”或“一个方面”意指结合该实施例描述的具体特征、结构或特性包括在本发明的至少一个实施例中。因此,本说明书通篇各处出现的短语“在一个实施例中”或“在实施例中”不一定都指同一个实施例。此外,具体特征、结构或特性可以在一个或多个实施例中以任何合适的方式组合。实例实例1:单分子标记。单分子标记过程使用聚合酶链反应方法为适应性免疫受体编码序列标记上独特条形码和通用引物。用来标记多个独立条形码的PCR反应使用QIAGEN多重PCR主混合物(QIAGEN部件编号206145,美国加利福尼亚州瓦伦西亚的凯杰公司(Qiagen,Valencia,CA))、10%Q-溶液(凯杰公司(QIAGEN))及300ng模板DNA。加入合并的引物,使最终反应液的聚集正向引物浓度为2μM,聚集反向引物浓度为2μM。正向引物由退火至V基因的核苷酸序列部分(退火至V基因的区段示于表2中)和5’端的通用引物(pGEXf,表3)构成。聚集引物列于表6中。由于这些引物具有更大的特异性,所以可在V引物的3’端与通用引物序列的5’端之间随机插入核苷酸。反向引物具有可退火至J基因区的一段核苷酸(表2)、在J引物5’端上由随机核苷酸构成的8bp条形码,以及在8bp随机条形码5’端上的通用引物(pGEXr,表3)。这些引物的例子列于表5中。由随机核苷酸构成的8bp条形码可能更短或更长,额外的碱基对增加了独特条形码的数量。采用7次循环PCR反应将核苷酸标签结合到分子上。热循环条件为:95℃5分钟,然后95℃30秒、68℃90秒、72℃30秒循环7次。循环后,将反应在72℃温度下保持10分钟。一旦抗原受体分子被标记上携带随机8bp标签的引物,就使用ExoSAP-IT(产品编号78200,美国加利福尼亚州圣克拉拉的昂飞公司(Affymetrix,SantaClara,CA))将任何剩余的引物破坏掉。ExoSAP-IT是得自昂飞公司(Affymetrix)的具有外切核酸酶I和虾碱性磷酸酶活性的产品;外切核酸酶I能破坏单链DNA,而虾碱性磷酸酶能降解dNTP。在该实例中使用了10μlPCR试剂和4μlExoSAP-IT。将反应液在37℃下温育15分钟,随后在80℃下温育15分钟使ExoSAP-IT失活。此时,分子被独特地标记上了条形码和通用引物。为了扩增标记的产物,用通用pGEX引物执行另一次PCR反应。该PCR反应使用QIAGEN多重PCR主混合物(QIAGEN部件编号206145,美国加利福尼亚州瓦伦西亚的凯杰公司(Qiagen,Valencia,CA))、10%Q-溶液(凯杰公司(QIAGEN))以及作为模板的6μl净化PCR反应液。将正向通用(pGEXf)引物加入混合物中,使其最终浓度为2μM,接着将反向通用引物(pgEXr)加入反应液中,使其最终浓度为2μM。为了对这些分子进行测序,使用pGEX引物掺入Illumina接头。反应条件与上文相同,不同的是将引物替换为加尾引物(下表7(SEQIDNO:5686-5877)。采用7次循环PCR反应将也包含8bp标签和6bp随机核苷酸组的Illumina接头结合到分子上。热循环条件为:95℃5分钟,然后95℃30秒、68℃90秒、72℃30秒循环7次。循环后,将反应在72℃温度下保持10分钟。一旦标记的分子被“加尾”了Illumina接头,它们就适于测序。对于该实例,在IlluminaHISEQTM测序平台上通过进入适应性免疫受体编码序列中的8bp随机物进行测序。经测序的分子包含8bp随机标签。用单个细胞的适应性免疫受体编码多核苷酸序列来扩增具有相同的CDR3和8bp随机标签序列的每个经测序的分子。表5示出了用于单分子测序的J引物(反向引物),表6示出了V引物(正向引物)。PCR方案很简短:用以上引物进行第一次PCR(循环5次),以便独特地标记每个分子;然后用通用引物(PGEX)进行第二次PCR(循环35次),从而扩增所述分子。这些反应之后是加尾Illumina接头的PCR反应。表5表6表7示例性加尾引物实例2:对适应性免疫受体编码序列进行单细胞标记该实例描述了通过RT-PCR对免疫球蛋白及T细胞受体重链和轻链编码序列进行单细胞标记。将从健康人类志愿者新鲜抽取的血液用作白细胞来源。获得100,000至300,000个白细胞需要的全血量不足1mL;使用1至3mL血液执行血细胞分离。将血液置于-1077(美国密苏里州圣路易斯的西格玛公司(Sigma,St.Louis,MO))上,根据供应商说明执行密度梯度离心,而从血液中分离出外周血单核细胞(PBMC)。使用全血CD45微珠(美国加利福尼亚州奥本的美天旎公司(MiltenyiBiotec,Auburn,CA)),遵循制造商的指导并基本按照Koehletal.(2003Leukemia17:232)(Koehl等人,2003年,《白血病》,第17卷,第232页)所述的过程,使CD45+造血细胞结合于抗CD45包被磁珠,而分离出CD45+造血细胞。将白细胞的细胞悬液置于磷酸盐缓冲盐水溶液(PBS)中洗涤,然后调节至每毫升1×106个细胞的浓度。将1至3μL(1至3×103个细胞)的等分试样分配到保持在预先冷冻过的板架中的冰上的96孔PCR多孔板的多个孔中。在填充所有板孔之后,立即将板密封并置于干冰上冷冻,使细胞裂解。在下文所述的逆转录制备步骤期间,将板保持在干冰上。使用Illumina测序用SMARTerTM超低量RNA试剂盒(美国加利福尼亚州山景城的Clontech公司(Clontech,MountainView,CA)),基本按照供应商的说明执行逆转录。将380μl稀释缓冲液与20μlRNA酶抑制剂(40U/μl)混合,制得储备反应缓冲液。然后将250μl反应缓冲液与100μl12μΜ的3’SmarterTMCDSII寡核苷酸(5’-Bio-AAGCAGTGGTATCAACGCAGAGTACT(30)NN-3’[SEQIDNO:5878]溶液混合,其中Bio为生物素部分;AAGCAGTGGTATCAACGCAGAGTAC[SEQIDNO:5879]为通用接头序列,T(30)(SEQIDNO:5880)为胸腺嘧啶残基的30聚体,N为任意核苷酸(A、C、G或T)。通过如下方式设置用于逆转录的第一步退火反应:将3.5μl含有3’SmarterTMCDSII寡核苷酸引物的反应缓冲液加入装有裂解细胞的96孔板的每个孔中,将板密封再将其置于72℃环境中温育3分钟,之后将其放回冰上的冷却架。将200μl的5x第一链缓冲液、25μl的100mM二硫苏糖醇(DTT)、100μl的dNTP(10mM)、25μl的RNA酶抑制剂(40U/μl)和100μl的逆转录酶合并,制得逆转录主混合物(450μl,供100次反应用)。制备96孔工作板,每孔装有1.0μl编有条形码的3’-SmartTMCDSII寡核苷酸。3’-SmartCDSII寡核苷酸序列为:5'-AAGCAGTGGTATCAACGCAGAGTACBBBBBBBBrGrGrG-P-3’[SEQIDNO:5881],其中AAGCAGTGGTATCAACGCAGAGTAC[SEQIDNO:5879]为通用接头序列;BBBBBBBB为8-核苷酸条形码(参见以下列表,了解条形码的例子);rG为核糖鸟嘌呤;P为3’磷酸阻断部分。表8.条形码列表(96个JS条形码):名称序列JS01CAAGGTCAJS02GCATAACTJS03CTCTGATTJS04TACGTACGJS05TACGCGTTJS06CTCAGTGAJS07TCTGATATJS08CATATGCTJS09CGTAATTAJS10ACGTACTCJS11CTTCTAAGJS12ACTATGACJS13GACGTTAAJS14ACAAGATAJS15GACTAAGAJS16GTGTCTACJS17TTCACTAGJS18AATCGGATJS19AGTACCGAJS20TTGCCTCAJS21TCGTTAGCJS22TATAGTTCJS23TGGCGTATJS24TGGACATGJS25AGGTTGCTJS26ATATGCTGJS27GTACAGTGJS28ATCCATGGJS29TGATGCGAJS30GTAGCAGTJS31GGATCATCJS32GTGAACGTJS33ATTAAGCGJS34TATTGGCGJS35CGATTACAJS36TGTCATCGJS37TATCAAGTJS38AGGCTTGAJS39GATAACCAJS40AATCCTGCJS41GTTATATCJS42ACACACGTJS43ATACGACTJS44ATCTTCGTJS45ACATGTATJS46TCCACAGTJS47CAGTCTGTJS48TCCATGTGJS49TCACTGCAJS50ATGGTCAAJS51CAAGTCACJS52TAGACGGAJS53CAGCTCTTJS54GAGCGATAJS55CTCGAGAAJS56ATGACACCJS57CTTCACGAJS58CTATAAGGJS59CGTAGAGTJS60ATAGATACJS61TCGTCGATJS62TAAGAATCJS63AATGACAGJS64AGCTAGTGJS65TGAGACCTJS66AGCGTAATJS67TAACCAAGJS68GATGGCTTJS69GCATCTGAJS70TTCCGGTAJS71GACACTCTJS72TTAAGCATJS73TGCTACACJS74TCAGCTTGJS75CATGTAGAJS76TTCGGAACJS77GCAATTCGJS78CAAGAGGTJS79TCGATTAAJS80GAATGGACJS81AGAATCAGJS82AACTGCCAJS83AAGTAACGJS84ACTCAATGJS85CCTAGTAGJS86CTGACGTTJS87TGCAGACAJS88AGTTGACCJS89GTCTCCTAJS90CTGCAATCJS91TGAGCGAAJS92TTGGACTGJS93AGCAATCCJS94CGAACTACJS95TTAATGGCJS96GCTTAGTA向装有1.0μl编有条形码的3’-SmartTMCDSII寡核苷酸的96孔工作板的每个孔中加入4.5μl主混合物,并且在退火反应完成后,将5.5μl包含编有条形码的3’-SmartTMCDSII寡核苷酸的主混合物从96孔工作板的每个孔转移到逆转录退火板的对应位置(相应)孔中。将逆转录退火板置于热循环仪上,以42℃下90分钟、然后70℃下10分钟的步骤运行程序。从每孔中的白细胞释放的所有多聚AmRNA转录分子在该温度分布下合成第一cDNA链。根据非限制性理论,在合成第一cDNA链之后,孔中的每个cDNA分子在5’端和3’端均含有通用接头序列,并且在5’端独特地标记上8个核苷酸的条形码。任选地,可在该步骤处将来自所有96个反应的编有条形码的cDNA分子合并,再重新等分到PCR板上,接着利用此PCR板PCR扩增免疫球蛋白或T细胞受体cDNA。执行该合并再分配步骤的结果是,在后续用适应性免疫受体编码(例如,IG或TCR)C区段基因特异性引物执行的PCR扩增反应中,基本上所有编有条形码的cDNA分子都具有基本上均匀的占比。接下来,利用固相可逆化固定纯化法(SPRI),按以下步骤分离逆转录/合成cDNA第一链的产物:将来自逆转录反应板中每个孔的内容物与25μlAmpureTMXPSPRI磁珠(美国加利福尼亚州布雷亚的贝克曼库尔特公司(Beckman-CoulterInc.,Brea,CA))的悬液混合,在室温下温育8分钟,然后使用MagnaBotTM磁力分离器(美国威斯康星州麦迪逊的普洛麦格公司(Promega,Madison,WI)),根据供应商的说明在室温下执行磁微珠分离。立即加入SPRI磁微珠固定的cDNA第一链,使用Advantage2TMPCR试剂(Clontech)根据制造商的说明执行多次5’RACE(快速扩增cDNA末端)PCR扩增反应。每次扩增反应都加入了50μlPCR主混合物,该PCR主混合物包含dNTP、UPM引物混合物、如本文别处所述的IG/TCR引物混合物,以及Advantage2TM聚合酶和PCR缓冲液。热循环条件为:95℃1分钟;95℃30秒、63℃30秒、72℃3分钟,循环30次;72℃7分钟;接着将反应保持在10℃温度下,直到作好Illumina测序准备。PCR引物序列为:Illumina测序文库制备通过翻转式离心96孔板将PCR产物合并,然后使用BeckmanCoulterAmpureTMXP微珠,根据供应商的说明纯化合并的产物,去除短于200至300bp的DNA片段。使用Caliper生物分析仪(美国康涅狄格州诺沃克的珀金埃尔默公司(PerkinElmer,Norwalk,CT)),通过毛细管电泳评估DNA纯度,确认大多数dsDNA的大小在600至700bp范围内。使用荧光测定法或通过A260紫外吸光度量化dsDNA产物。使用1μg经纯化的DNA作为Illumina样品制备方案(美国加利福尼亚州圣地亚哥的亿明达公司(IlluminaInc.,SanDiego,CA))的输入物,根据IlluminaDNA样品制备指南(部件编号15026486Rev.C,2012年7月,美国加利福尼亚州圣地亚哥的亿明达公司(Illumina,Inc.,SanDiego,CA))构建测序文库。该方案生成的测序文库可使用Illumina测序仪、2000测序仪和2500测序仪上的末端配对流通池完成测序。在利用试剂盒v2的Illumina测序仪上,根据测序方案执行500次循环Illumina测序。试剂盒中的化学品提供最多能在仪器上完成525次循环测序的成套试剂,而且提供足够用于251次循环配对末端运行的试剂,再加上两个八次循环索引读段。Illumina测序方案描述于试剂盒v2试剂制备指南(ReagentKitv2ReagentPrepGuide),部件编号15034097Rev.B,2012年10月(美国加利福尼亚州圣地亚哥的亿明达公司(IlluminaInc.,SanDiego,CA))。待测序的DNA靶标的结构示意图示于图6中(其中Ig重链用作例子)。实例3:T细胞受体α和β序列的高通量配对提供了实例,以便将本发明的高通量配对方法应用于T细胞受体(TCR)蛋白质的α多肽链和β多肽链配对。决定αβT细胞的抗原特异性的αβT细胞受体(TCR)蛋白质是由以下两种肽构成的异源二聚体:较长的β链(TCRB)和较短的α链(TCRA)(1-6)。最近已开发出高通量测序分析法,用于对TCR的α和β链及免疫球蛋白的重链和轻链执行谱型分析,这种分析法可用于多种诊断应用(7-15),包括检测癌克隆及测量淋巴性恶性肿瘤中的微小残留病的临床诊断应用(13、16-20)。然而,目前高通量方法每次只能对TCR的一条链测序。为了重建T细胞受体以便用于功能分析、治疗用途或对受体-抗原结合建模,必须成对识别来自完整TCR的α链和β链。已多次尝试使用单细胞技术将α链和β链配对。一种方法是分离独立的B细胞或T细胞,采用桥式PCR物理连接重链和轻链,再测序(参见下文的参考文献21-26)。或者,可在单细胞水平上使重链和轻链带上条形码(参见参考文献25、27-31)。虽然多种单细胞方法已得到明显改进,但仍面临技术挑战,且通量有限。这些方法还需要完好无损的单细胞,这让评估组织或实体瘤中的浸润性T细胞变得困难。本发明提供了不使用单细胞就可将α链序列与β链序列高通量配对的方法,此方法采用组合学途径而非物理分离来将α链和β链配对。作为首个实证,申请人将该技术应用于来自健康供体的外周血,在单次实验中就识别出了约35,000个配对的αTCR基因序列和βTCR基因序列。此方法还可应用于来自受试者的组织样品,或血液样品和组织样品两者。本发明的方法依赖于这样的观察结果:T细胞的每个克隆群体的重排TCRA核苷酸序列和重排TCRB核苷酸序列几乎都是独特的。不同TCR序列通过如下方式出现:在淋巴细胞发育期间,体细胞中基因区段发生重组,以及在V-J、V-D和D-J接合部处发生核苷酸的模板独立缺失或插入(32)。这种不同寻常的多样性意味着编码特异性T细胞克隆的TCRA链和TCRB链的mRNA通常只存在于包括该克隆的细胞群中。申请人通过如下方式来利用这种极大的多样性:将T细胞的样品分为多个亚群,然后对TCRAmRNA分子和TCRBmRNA分子测序,确定每个亚群中是否存在每种TCR链。来自一个克隆的TCRA序列和TCRB序列应当在T细胞的相同亚群中见到,而且只在这些亚群中见到。在一些实施例中,此方法可涉及从样品的细胞中提取基因组DNA而非mRNA,用于扩增特异性适应性免疫受体异源二聚体的多肽链。将TCRA链与TCRB链配对于是变成了统计学问题:要宣称出现了独特配对,就必须证实给定克隆占用与另一克隆相同的T细胞亚群集合的可能性非常小。对于使用本发明方法的实验中的许多克隆来说,给定克隆占用与另一克隆相同的T细胞亚群集合的概率接近零。图7示出了占用T细胞亚群的数量与共用亚群的概率的关系曲线图。对于使用本发明方法的模拟实验,其中T细胞被分为各包含70,000个T细胞的96个亚群,该曲线图给出了存在于给定数量亚群(x轴)中的任何克隆将出现于完全相同的亚群中的概率(y轴)。本发明的方法提供了根据初始样品中克隆的频率、每个亚群中的T细胞总数以及所形成的亚群的数量来将该克隆的TCRA序列和TCRB序列配对的方式。最后两个参数是实验设计的一部分,可调整这两个参数,来选择哪个范围中的T细胞克隆频率将被准确配对。图8示出了根据本发明实施例的本发明方法的示意图。在图8A中,将固定数量的T细胞(例如,70,000个外周血单核细胞(PBMC))随机分配到96孔板的每个孔中,形成96个不同亚群。提取总细胞RNA,再由孔中的RNA逆转录成cDNA。在每个孔内对TCRA序列和TCRB序列进行PCR扩增,并将孔特异性寡核苷酸DNA条形码连接到经扩增的受体分子上。在图8B中,将编有条形码的TCRA扩增子和TCRB扩增子合并到一起。在图8C中,在相同文库中始终观察到的成对TCRA/TCRB序列代表由同一细胞的克隆携带的序列。接下来,将经扩增的分子合并,执行高通量DNA测序,读取每条链的受体序列和条形码。使用经测序的条形码将受体序列分配给初始的孔,另外,我们通过寻找TCRA序列识别出推定的成对TCRA/TCRB序列,所述TCRA序列偶然与特异性TCRB序列共用的孔比预期多得多。这涉及计算逆多重化以将每个TCR序列定位回其所来源的孔。免疫组库是高度多样的,并且两个克隆将共用孔模式的概率极小,因此可推测共用孔模式的任一个TCRA/TCRB对来自同一克隆。为了证实申请人提出的方法具有前所未有的通量,我们取样了960万个人类PBMC,并将它们等量细分到96孔微量滴定板的各孔中。每个孔因此容纳100,000个PBMC,其中预计约70,000个为T细胞。在提取RNA,再合成并过滤cDNA之后,我们执行的多次测序产生了29800万个读段,平均覆盖率为每个T细胞15个读段。(详情参见补充材料)。我们将每条TCRA序列的孔占用模式与每条TCRB序列的孔占用模式比较,识别出了推定的同源对。孔占用模式也可称为容器占用模式。在一些实施例中,将每条TCRB序列的孔占用模式与每条TCRA序列的孔占用模式比较。与给定TCRA产生最小p值的TCRB被标记为可能的配对伴侣。于是凭借计算置换观察到的孔模式,就估计出了推定的TCRA/TCRB对集合的假发现率(FDR)。该程序量化了配对的必然性,同时容许实验效率低下,这将在下文更详细描述。下文提供了有关计算FDR的说明。实验结果示于图9A和图9B中。FDR为1%时(在图9A中以虚线示出),发现了来自25,000个不同克隆的34,763对TCRA和TCRB序列。图9A示出了采用96孔和70,000个T细胞/孔的实验的假发现率曲线。FDR曲线为L型,意味着大多数TCRA/TCRB对从背景噪声中清晰地显出。以极高置信度判定了这些TCRA/TCRB对中的大多数。图9B示出了该配对实验的完成度。灰色(浅色阴影)条表示随孔占用数而变的观察到的TCRB序列的总数,黑色阴影条表示这些TCRB序列之中有多少序列以不到1%的FDR配对。我们已成功使占用15-70个孔的超过85%的TCRB序列配对。剩下的序列不能以高置信度配对,可能是因为较低的TCRA和TCRBmRNA水平会导致序列丢失。图10示出了用于该实验的示例性工作流程。在该示例性工作流程中,从抗凝血液开始(1),用Ficoll梯度液分离出PBMC(2),将PBMC细胞等分到96孔板的每个孔中(3),分离96孔板中的RNA(4),合成cDNA(5),用基因特异性PCR引物扩增TCRA序列(6),用基因特异性PCR引物扩增TCRB序列(7),运行巢式PCR以将测序接头和孔特异性条形码添加到TCRA序列(8),运行巢式PCR以将测序接头和孔特异性条形码添加到TCRA序列(9),最后将扩增子合并,得到测序文库(10)。图11示出了孔占用数与配对的TCRB序列的分率的关系。图11示出了来自采用96孔和70,000个T细胞/孔的实验的TCRB克隆的配对收率(配对序列的分率)。按照其中观察到TCRB克隆的孔的数量(x轴)将TCRB克隆分类,并计算出以低于1%的FDR配对的这些序列的分率(y轴)。为了验证实验设计和统计框架,我们执行了图12中概述的样品混合实验,由此直接测得了FDR。图12示出了双受试者验证实验的示意图。从标记为‘X’和‘Y’的两名健康成人受试者采集外周血。使用深度免疫测序来表征每名受试者的TCRA和TCRB组库(14、15、33)。然后将来自这两名受试者的PBMC混合,使用所得的混合物执行本发明的方法。真阳性对必须包括来自同一名受试者的TCRA和TCRB,而大约一半的假阳性结果将为跨受试者的TCRA/TCRB对。我们从这些数据中识别出受试者X特有的652,027个TCRA序列和703,909个TCRB序列,以及受试者Y特有的395,476个TCRA序列和433,086个TCRB序列。接下来,我们将来自这两名受试者每者的120万个PBMC合并,将混合样品以25,000个PBMC(约17,500个T细胞)/孔的密度分配于96个孔中,然后对混合样品执行本发明的方法。真实配对必须包括来自单名受试者的独特组库的TCRA和TCRB,而大约一半错误配对应包括来自一名受试者的TCRA序列和来自另一名受试者的TCRB序列,因此该实验直接评估了识别出的TCRA/TCRB对的可信度,以及所述方法控制错误配对的比率的能力。样品混合实验的FDR分析结果示于图13中,其中我们将所有的TCRA/TCRB对分为三类:(i)两个TCR序列都为受试者X独有的TCRA/TCRB对(“X/X”);(ii)两个TCR序列都为受试者Y独有的TCRA/TCRB对(“Y/Y”);(iii)一个序列为受试者X独有,另一个序列为受试者Y独有的TCRA/TCRB对(“X/Y”)。图13A中的垂直虚线示出了估计的FDR<1%的截断值。在1%的预测FDR处,我们观察到1,115个X/X对、706个Y/Y对和7个X/Y对。在该实验中,检测到的TCRA和TCRB对比第一个实验中要少,原因是更少T细胞被分配到每个孔,这使灵敏度范围朝更常见克隆偏移。如果跨供体对占错误配对的一半,可以预知,该配对阈值处的FDR将为1.06%,这显示与本发明方法作出的预测非常吻合。图13B将该错误配对分析扩展到0.1%至20%范围内的预测FDR值。在整个该范围内,预测值都紧密跟随经验值,这表明我们的分析框架提供了有效的FDR估值,对于不同应用,可调整这些估值来平衡特异性和灵敏度。在图14A至14D中,示出了mRNA子样品中及各TCRA/TCRB对内的孔丢失率。图14A示出了由使用70,000个T细胞/孔的本文所述方法的实验得出的mRNA的第一子样品中的孔丢失率。TCRA的中值丢失率(垂直虚线)为20%,TCRB的中值丢失率为14%。图14B示出了由使用本文所述方法的实验得出的mRNA的第二子样品中的孔丢失率。TCRA的中值丢失率为24%,TCRB的中值丢失率为21%。图14C示出了由mRNA的第一子样品中确信配对的序列(FDR<1%)估计的孔丢失率(与图14A中相同的数据)。TCRA的中值丢失率为14%,TCRB的中值丢失率为10%,这些中值丢失率从图A中的估值向下偏倚。图14D示出了由mRNA的全部合并样品中确信配对的序列(FDR<1%)估计的丢失率,也就是说,我们发现约35,000个TCRA/TCRB对时所用的数据。TCRA的中值丢失率为7%,TCRB的中值丢失率为3%。在考虑偏倚之后,我们预计真实中值丢失率分别为10%和5%。图15示出了随克隆频率和输入T细胞的数量而变的模拟孔占用数。使用本发明方法的实验中的关键设计参数是分配至每个孔的T细胞的数量。图15示出了采用96孔板及在100-100,000个细胞/孔范围内的T细胞输入(x轴)的模拟实验的结果。不同颜色描绘了具有不同组库频率的克隆,并且该曲线图示出了这些克隆在我们的模拟实验中占用的孔的数量。为了从不同频带捕获克隆,可以简单地改变使用本发明方法的实验中输入T细胞的数量。也可以凭借改变板上各孔的输入T细胞的数量,在单个实验中捕获多个频带。最后,我们通过用JurkatE6-1T-ALL细胞系执行加标(spike-in)实验,证实了我们的方法能够使来自已知克隆样品的TCR链正确配对,对于所述JurkatE6-1T-ALL细胞系来说,TCRBV、D、J和CDR3序列是已知的(34)。我们以70,000个PBMC中不到1个的靶标频率(以约1/150,000测量)将Jurkat细胞加标到大量PBMC样品中,再以约50,000个PBMC/孔的密度将样品细胞分配到96孔微量滴定板中,然后对PBMC和加标序列执行盲法配对分析。在21个孔中观察到已知的Jurkat序列,这些孔中有14个孔包含JurkatTCRA和JurkatTCRB两者。该重叠水平产生2.6×10-11的p值,该p值远低于我们的FDR阈值,故相比于所示任一Jurkat序列与任何非Jurkat序列的配对(下一最小p值为5.2×10-5),更有力地证实配对成功。受试者和样品制备在该实例中,研究供体为44岁和51岁的两名健康男性。在获取知情同意书后,从每位供体抽取静脉血到10mlEDTA血液试管(美国新泽西州富兰克林湖的碧迪公司(BecktonDickinson,FranklinLakes,NJ))中。用FicollHistopaque梯度液(美国密苏里州圣路易斯的西格玛公司(Sigma,St.Louis,MO))分离新鲜抗凝血液,再抽吸外周血单核细胞(PBMC)层。使用1xRBC裂解溶液(美国加利福尼亚州奥本的美天旎公司(MiltenyiBiotec,Auburn,CA))来裂解红血细胞。将细胞置于pH7.2的1xPBS(Ambion,美国加利福尼亚州福斯特城的生命技术公司(LifeTechnologies,FosterCity,CA))中洗涤,再重悬于RNAlater溶液(美国加利福尼亚州瓦伦西亚的凯杰公司(Qiagen,Valencia,CA))中。使用TC-20细胞计数仪(美国加利福尼亚州赫拉克勒斯的伯乐公司(Bio-Rad,Hercules,CA))对细胞进行计数,然后使用RNAlater溶液调节细胞悬液的浓度。将细胞悬液保存在4℃,直到使用为止。在等分细胞之前,将细胞悬液调节到500或2,000个细胞/μlRNAlater溶液的浓度,再利用BiomekFX液体处理器(美国马萨诸塞州丹弗斯的贝克曼库尔特公司(BeckmanCoulter,Danvers,MA))将多份50μl等分试样分配到0.8ml深孔板(美国加利福尼亚州联合市的爱思进公司(Axygen,UnionCity,CA))的多个孔中。采用将等体积细胞悬液从常用均质细胞悬液贮存器分配到96孔板上的机器人式方法,确保相似数量的细胞被置于每个板孔中。被置于每个孔中的单核细胞的最终数量为:初始配对实验100,000个(图9中的结果),样品混合实验25,000个(图12示意图,以及图13中的结果),JurkatT-ALL加标实验50,000个。RNA分离和逆转录通过如下方式裂解细胞:加入400μl裂解缓冲液RLTPlus(凯杰公司(Qiagen)),在室温下混合并温育30分钟。采用自动化方案在QIAsymphony实验室机器人(凯杰公司(Qiagen))上分离总细胞RNA。将RNA洗脱到96深孔板中的50μl无RNA酶水中,立即保存在-80℃环境中。使用CaliperRNAPico灵敏度测定法(美国马萨诸塞州沃尔瑟姆的珀金埃尔默公司(PerkinElmer,Waltham,MA))评估每份RNA样品的质量和数量。使用SuperscriptVILOcDNA合成试剂盒(美国加利福尼亚州卡尔斯巴德的英杰公司(Invitrogen,Carlsbad,CA)),根据制造商的方案将15微升(等于4至5ngRNA)的RNA逆转录成单链cDNA。在分离RNA和逆转录反应期间,使孔种类与装有96份细胞悬液等分试样的初始来源板保持一致。多重PCR和条形码使用TCRA或TCRB基因特异性引物,对cDNA板的两份96孔板平行样执行多重PCR,该多重PCR针对TCRA和TCRB使用多种V基因特异性引物,针对每种同种型使用单种C区段反向引物(参见表S1和表S2,获得PCR引物的列表)。将多重PCRPlus试剂(凯杰公司(Qiagen))用于50μl反应液和按下列条件执行的35次循环PCR扩增:95℃变性300秒,然后90℃变性30秒、60℃退火90秒、72℃延伸90秒循环35次。使用SPR1Select磁珠(贝克曼库尔特公司(BeckmanCoulter))纯化PCR产物,再将每个PCR反应的5μl等分试样用于巢式PCR。八次循环的第二次巢式PCR在扩增子的每个末端掺入Illumina末端配对接头序列,随后就可将样品置于IlluminaMiSeq或HiSeq测序仪(美国加利福尼亚州圣地亚哥的亿明达公司(Illumina,SanDiego,CA))中测序。另外,如之前所述将孔特异性八碱基DNA条形码掺入每份PCR产物中(33)。参见表S3获得条形码的完整列表。在TCRA和TCRB特异性PCR及后续纯化期间,以及在设置巢式PCR反应期间,使板取向与初始来源板保持一致。第二次巢式PCR用特异性条形码为每个板上每个孔中的DNA编上条形码。条形码1-96用于TCRB扩增子孔,条形码97-192用于TCRA扩增子孔。在PCR扩增之后,将所有TCRA孔和所有TCRB孔(分别有96个孔)中产物合并成两份5μl等分试样,得到两个DNA样品库。用荧光测定法确定每个库的DNA浓度,再使用CaliperDNA1K测定法(铂金埃尔默公司(PerkinElmer))分离DNA产物,验证正确的扩增子大小且不存在过剩引物和引物二聚体。然后将这两个库(TCRA和TCRB)以等摩尔比率合并成最终测序文库样品,该样品现在包含192种分别编有条形码的DNA物质。高通量测序根据上述方法,利用细胞,每个起始96孔板制备一个测序文库。按照Illumina方案将测序文库稀释、变性并上样到IlluminaHiSeq2500测序仪流通池中,采用快速运行格式执行机载(onboard)聚类生成及边合成边测序。该运行格式使用单次读段测序(15次循环的索引读段,然后是150次循环的对V(D)JC受体结构域测序),每运行一次测序,便分析完毕来自一个供体的DNA。分别用对TCRA和TCRB具有特异性的两种测序引物,从TCRA恒定区和TCRB恒定区引发TCR测序读段。这些引物使来自J-C剪接位点的14个碱基退火,由此,我们便不仅能够确认同种型和孔特异性DNA条形码,还能确认序列是TCRA还是TCRB。高通量测序方法在上文及U.S.A.N.13/217,126、U.S.A.N.12/794,507、PCT/US2011/026373或PCT/US2011/049012中有详细描述,这些专利以引用的方式全文并入本文。T-all克隆加标我们购买了人类T细胞白血病(T-ALL)细胞系JurkatCloneE6-1(TIB-152TM)作为配对的阳性对照,已知该细胞系为αβTCR阳性(35)。JurkatTCRA和TCRB的序列是已知的,还通过深度测序确认了这些序列(34)。在37℃和5%CO2的环境中,在T25烧瓶中的补充有10%胎牛血清(美国马萨诸塞州沃尔瑟姆的飞世尔科技公司(FischerScientific,Waltham,MA))的HyCloneRPMI1640培养基(美国加利福尼亚州圣何塞的赛默科技公司(ThermoScientific,SanJose,CA))中繁殖Jurkat细胞。将收获的细胞稀释,再把大约450个Jurkat细胞加入3000万个PBMC的样品中,使Jurkat克隆以1.5×10-5的频率存在于PBMC样品中,或者说使96孔板的每个孔中装有0.75个Jurkat细胞。将细胞悬液浓度调节至每μl1000个细胞,然后以每孔50μl的体积将细胞悬液机器人式分配到96孔板上。在分配样品细胞悬液之后,板上每个孔内包含50,000个PBMC。然后按图7中所述的本发明标准实验工作流程处理样品板。基于泊松统计,预计在加标Jurkat克隆的该水平下,51个孔将包含已知的TCRαβ对序列。孔中的JurkatTCRαβ序列的丰度应当与TCR受体mRNA表达水平成比例。至少一篇文献报道比较了外周血CD4+T细胞与JurkatE6-1细胞的TCR转录物的数量。作者使用半定量PCR证实,Jurkat细胞中TCR转录物的丰度并不比来自从健康供体血液获得的CD4+细胞的TCR转录物的丰度高(36)。对TCRBcDNA执行下一代测序比较了所有T细胞亚群的TCRB链转录水平,确定天然CD8T细胞亚群、经活化的CD8T细胞亚群和记忆CD8T细胞亚群的这种转录水平相似,都在每个细胞3至12个TCRB转录物的范围内(37)。我们以文献数据为根据,预计孔中存在的Jurkat克隆序列的水平将低于源自PBMC的TCR组库序列的水平。Jurkat克隆的TCRA和TCRBmRNA的相对表达量是会影响实验回收预期的克隆TCRαβ对的能力的另一种生物特性。已有报道称α链和β链的转录活性在T细胞的胸腺内个体发生期间受到调节,并取决于胸腺分化的阶段不同而差异明显(38)。示出了来源于胸腺发育各阶段的肿瘤细胞系来证实相对表达差异。因此,预计作为“第II阶段T细胞”的Jurkat细胞系的TCRA表达水平比TCRB表达水平低(39)。双供体实验为了直接测量配对假阳性率,我们使用如上所述的标准技术从两名健康成人的静脉血中分离出PBMC。将细胞悬液保存在RNAlater溶液中(RNAlater溶液是已知能保护RNA免于降解并保留其中保存的组织或细胞样品的mRNA表达谱的基于铵的高盐RNA防腐剂(40)),保护新鲜分离的PBMC中的细胞RNA。使用机器人式液体处理器将细胞悬液样品分配到微量滴定板的96个孔中。取决于实验设置,放入每个孔中的细胞数量在25,000个与100,000个之间变化。因此,整个板中在细胞计数方面的孔与孔变异性取决于液体处理器的移液精度,估计变异系数(CV)低于25%。我们以来自两名健康成人的PBMC样品为起始物,将细胞均匀分配到三块96孔板的各孔中。第一块板包含来自供体X的细胞,第二块板包含来自供体Y的细胞,第三块板包含通过以1:1比率混合来自这两名供体的样品而制备的细胞。首先将接受分析的供体细胞样品的每块96孔板作为一个单位,通过分离总细胞RNA和合成cDNA这两个工作流程步骤来处理(所有96个孔并行处理)。然后复制所有96个cDNA库,用于在两块独立的96孔板上执行TCRAcDNA和TCRBcDNA的特异性PCR扩增,同时保留孔坐标。我们设计了包含通用接头互补序列的正向V基因和反向C区段PCR引物,这些PCR引物使我们能够在后续有限循环PCR反应中将编有独特条形码的巢式寡核苷酸接头连接到每个DNA模板。在第二次PCR中,每块96孔板中的所有孔特异性TCRA和TCRB扩增子都被标记上192种八核苷酸条形码中的一种。另外,第二次PCR还将Illumina特异性接头掺入扩增子中,这样,我们便能将所有192个孔的产物合并成一个测序文库,继而用HiSeq2500测序。凭借对TCRA和TCRBPCR产物的设计,我们就能如前所述那样对经扩增的cDNA文库进行测序(33)。简言之,首先是较短的15循环读段确定了独特的孔条形码,然后是跨越V(D)JC结构域的150循环测序读段。第二读段的测序引物位于TCRA和TCRB的C区段中,即,来自J-C剪接接合部的14个核苷酸。由于读段长度为150个碱基,这使我们能够解析所有V、D(就TCRB而言)和J基因。TCRA或TCRB同种型中C区段基因的残余14个碱基具有特异性,从而证实读段属于任一同种型,并与孔条形码无关。适应性免疫受体配对的统计分析对于所考虑的每个适应性免疫受体对,我们计算了受体序列所共用的孔数的p值。举例来说,就TCRA/TCRB配对来说,我们认为TCRA序列占用Na个孔,TCRB序列占用Nβ个孔。如果这些序列共用Naβ个孔,那么,N*α=Nα-Nαβ个孔仅被TCRA序列占用,N*β=Nβ-Nαβ个孔仅被TCRB序列占用。假使每个孔包含相同数量的T细胞,那么偶然观察到这种孔共用量的概率取决于孔总数Ntot及边际孔计数Nα和Nβ。我们通过对所有孔构型具有相同边际计数及所占用的孔具有相等或更大重叠的概率求和,获得了推定的TCR对的p值。采用这种方式计算p值有两个目的:其一,考虑了不同数量孔中的TCR序列具有不同的偶然重叠概率这一事实,其二,不需要所占用的孔完全重叠就能捕获与偶然配对的偏差。孔不完全重叠是TCR对的成员间的常见现象,原因是每个细胞中有较少数量的mRNA转录物可能导致孔丢失(参见下文有关“mRNA子样品中及各对内的孔丢失率”的那部分),使得有必要使用考虑了数据的该特征的统计学结果。可对其他TCR或IG配对(比如TCRG/TCRD配对,IGH与IGK或IGL配对)执行这种分析。模拟零p值分布与采用生物学数据分析的许多应用不同,使用本发明方法分析时,p值的零分布既不连续,也不均匀。离散的孔占用模式导致离散的p值,并且选择每个TCR序列的p值作为与可能的配对伴侣的多次比较中见到的最小值,这使零分布朝较小p值偏斜。具有不同孔占用数的TCR克隆也具有不同零分布:在占用中等数量孔的克隆中可观察到更为极端的p值,而且此类孔往往还涉及可能生成极端p值的次数相对少的比较。为了将这些独特性考虑在内,我们使用本发明的置换算法对使用本发明方法的实验的结构进行完全建模。指定一个基因座(通常为TCRA)为“查询”基因座,指定另一个基因座为“靶”基因座。首先对占用i个孔的靶序列数量Ti进行计数,此处i=1、…、96。一种置换涉及下列步骤:对于每种占用水平i,在[0,1]中取样Ti随机数量。表示最大取样数量γi。对于每种占用水平j,在占用i和j个孔的序列的累积分布函数中使用γi确定共用孔的数量Nij。该值代表j个孔中的序列将与采样Ti随机构型后i个孔中的最佳序列共用的孔的最大数量。使用Nij计算占用水平j的p值δj。如果δj小于迄今为止在水平j处见到的最小p值,则将其保存。一旦我们对所占用的靶孔i的每一数量完成了这些步骤,便会获得查询孔j的每一可能数量的最小p值δj。这些值被保存为一种置换的结果,它们直接类似于在将给定查询序列与靶序列集合(如本发明的实验中见到的集合)进行比较时见到的最小p值。申请人获得的结果是基于每次实验运行10,000种置换。假发现率估值在一个实例中,使用上述模拟零分布来估计示例性实验中的假发现率。每种孔占用水平处的克隆具有不同的零p值分布,因此这些克隆也需要单独的FDR估值。该方法遵照Bancroft等人提出的方法(41),他们开发了一种用顺序置换p值估计假发现率的方法。与高通量配对实验中的p值相似,顺序置换p值也是离散且不均匀的,因此Bancroft等人的方法直接适用于我们的情形。在一个实施例中,此方法的步骤如下:对于每种孔占用水平i=1、…、96,构建尽可能多的概率比零点至少低0.05的分区(bin)。该过程考虑到了离散分布中的不连续性,这种不连续性可能导致对真实零假设数量的估值过于保守。对于每种孔占用水平i,采用Bancroft等人提出的第2定理估计观察到的多个p值的零假设数量moi,该数量与基于直方图的估计量mo相关。以常规方式针对占用水平i处的p值,计算任何拒绝阈值a处的FDR。为了获得实验范围的FDR估值如图9和图13中示出的估值,对整个占用水平的预期第一类型错误和总配对判定的数量分别求和,再将前一总和除以后一总和。当观察到的p值的数量较少时(用少量TCR序列获得占用水平可能出现这种情况),m0的估值可能不稳定。为了获得更好的估值,我们将具有较少序列计数和相似零分布的占用水平合并。可使用其他方法确定FDR。在一个实施例中,确定独特的第一适应性免疫受体扩增子序列和独特的第二适应性免疫受体扩增子序列的可能错误配对的假发现率估值包括:计算所述样品中识别出的所述多个推定的同源对每个的p值;将所述多个推定的同源对所有的p值与预期p值分布进行比较,其中计算得到的预期p值分布用来代表不存在真实同源对的实验;然后针对每个推定的同源对,确定假阳性结果的预期比例,由此确定等于或低于所述推定同源对的p值的所有p值,用来代表真实同源配对。mRNA子样品中及各对内的孔丢失率本发明的配对方法取决于可靠地检测来自其所占用的每个孔中的T细胞克隆的TCRA序列和TCRB序列。给定的T细胞通常只会携带每种TCR重排的一些mRNA拷贝(可能只有5到10个),另外,实验效率低下可能导致无法在孔的子集中观察到任一种基因座。为了表征这种孔丢失率,用每孔70,000个T细胞构建实验,再两次独立地从板上的相同RNA库抽取RNA。通过分别处理每次抽取的RNA,表征了由实验设计导致的不完全mRNA采样的效果,这种由实验设计导致的不完全mRNA采样代替了因实验效率低下导致的不完全mRNA采样。以下章节描述了这些mRNA子样品中的孔丢失率。使用配对序列的孔重叠数来估计主要分析过程中用到的经测序mRNA的完整集合中的丢失率。图13示出了该实验中这两份mRNA子样品每一份的估计孔丢失率。在这些分图中,通过将每个TCR序列在一次RNA抽取中占用的孔的数量与来自两次抽取的组合数据进行比较,确定了每个TCR序列的孔丢失率。该分割实验的中值丢失率为:针对TCRA(Y/Y),在20%至24%的范围内;针对TCRB(X/X),在14%至21%的范围内。这些子样品的孔丢失率很高,突显了在实验过程中从细胞充分采集mRNA库样品的重要性。TCRB的丢失率往往低于TCRA的丢失率,这可能反映每个细胞中TCRBmRNA分子的数量居多。图14A至图14D示出了使用不同孔丢失率估计量的结果。在给定实验中,我们通过比较确信配对的(FDR<1%)序列内的孔重叠,计算出丢失率。在TCR对的一个成员中见到但没有在另一个成员中见到的孔被视为缺失。图14C示出了将该估计量应用于图14A所用的mRNA子样品的结果。可以预知,由于孔丢失导致检测TCR对的能力下降,所以基于配对的估值向下偏倚。对TCRA和TCRB两者来说,配对序列的表观丢失率(图14C)比同完整mRNA样品相比见到的丢失率(图14A)低30%。在确定了基于配对的估计量中的偏倚后,我们使用该方法估计了来自高通量配对实验的完整mRNA样品中的孔丢失率。结果示于图14D中。此时的中值丢失率比用于图14C的mRNA子集的中值丢失率低30%至50%,这进一步证实了穷尽采集mRNA库样品非常重要。如果假设图14D中的丢失率比真实丢失率低30%,那么,TCRA的最终中值丢失率为10%,TCRB的最终中值丢失率为5%。尽管一些TCR序列仍实质存在丢失率,我们也确信能够在该实验中分配大约35,000个TCR对,这证实了本发明的方法足够稳健。实例4:将高通量配对应用于适应性免疫受体异源二聚体多肽序列在上文的实例中,本发明方法的应用集中于该方法使TCR序列以高通量准确配对的能力,但本发明方法的作用不限于此,其还是一种能够以多种方式拓宽免疫组库分析范围的灵活工具。举例来说,只是改变每孔中输入T细胞的数量来调整所述方法,就能够使同源TCR链以任何所需频率范围内的频率配对。也可以凭借使板上孔子集中的输入T细胞数量分层,在单个实验中分析测定来自多个频带的同源对。如上所述,此方法可用于使TCR序列或IG序列以高通量准确配对。譬如,本发明的方法可用于使适应性免疫受体异源二聚体的第一多肽链即TCRα(TCRA)链与适应性免疫受体异源二聚体的第二多肽即TCRβ(TCRB)链配对。另外,本发明的方法可用于使适应性免疫受体异源二聚体的第一多肽即TCRγ(TCRG)链与适应性免疫受体异源二聚体的第二多肽即TCRδ(TCRD)链配对。又如,本发明的方法可用于使适应性免疫受体异源二聚体的第一多肽即免疫球蛋白重链(IGH)与适应性免疫受体异源二聚体的第二多肽(选自免疫球蛋白轻链IGL或IGK)配对。所述方法提供了用于识别多个同源对的步骤,所述同源对包含第一多肽和第二多肽,这两种多肽形成了适应性免疫受体异源二聚体,所述适应性免疫受体异源二聚体包含来自样品中的单个克隆的T细胞受体(TCR)或免疫球蛋白(IG),所述样品包含来自哺乳动物受试者的多个淋巴样细胞。如上所述,所述方法包括如下步骤:将多个淋巴样细胞分配到多个容器中,让每个容器装有多个淋巴样细胞;对由得自所述多个淋巴样细胞的mRNA分子逆转录产生的cDNA分子执行多重PCR,而在所述多个容器中生成扩增子的文库。所述扩增子的文库包含:i)编码第一多肽的多个第一适应性免疫受体扩增子,每个这种扩增子包含独特可变(V)区编码序列、独特J区编码序列或者独特J区编码序列与独特C区编码序列两者、至少一个条形码序列、至少一个通用接头序列以及测序平台标签序列,和ii)编码第二多肽的多个第二适应性免疫受体扩增子,每个这种扩增子包含独特V区编码序列、独特J区编码序列或者独特J区编码序列与独特C区编码序列两者、至少一个条形码序列、至少一个通用接头序列以及测序平台标签序列。所述方法还包括用于对扩增子文库执行高通量测序,从而获得多个第一和第二适应性免疫受体扩增子序列的数据集的步骤。另外,所述方法包括通过将每个独特第一适应性免疫受体扩增子序列分配至一个或多个容器来确定每个独特第一适应性免疫受体扩增子序列的容器占用模式,以及通过将每个独特第二适应性免疫受体扩增子序列分配至一个或多个容器来确定每个独特第二适应性免疫受体扩增子序列的容器占用模式,其中所述独特第一或第二适应性免疫受体扩增子序列中的每个条形码序列与具体容器相关。对于独特第一和第二适应性免疫受体扩增子序列形成推定同源对的每种可能配对来说,所述方法涉及计算观察到容器占用模式、或偶然观察到比预期高的任意共用容器比例的统计概率,前提是第一和第二适应性免疫受体扩增子序列不源自淋巴样细胞的相同克隆群体;随后基于分数比预定可能性截断值低的统计概率来识别多个推定的同源对。然后,对于每个识别出的推定同源对,确定独特第一适应性免疫受体扩增子序列和独特第二适应性免疫受体扩增子序列的可能错误配对的假发现率估值。所述方法包括基于所述统计概率和所述假发现率估值,将独特第一适应性免疫受体序列和独特第二适应性免疫受体序列的多个同源对识别为编码所述样品中的所述适应性免疫受体的真实同源对的步骤。在一些实施例中,统计分数可以是计算得到的将独特第一适应性免疫受体扩增子序列和独特第二适应性免疫受体扩增子序列配成每个推定同源对的p值。在一个实施例中,计算统计分数包括计算独特第一和第二适应性免疫受体扩增子序列应当共同占用与观察到的它们实际共同占用的容器数相比一样多或更多的容器数的概率,计算假定没有真实同源配对,并给定了由所述独特第一适应性免疫受体扩增子序列占用的容器数和由所述独特第二适应性免疫受体扩增子序列占用的容器数。实质上,在给定任何两个适应性免疫受体序列的情况下,所述方法分析这两个序列是否偶然共同出现在比预期更多的容器中。在给定总计有N个容器、在总计X个容器中观察到第一适应性免疫受体序列(A)、在总计Y个容器中观察到第二适应性免疫受体序列(B),并且在Z个容器中同时观察到适应性免疫受体序列(A)和(B)的前提下,所述方法规定,给定的序列(A)在N个容器中的X个中被发现(X/N),序列(B)在N个容器中的Y个中被发现(Y/N),计算结果考虑了这两个序列在Z个或更多个容器中被同时发现的概率。在一些实施例中,所观察到的A与B序列之间重叠容器的数量可偶然出现的概率越低,它们并非因偶然而是由于真实同源配对而共同出现的可能性就越高。接下来,基于统计概率来识别具有高配对可能性的多个推定的同源对可包括对于每个独特第一适应性免疫受体扩增子序列而言,识别具有匹配的最低p值分数的独特第二适应性免疫受体扩增子序列,或者对于每个独特第二适应性免疫受体扩增子序列而言,寻找具有匹配的最低p值分数的独特第一适应性免疫受体扩增子序列。在其他实施例中,确定假发现率估值包括:计算样品中识别出的所述多个推定的同源对每个的p值;将所述多个推定的同源对所有的p值与预期p值分布进行比较,其中所述计算得到的预期p值分布用来代表不存在真实同源对的实验;然后针对每个推定的同源对,确定假阳性结果的预期比例,由此确定等于或低于所述推定同源对的p值的所有p值,用来代表真实同源配对。在某些实施例中,计算预期p值分布包括:置换容器,其中已在无真实同源对的其他方面相同的实验中观察到每个第一和第二适应性免疫受体序列,以及计算与每个推定的同源对相关的p值的分布。所述方法包括通过选择具有低于阈值的p值的多个推定的同源对,将独特第一和第二适应性免疫受体序列的多个同源对识别为真实同源对,所述阈值是基于所述假发现率估值计算的。在一个实施例中,识别出的独特第一和第二适应性免疫受体扩增子序列的同源对具有小于1%的假发现率估值。在其他实施例中,识别出的独特第一和第二适应性免疫受体扩增子序列的同源对具有小于2%、3%、4%、5%、6%、7%、8%、9%或10%的假发现率估值。所述方法还可包括在足以促进得自所述多个淋巴样细胞的mRNA分子逆转录的条件及持续时间下,使所述多个容器中的每一个与第一逆转录引物组接触。在某些实施例中,(A)第一寡核苷酸逆转录引物组包括的引物能够逆转录编码所述多个第一和第二适应性免疫受体多肽的多个mRNA序列,从而生成多个第一和第二逆转录的适应性免疫受体cDNA扩增子,其中编码第一适应性免疫受体多肽的所述多个第一逆转录的适应性免疫受体cDNA扩增子包含1)独特V区编码基因序列和2)独特J区编码基因序列,或者独特J区编码基因序列和独特C区编码基因序列两者,并且其中编码第二适应性免疫受体多肽的所述多个第二逆转录的适应性免疫受体cDNA扩增子包含1)独特V区编码基因序列和2)独特J区编码基因序列,或者独特J区编码基因序列和独特C区编码基因序列两者。然后在第二反应中扩增第一和第二逆转录的适应性免疫受体cDNA扩增子。所述反应从如下步骤开始:在足以促进对所述第一和第二逆转录的适应性免疫受体cDNA扩增子进行多重PCR扩增的条件和持续时间下,使所述多个容器中的每一个与第二(B)和第三(C)寡核苷酸引物组接触。在一些方面,(B)第二寡核苷酸引物组包括能够扩增所述多个第一逆转录的适应性免疫受体cDNA扩增子的正向引物和反向引物,其中所述正向引物和反向引物各自能够杂交于第一逆转录的适应性免疫受体cDNA扩增子。第二寡核苷酸引物组中的每对正向引物和反向引物能够扩增所述第一逆转录的适应性免疫受体cDNA扩增子。第二寡核苷酸引物组中的正向引物包含第一通用接头序列和与V区编码基因序列互补的区域。第二寡核苷酸引物组中的反向引物包含第二通用接头序列和与J区编码基因序列或C区编码基因序列互补的区域。(C)第三寡核苷酸引物组包括能够扩增所述多个逆转录的第二适应性免疫受体cDNA扩增子的正向引物和反向引物。第三寡核苷酸引物组中的每对正向引物和反向引物能够扩增所述第二逆转录的适应性免疫受体cDNA扩增子。在一个方面,第三寡核苷酸引物组中的正向引物包含第一通用接头序列和与V区编码基因序列互补的区域。第三寡核苷酸引物组中的反向引物包含第二通用接头序列,以及与J区编码基因序列互补或与C区编码基因序列互补的区域。所述方法还包括生成i)多个第三适应性免疫受体扩增子,每个这种扩增子包含独特V区编码基因序列或其互补序列、独特J区编码基因序列或其互补序列,或者独特J区编码基因序列和独特C区编码基因序列两者或这两者的互补序列,以及第一和第二通用接头序列,和ii)多个第四适应性免疫受体扩增子,每个这种扩增子包含独特V区编码基因序列或其互补序列、独特J区编码基因序列或其互补序列,或者独特J区编码基因序列和独特C区编码基因序列两者或这两者的互补序列,以及第一和第二通用接头序列。然后用另外的引物扩增所述多个第三适应性免疫受体扩增子和所述多个第四适应性免疫受体扩增子。所述方法包括在足以促进对所述多个第三和第四适应性免疫受体扩增子进行第二次多重PCR扩增的条件和持续时间下,使所述多个容器中的每一个与第四(D)寡核苷酸引物组和第五(E)寡核苷酸引物组接触。在一个实施例中,(D)第四寡核苷酸引物组包括能够扩增所述多个第三适应性免疫受体扩增子的正向引物和反向引物,其中所述正向引物和反向引物各自能够杂交于第三适应性免疫受体扩增子。第四寡核苷酸引物组中的每对正向引物和反向引物能够扩增所述第三适应性免疫受体扩增子。第四寡核苷酸引物组中的所述正向引物包含测序平台标签序列和与所述多个第三适应性免疫受体扩增子中的第一通用接头序列互补的区域,所述反向引物包含测序平台标签序列和与所述多个第三适应性免疫受体扩增子中的第二通用接头序列互补的区域。在另一个实施例中,第四寡核苷酸引物组中的所述正向引物和反向引物中的一者或两者包含独特条形码序列,所述条形码序列与其中引入了第四寡核苷酸引物组的容器相关联。(E)第五寡核苷酸引物组包括能够扩增所述多个第四适应性免疫受体扩增子的正向引物和反向引物,其中所述正向引物和反向引物各自能够杂交于所述第四适应性免疫受体扩增子。所述第四寡核苷酸引物组中的每对正向引物和反向引物能够扩增所述多个第四适应性免疫受体扩增子。第五寡核苷酸引物组中的所述正向引物包含测序平台标签序列和与所述多个第四适应性免疫受体扩增子中的第一通用接头序列互补的区域,第五寡核苷酸引物组中的所述反向引物包含测序平台标签序列和与所述多个第四适应性免疫受体扩增子中的第二通用接头序列互补的区域。第四寡核苷酸引物组的所述正向引物和反向引物中的一者或两者包含独特条形码序列,所述条形码序列与其中引入了所述第四寡核苷酸引物组的容器相关联,从而生成包含所述多个第一适应性免疫受体扩增子和所述多个第二适应性免疫受体扩增子的扩增子文库。接下来,所述方法包括将来自所述多个容器的扩增子文库合并成用于测序的混合物。高通量测序方法在上文及U.S.A.N.13/217,126、U.S.A.N.12/794,507、PCT/US2011/026373或PCT/US2011/049012中有详细描述,这些专利以引用的方式全文并入本文。在一个方面,所述多个第一适应性免疫受体扩增子包含C区编码序列。在一些方面,所述多个第二适应性免疫受体扩增子包含C区编码序列。在一些情况下,所述样品包括血液样品。在另一个实施例中,所述样品包括组织样品。在某些实施例中,所述样品包括样品纯化或培养的人类淋巴样细胞。在其他实施例中,所述容器包含至少104个淋巴样细胞。在另一个实施例中,所述样品包含至少104个细胞。所述方法适用于如上所述的各种适应性免疫受体基因座,诸如TCRα(TCRA)链与TCRβ(TCRB)链的配对、TCRγ(TCRG)链与TCRδ(TCRD)链的配对、或免疫球蛋白重链(IGH)与免疫球蛋白轻链IGL或IGK的配对。在适应性免疫受体异源二聚体的第一多肽为IGH链,而适应性免疫受体异源二聚体的第二多肽为IGL和IGK两者的情况下,使用三种不同的扩增引物组,包括:针对IGH的第一寡核苷酸扩增引物组、针对IGK的第二寡核苷酸扩增引物组以及针对IGL的第三寡核苷酸扩增引物组。因此,我们发现本发明的方法和组合物可用于免疫学、医学及治疗法开发方面的许多应用。本发明的方法提供了研究所选免疫受体的集合的一级序列与促成其选择的靶标(和表位)之间的联系的机会。本发明的方法可用于设计实验和控制变量(例如,HLA类型),具体地讲,用于从肿瘤浸润性淋巴细胞识别出关键TCR,用于建立常规或实验性疫苗接种反应的新标准,用于对公众暴露和共有反应进行流行病学分析。本发明的方法还提供了有关每条独立链对给定反应的相对贡献的信息。另外,我们的方法提供了有关是否可能存在控制具体免疫反应的物理TCR链属性的数据。例如,就针对给定类型抗原激发作出的给定类型反应来说,有关一条或两条链的长度或生物物理参数的限值。本发明的方法可用标准实验室用品和设备来执行,无需专门技术,而且起始样品类型的可能范围相当宽泛(肿瘤样品、分选的细胞、悬浮的细胞等)。该技术被设计为可规模化,并可供多种实验室利用。重要的是应认识到,本发明的方法可应用于而且能同样有效地作用于TCRγ/δ,及有效地用于连接免疫球蛋白的重链和轻链(IGH与IGK或IGL)。考虑到本发明方法在单克隆抗体开发方面的实际意义,以及对体液免疫反应的普遍重要性,该方法有可能成为用于生物医学发现的重要技术。可将上述各种实施例组合,提供另外的实施例。本说明书中提及的所有美国专利、美国专利申请公布、美国专利申请、美国以外国家专利、美国以外国家专利申请和非专利出版物均全文以引用方式并入本文。如有必要,可修改上述实施例的各方面以便采用各专利、专利申请和出版物的概念,从而提供另外的实施例。可按照以上具体实施方式对上述实施例作出这些和其他更改。一般来讲,在以下权利要求书中,所用的术语不应解释为将权利要求限制于本说明书和权利要求书中所公开的具体实施例,而应解释为包括所有可能的实施例,连同此类权利要求赋予其权利的等同物的整个范围。因此,权利要求书不受公开内容限制。表格表S1。TCRADV和TCRBV基因正向PCR引物的序列表S2。TCRAC和TCRBC基因反向PCR引物的序列表S3。DNA条形码的序列参考文献1.N.R.Gascoigne,Y.-h.Chien,D.M.Becker,J.Kavaler,Μ.M.Davis,GenomicorganizationandsequenceofT-cellreceptorβ-chainconstant-andjoining-regiongenes.Nature310,387-391(1984)(N.R.Gascoigne、Y.-h.Chien、D.M.Becker、J.Kavaler、Μ.M.Davis,T细胞受体β链恒定区和连接区基因的基因组组构和序列,《自然》,第310卷,第387-391页,1984年)。2.G.Siu,S.P.Clark,Y.Yoshikai,M.Malissen,Y.Yanagi,E.Strauss,T.W.Mak,L.Hood,ThehumanTcellantigenreceptorisencodedbyvariable,diversity,andjoininggenesegmentsthatrearrangetogenerateacompleteVgene.Cell37,393-401(1984)(G.Siu、S.P.Clark、Y.Yoshikai、M.Malissen、Y.Yanagi、E.Strauss、T.W.Mak、L.Hood,人类T细胞抗原受体由重排生成完整V基因的可变基因区段、多样性基因区段和连接基因区段编码,《细胞》,第37卷,第393-401页,1984年)。3.Y.Yoshikai,S.P.Clark,S.Taylor,U.Sohn,B.I.Wilson,M.D.Minden,T.W.Mak,Organizationandsequencesofthevariable,joiningandconstantregiongenesofthehumanT-cellreceptorα-chain.(1985)(Y.Yoshikai、S.P.Clark、S.Taylor、U.Sohn、B.I.Wilson、M.D.Minden、T.W.Mak,人类T细胞受体α-链的可变区基因、连接区基因和恒定区基因的组构和序列,1985年)。4.B.Toyonaga,Y.Yoshikai,V.Vadasz,B.Chin,T.W.Mak,Organizationandsequencesofthediversity,joining,andconstantregiongenesofthehumanT-cellreceptorbetachain.ProceedingsoftheNationalAcademyofSciences82,8624-8628(1985)(B.Toyonaga、Y.Yoshikai、V.Vadasz、B.Chin、T.W.Mak,人类T细胞受体β链的多样性区基因、连接区基因和恒定区基因的组构和序列,《美国国家科学院院刊》,第82卷,第8624-8628页,1985年);在线发布,1985年12月1日电子出版5.M.M.Davis,P.J.Bjorkman,T-cellantigenreceptorgenesandT-cellrecognition.Nature334,(1988)(M.M.Davis、P.J.Bjorkman,T细胞抗原受体基因和T细胞识别,《自然》,第334卷,1988年)。6.J.M.K.Slifka,I.Messaoudi,ThemanyimportantfacetsofT-cellrepertoirediversity.NatureReviewsImmunology4,123-132(2004)(J.M.K.Slifka、I.Messaoudi,T细胞组库多样性的许多重要方面,《自然评论:免疫学》,第4卷,第123-132页,2004年)。7.H.S.Robins,P.V.Campregher,S.K.Srivastava,A.Wacher,C.J.Turtle,O.Kahsai,S.R.Riddell,E.H.Warren,C.S.Carlson,ComprehensiveassessmentofT-cellreceptorbeta-chaindiversityinalphabetaTcells.Blood114,4099-4107(2009)(H.S.Robins、P.V.Campregher、S.K.Srivastava、AWacher、C.J.Turtle、O.Kahsai、S.R.Riddell、E.H.Warren、C.S.Carlson,αβT细胞中的T细胞受体β链多样性的综合评估,《血液》,第114卷,第4099-4107页,2009年);在线发布,11月5日电子出版(10.1182/blood-2009-04-217604)。8.R.L.Warren,B.H.Nelson,R.A.Holt,ProfilingmodelT-cellmetagenomeswithshortreads.Bioinformatics25,458-464(2009)(R.L.Warren、B.H.Nelson、R.A.Holt,用短读段对模型T细胞宏基因组进行谱图分析,《生物信息学》,第25卷,第458-464页,2009年)。9.J.Glanville,W.Zhai,J.Berka,D.Telman,G.Huerta,G.R.Mehta,LNi,L.Mei,P.D.Sundar,G.M.Day,D.Cox,A.Rajpal,J.Pons,Precisedeterminationofthediversityofacombinatorialantibodylibrarygivesinsightintothehumanimmunoglobulinrepertoire.ProceedingsoftheNationalAcademyofSciencesoftheUnitedStatesofAmerica106,20216-20221(2009)(J.Glanville、W.Zhai、J.Berka、D.Telman、G.Huerta、G.R.Mehta、LNi、L.Mei、P.D.Sundar、G.M.Day、D.Cox、A.Rajpal、J.Pons,凭借精密测定组合抗体文库的多样性能深入了解人类免疫球蛋白组库,《美国国家科学院院刊》,第106卷,第20216-20221页,2009年);在线发布,12月1日电子出版(10.1073/pnas.0909775106)。10.S.D.Boyd,E.L.Marshall,J.D.Merker,J.M.Maniar,L.N.Zhang,B.Sahaf,C.D.Jones,B.B.Simen,B.Hanczaruk,K.D.Nguyen,MeasurementandclinicalmonitoringofhumanlymphocyteclonalitybymassivelyparallelVDJpyrosequencing.Sciencetranslationalmedicine1,12ra23-12ra23(2009)(S.D.Boyd、E.L.Marshall、J.D.Merker、J.M.Maniar、L.N.Zhang、B.Sahaf、C.D.Jones、B.B.Simen、B.Hanczaruk、K.D.Nguyen,通过大规模并行VDJ焦磷酸测序对人类淋巴细胞克隆性进行测量和临床监测,《科学转化医学》,第1卷,第12ra23-12ra23页,2009年)。11.H.S.Robins,S.K.Srivastava,P.V.Campregher,C.J.Turtle,J.Andriesen,S.R.Riddell,C.S.Carlson,E.H.Warren,OverlapandeffectivesizeofthehumanCD8+Tcellreceptorrepertoire.Sciencetranslationalmedicine2,47ra64(2010)(H.S.Robins、S.K.Srivastava、P.V.Campregher、C.J.Turtle、J.Andriesen、S.R.Riddell、C.S.Carlson、E.H.Warren,人类CD8+T细胞受体组库的重叠和有效大小,《科学转化医学》,第2卷,第47ra64页,2010年)。12.R.L.Warren,J.D.Freeman,T.Zeng,G.Choe,S.Munro,R.Moore,J.R.Webb,R.A.Holt,ExhaustiveT-cellrepertoiresequencingofhumanperipheralbloodsamplesrevealssignaturesofantigenselectionandadirectlymeasuredrepertoiresizeofatleast1millionclonotypes.Genomeresearch21,790-797(2011)(R.L.Warren、J.D.Freeman、T.Zeng、G.Choe、S.Munro、R.Moore、J.R.Webb、R.A.Holt,人类外周血样品的穷尽T细胞组库测序揭示了抗原选择的识别标志以及直接测得的至少一百万种克隆型的组库大小,《基因组研究》,第21卷,第790-797页,2011年)。13.H.Robins,C.Desmarais,J.Matthis,R.Livingston,J.Andriesen,H.Reijonen,C.Carlson,G.Nepom,C.Yee,K.Cerosaletti,Ultra-sensitivedetectionofrareTcellclones.JournalofImmunologicalMethods,(201l)10.1016/j.jim.2011.09.001)(H.Robins、C.Desmarais、J.Matthis、R.Livingston、J.Andriesen、H.Reijonen、C.Carlson、G.Nepom、C.Yee、K.Cerosaletti,罕见T细胞克隆的超灵敏检测,《免疫学方法杂志》,2011年,10.1016/j.jim.2011.09.001)。14.A.M.Sherwood,C.Desmarais,R.J.Livingston,J.Andriesen,M.Haussier,C.S.Carlson,H.Robins,DeepsequencingofthehumanTCRgammaandTCRbetarepertoiressuggeststhatTCRbetarearrangesafteralphabetaandgammadeltaTcellcommitment.Sciencetranslationalmedicine3,90ra61(2011)(A.M.Sherwood、C.Desmarais、R.J.Livingston、J.Andriesen、M.Haussier、C.S.Carlson、H.Robins,人类TCRγ和TCRβ组库的深度测序表明在αβ和γδT细胞定型后TCRβ发生重排,《科学转化医学》,第3卷,第90ra61页,2011年);在线发布,7月6日电子出版(10.1126/scitranslmed.3002536)。15.K.Larimore,M.W.McCormick,H.S.Robins,P.D.Greenberg,ShapingofhumangermlineIgHrepertoiresrevealedbydeepsequencing.Journalofimmunology189,3221-3230(2012)(K.Larimore,M.W.McCormick,H.S.Robins,P.D.Greenberg,由深度测序揭示人类生殖系IgH组库的成形过程,《免疫学杂志》,第189卷,第3221-3230页,2012年);在线发布,9月15日电子出版(10.4049/jimmunol.l201303)。16.M.Faham,J.Zheng,M.Moorhead,V.E.Carlton,P.Stow,E.Coustan-Smith,C.H.Pui,D.Campana,Deep-sequencingapproachforminimalresidualdiseasedetectioninacutelymphoblasticleukemia.Blood120,5173-5180(2012)(M.Faham、J.Zheng、M.Moorhead、V.E.Carlton、P.Stow、E.Coustan-Smith、C.H.Pui、D.Campana,用于检测急性成淋巴细胞性白血病的微小残留病的深度测序方法,《血液》,第120卷,第5173-5180页,2012年);在线发布,12月20日电子出版(10.1182/blood-2012-07-444042)。17.D.Wu,A.Sherwood,J.R.Fromm,S.S.Winter,K.P.Dunsmore,M.L.Loh,H.A.Greisman,D.E.Sabath,B.L.Wood,H.Robins,High-throughputsequencingdetectsminimalresidualdiseaseinacuteTlymphoblasticleukemia.Sciencetranslationalmedicine4,134ral63-134ral63(2012)(D.Wu、A.Sherwood、J.R.Fromm、S.S.Winter、K.P.Dunsmore、M.L.Loh、H.A.Greisman、D.E.Sabath、B.L.Wood、H.Robins,高通量测序检测急性T成淋巴细胞性白血病的微小残留病,《科学转化医学》,第4卷,第134ral63-134ral63页,2012年)。18.S.A.Grupp,M.Kalos,D.Barrett,R.Aplenc,D.L.Porter,S.R.Rheingold,D.T.Teachey,A.Chew,B.Hauck,J.F.Wright,Chimericantigenreceptor-ModifiedTcellsforacutelymphoidleukemia.NewEnglandJournalofMedicine368,1509-1518(2013)(S.A.Grupp、M.Kalos、D.Barrett、R.Aplenc、D.L.Porter、S.R.Rheingold、D.T.Teachey、A.Chew、B.Hauck、J.F.Wright,急性淋巴细胞白血病的嵌合抗原受体修饰的T细胞,《新英格兰医学杂交》,第368卷,第1509-1518页,2013年)。19.W.-K.Weng,R.Armstrong,S.Arai,C.Desmarais,R.Hoppe,Y.H.Kim,MinimalResidualDiseaseMonitoringwithHigh-ThroughputSequencingofTCellReceptorsinCutaneousTCellLymphoma.Sciencetranslationalmedicine5,214ral71-214ral71(2013)(W.-K.Weng、R.Armstrong、S.Arai、C.Desmarais、R.Hoppe、Y.H.Kim,通过对皮肤T细胞淋巴瘤中的T细胞受体进行高通量测序来监测微小残留病,《科学转化医学》,第5卷,第214ral71-214ral71页,2013年)。20.M.L.Davila,I.Riviere,X.Wang,S.Bartido,J.Park,K.Curran,S.S.Chung,J.Stefanski,O.Borquez-Ojeda,M.Olszewska,EfficacyandToxicityManagementof19-28zCARTCellTherapyinBCellAcuteLymphoblasticLeukemia.Sciencetranslationalmedicine6,224ra225-224ra225(2014)(M.L.Davila、I.Riviere、X.Wang、S.Bartido、J.Park、K.Curran、S.S.Chung、J.Stefanski、O.Borquez-Ojeda、M.Olszewska,用于B细胞急性成淋巴细胞性白血病的19-28zCART细胞疗法的疗效与毒性管理,《科学转化医学》,第6卷,第224ra225-224ra225页,2014年)。21.M.Embleton,G.Gorochov,P.T.Jones,G.Winter,In-cellPCRfrommRNA:amplifyingandlinkingtherearrangedimmunoglobulinheavyandlightchainV-geneswithinsinglecells.Nucleicacidsresearch20,3831-3837(1992)(M.Embleton、G.Gorochov、P.T.Jones、G.Winter,从mRNA开始的细胞内PCR:在单个细胞内扩增并连接重排免疫球蛋白的重链和轻链V基因,《核酸研究》,第20卷,第3831-3837页,1992年)。22.P.-J.Meijer,P.S.Andersen,M.HaahrHansen,L.Steinaa,A.Jensen,J.Lantto,M.B.Oleksiewicz,K.Tengbjerg,T.R.Poulsen,V.W.Coljee,Isolationofhumanantibodyrepertoireswithpreservationofthenaturalheavyandlightchainpairing.Journalofmolecularbiology358,764-772(2006)(P.-J.Meijer、P.S.Andersen、M.HaahrHansen、L.Steinaa、A.Jensen、J.Lantto、M.B.Oleksiewicz、K.Tengbjerg、T.R.Poulsen、V.W.Coljee,在保留天然重链和轻链配对的前提下将人类抗体组库分离,《分子生物学杂志》,第358卷,第764-772页,2006年)。23.N.Chapal,M.Bouanani,M.Embleton,I.Navarro-Teulon,M.Biard-Piechaczyk,B.Pau,S.Peraldi-Roux,In-cellassemblyofscFvfromhumanthyroid-infiltratingBcells.Biotechniques23,518-524(1997)(N.Chapal、M.Bouanani、M.Embleton、I.Navarro-Teulon、M.Biard-Piechaczyk、B.Pau、S.Peraldi-Roux,来自人类甲状腺浸润B细胞的scFv的细胞内组装,《生物技术》,第23卷,第518-524页,1997年)。24.S.M.Kim,L.Bhonsle,P.Besgen,J.Nickel,A.Backes,K.Held,S.Vollmer,K.Dommair,J.C.Prinz,AnalysisofthepairedTCRalpha-andbeta-chainsofsinglehumanTcells.PloSone7,e37338(2012)10.1371/joumal.pone.0037338)(S.M.Kim、L.Bhonsle、P.Besgen、J.Nickel、A.Backes、K.Held、S.Vollmer、K.Dommair、J.C.Prinz,单个人类T细胞的配对TCRα-链和β-链的分析,《公共科学图书馆-综合》,第7卷,第e37338页,2012年,10.1371/joumal.pone.0037338)。25.B.J.DeKosky,G.C.Ippolito,R.P.Deschner,J.J.Lavinder,Y.Wine,B.M.Rawlings,N.Varadarajan,C.Giesecke,T.Domer,S.F.Andrews,P.C.Wilson,S.P.Hunicke-Smith,C.G.Willson,A.D.Ellington,G.Georgiou,High-throughputsequencingofthepairedhumanimmunoglobulinheavyandlightchainrepertoire.Naturebiotechnology31,166-169(2013)(B.J.DeKosky、G.C.Ippolito、R.P.Deschner、J.J.Lavinder、Y.Wine、B.M.Rawlings、N.Varadarajan、C.Giesecke、T.Domer、S.F.Andrews、P.C.Wilson、S.P.Hunicke-Smith、C.G.Willson、A.D.Ellington、G.Georgiou,配对人类免疫球蛋白重链和轻链组库的高通量测序,《自然生物技术》,第31卷,第166-169页,2013年);在线发布,2月电子出版(10.1038/nbt.2492)。26.M.A.Turchaninova,Ο.V.Britanova,D.A.Bolotin,M.Shugay,E.V.Putintseva,D.B.Staroverov,G.Sharonov,D.Shcherbo,I.V.Zvyagin,I.Z.Mamedov,C.Linnemann,T.N.Schumacher,D.M.Chudakov,PairingofT-cellreceptorchainsviaemulsionPCR.Europeanjournalofimmunology43,2507-2515(2013)10.1002/eji.201343453)(M.A.Turchaninova、Ο.V.Britanova、D.A.Bolotin、M.Shugay、E.V.Putintseva、D.B.Staroverov、G.Sharonov、D.Shcherbo、I.V.Zvyagin、I.Z.Mamedov、C.Linnemann、T.N.Schumacher、D.M.Chudakov,经由乳液PCR使T细胞受体链配对,《欧洲免疫学杂志》,第43卷,第2507-2515页,2013年,10.1002/eji.201343453)。27.X.Sun,M.Saito,Y.Sato,T.Chikata,T.Naruto,T.Ozawa,E.Kobayashi,H.Kishi,A.Muraguchi,M.Takiguchi,UnbiasedAnalysisofTCRα/βChainsattheSingle-CellLevelinHumanCD8+T-CellSubsets.PloSone7,e40386(2012)(X.Sun、M.Saito、Y.Sato、T.Chikata、T.Naruto、T.Ozawa、E.Kobayashi、H.Kishi、A.Muraguchi、M.Takiguchi,单细胞水平无偏分析人类CD8+T细胞亚群中的TCRα/β链,《公共科学图书馆-综合》,第7卷,第e40386页,2012年)。28.S.-M.Kim,L.Bhonsle,P.Besgen,J.Nickel,A.Backes,K.Held,S.Vollmer,K.Dommair,J.C.Prinz,AnalysisofthepairedTCRα-andβ-chainsofsinglehumanTcells.PloSone7,e37338(2012)(S.-M.Kim、L.Bhonsle、P.Besgen、J.Nickel、A.Backes、K.Held、S.Vollmer、K.Dommair、J.C.Prinz,单个人类T细胞的配对TCRα-链与β-链分析,《公共科学图书馆-综合》,第7卷,第e37338页,2012年)。29.P.Dash,J.L.McClaren,T.H.OguinIII,W.Rothwell,B.Todd,M.Y.Morris,J.Becksfort,C.Reynolds,S.A.Brown,P.C.Doherty,PairedanalysisofTCRαandTCRβchainsatthesingle-celllevelinmice.TheJournalofclinicalinvestigation121,288(2011)(P.Dash、J.L.McClaren、T.H.OguinIII、W.Rothwell、B.Todd、M.Y.Morris、J.Becksfort、C.Reynolds、S.A.Brown、P.C.Doherty,单细胞水平配对分析小鼠体内的TCRα链与TCRβ链,《临床研究杂志》,第121卷,第288页,2011年)。30.C.E.Busse,I.Czogiel,P.Braun,P.F.Amdt,H.Wardemann,Single-cellbasedhigh-throughputsequencingoffull-lengthimmunoglobulinheavyandlightchaingenes.Europeanjournalofimmunology44,597-603(2014)(C.E.Busse、I.Czogiel、P.Braun、P.F.Amdt、H.Wardemann,全长免疫球蛋白重链和轻链基因的基于单细胞的高通量测序,《欧洲免疫学杂志》,第44卷,第597-603页,2014年);在线发布,2月电子出版(10.1002/eji.201343917)。31.Y.-C.Tan,L.K.Scalfone,S.Kongpachith,C.-H.Ju,X.Cai,T.M.Lindstrom,J.Sokolove,W.H.Robinson,SequencingAntibodyRepertoiresProvidesEvidenceforOriginalAntigenicSinShapingtheAntibodyResponsetoInfluenzaVaccination.ClinicalImmunology,(2014)(Y.-C.Tan、L.K.Scalfone、S.Kongpachith、C.-H.Ju、X.Cai、T.M.Lindstrom、J.Sokolove、W.H.Robinson,对抗体组库测序提供了有关原始抗原痕迹使对流感疫苗的抗体反应成形的证据,《临床免疫学》,2014年)。32.J.D.Ashwell,A.Weissman,inClinicalImmunology,PrinciplesandPractice,R.R.Rich,Ed.(MosbyInternationalLimited,London,2001),chap.5,pp.5.1-5.19(J.D.Ashwell,A.Weissman,载于《临床免疫学:原理和实践》,R.R.Rich编辑,莫斯比国际有限公司,伦敦,2001年,第5章,第5.1-5.19页)。33.C.S.Carlson,R.O.Emerson,A.M.Sherwood,C.Desmarais,M.W.Chung,J.M.Parsons,M.S.Steen,M.A.LaMadrid-Hemnannsfeldt,D.W.Williamson,R.J.Livingston,D.Wu,B.L.Wood,M.J.Rieder,H.Robins,UsingsynthetictemplatestodesignanunbiasedmultiplexPCRassay.Naturecommunications4,2680(2013)10.1038/ncomms3680)(C.S.Carlson、R.O.Emerson、A.M.Sherwood、C.Desmarais、M.W.Chung、J.M.Parsons、M.S.Steen、M.A.LaMadrid-Hemnannsfeldt、D.W.Williamson、R.J.Livingston、D.Wu、B.L.Wood、M.J.Rieder、H.Robins,使用合成模板设计无偏多重PCR测定,《自然通讯》,第4卷,第2680页,2013年,10.1038/ncomms3680)。34.http://www.imgt.org/IMGTrepertoire/Probes/Rearrangements%20and%20junctions/human/Hu_TRrea.html。35.Y.Sandberg,B.Verhaaf,E.J.vanGastel-Mol,I.L.Wolvers-Tettero,J.deVos,R.A.Macleod,J.G.Noordzij,W.A.Dik,J.J.vanDongen,A.W.Langerak,HumanT-celllineswithwell-definedT-cellreceptorgenerearrangementsascontrolsfortheBIOMED-2multiplexpolymerasechainreactiontubes.Leukemia21,230-237(2007)(Y.Sandberg、B.Verhaaf、E.J.vanGastel-Mol、I.L.Wolvers-Tettero、J.deVos、R.A.Macleod、J.G.Noordzij、W.A.Dik、J.J.vanDongen、A.W.Langerak,具有定义明确的T细胞受体基因重排的人类T细胞系作为BIOMED-2多重聚合酶链反应试管的对照,《白血病》,第21卷,第230-237页,2007年);在线发布,2月电子出版(10.1038/sj.leu.2404486)。36.H.P.Bonarius,F.Baas,E.B.Remmerswaal,R.A.vanLier,LJ.tenBerge,P.P.Tak,N.deVries,MonitoringtheT-cellreceptorrepertoireatsingle-cloneresolution.PloSone1,e55(2006)10.1371/joumal.pone.0000055)(H.P.Bonarius、F.Baas、E.B.Remmerswaal、R.A.vanLier、LJ.tenBerge、P.P.Tak、N.deVries,以单克隆解析水平监测T细胞受体组库,《公共科学图书馆-综合》,第1卷,第e55页,2006年,10.1371/joumal.pone.0000055)。37.M.Klinger,K.Kong,M.Moorhead,L.Weng,J.Zheng,M.Faham,Combiningnext-generationsequencingandimmuneassays:anovelmethodforidentificationofantigen-specificTcells.PloSone8,e74231(2013)10.1371/joumal.pone.0074231)(M.Klinger、K.Kong、M.Moorhead、L.Weng、J.Zheng、M.Faham,下一代测序与免疫测定法相结合:一种识别抗原特异性T细胞的新颖方法,《公共科学图书馆-综合》,第8卷,第e74231页,2013年,10.1371/joumal.pone.0074231)。38.H.D.Royer,D.Ramarli,O.Acuto,T.J.Campen,E.L.Reinherz,GenesencodingtheT-cellreceptoralphaandbetasubunitsaretranscribedinanorderedmannerduringintrathymicontogeny.ProceedingsoftheNationalAcademyofSciences82,5510-5514(1985)(H.D.Royer、D.Ramarli、O.Acuto、T.J.Campen、E.L.Reinherz,在胸腺内个体发生期间以有序方式转录编码T细胞受体α和β亚基的基因,《美国国家科学院院刊》,第82卷,第5510-5514页,1985年)。39.R.N.Sangster,J.Minowada,N.Suciu-Foca,M.Minden,T.W.Mak,Rearrangementandexpressionofthealpha,beta,andgammachainTcellreceptorgenesinhumanthymicleukemiacellsandfunctionalTcells.TheJournalofexperimentalmedicine163,1491-1508(1986)(R.N.Sangster、J.Minowada、N.Suciu-Foca、M.Minden、T.W.Mak,人类胸腺白血病细胞和功能性T细胞中的α、β和γ链T细胞受体基因的重排与表达,《实验医学杂志》,第163卷,第1491-1508页,1986年);在线发布,1986年6月1日电子出版(10.1084/jem.163.6.1491)。40.D.Chowdary,J.Lathrop,J.Skelton,K.Curtin,T.Briggs,Y.Zhang,J.Yu,Y.Wang,A.Mazumder,PrognosticgeneexpressionsignaturescanbemeasuredintissuescollectedinRNAlaterpreservative.Thejournalofmoleculardiagnostics8,31-39(2006)(D.Chowdary、J.Lathrop、J.Skelton、K.Curtin、T.Briggs、Y.Zhang、J.Yu、Y.Wang、A.Mazumder,可测量收集在RNAlater防腐剂中的组织的预后基因表达识别标志,《分子诊断学杂志》,第8卷,第31-39页,2006年)。41.T.Bancroft,C.Du,D.Nettleton,EstimationofFalseDiscoveryRateUsingSequentialPermutationp-Values.Biometrics69,1-7(2013)(T.Bancroft、C.Du、D.Nettleton,使用顺序置换p值估计假发现率,《生物统计学》,第69卷,第1-7页,2013年)。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1