CRISPR/Cas9介导的大片段DNA拼接方法与流程
本发明涉及微生物合成生物学、基因组工程和分子生物学领域,具体涉及CRISPR/Cas9介导的大片段DNA拼接方法。
背景技术:
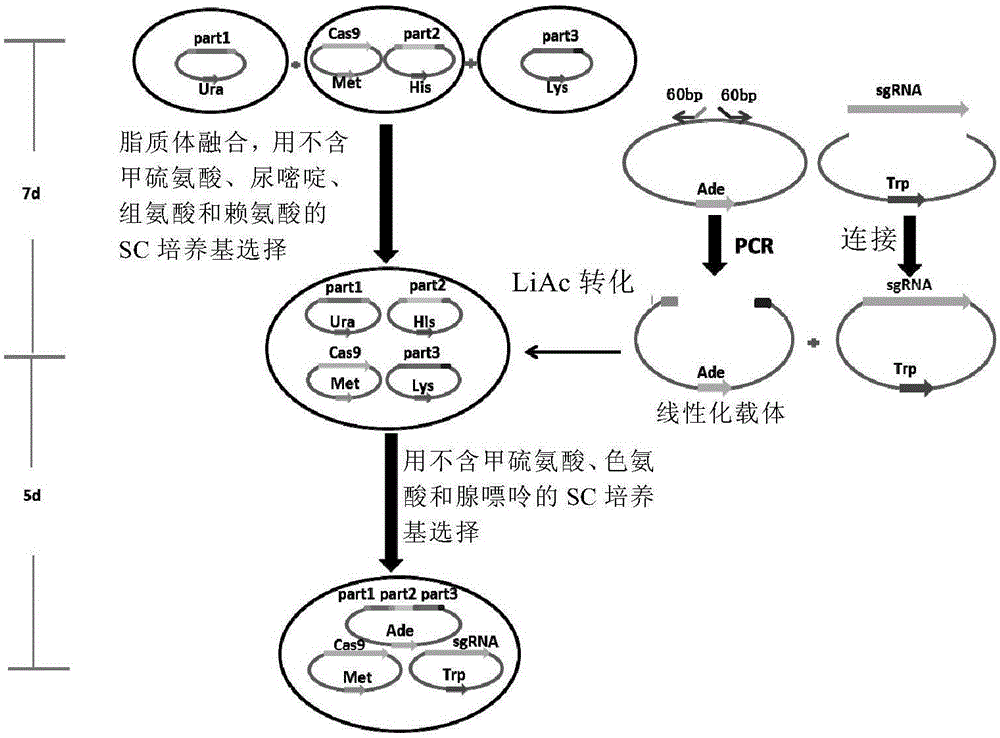
:合成生物学是一个新兴的研究领域。2010年,美国科学家Venter及其研究团队报道了世界上首个人造生命〔Gibson,D.G等,Creationofabacterialcellcontrolledbyachemicallysynthesizedgenome,Science,2010,329(5987):p.52-6〕,这一举世瞩目的研究成果使得合成生物学成为现代生命科学的研究热点。DNA合成、大片段拼接和基因组移植技术的发展是合成生物学研究中重要的技术基础。Venter及其研究团队发展的DNA体外拼接〔Gibson,D.G.等,Completechemicalsynthesis,assembly,andcloningofaMycoplasmagenitaliumgenome,Science,2008,319(5867):p.1215-20〕和体内拼接〔Gibson,D.G等,2010,同前〕技术被广泛应用于合成生物学研究中。DNA体外拼接的原理是将末端具有同源序列的DNA经3’-5’核酸外切酶处理,产生的单链突出端经退火复性后可以将多个DNA片段拼接在一起,而单链的缺刻可以被DNA聚合酶和DNA连接酶修复,从而形成完整的拼接片段。DNA体内拼接体系是利用酵母体内高效的同源重组系统,将末端具有同源序列的DNA片段连同线性化的载体转入酵母体内进行拼接。通过这两个技术,Venter等〔Gibson,D.G.等,2008,同前〕已经成功拼接装配了580kb的生殖道支原体(Mycoplasmagenitalium)基因组。不仅如此,他们还全合成了1.08Mbp的丝状支原体(M.mycoides)基因组JCVI-syn1.0,并移植导入到山羊支原体(M.capricolum)细胞中,获得了由合成的丝状支原体基因组控制的细胞,首次成功实现了人造生命〔Gibson,D.G等,2010,同前〕。随后,2014年Boeke等领导的研究团队人工合成了具有功能的酿酒酵母(Saccharomycescerevisiae)III号染色体〔Annaluru,N.等,Totalsynthesisofafunctionaldesignereukaryoticchromosome,Science,2014,344(6179):p.55-8〕,这是第一个人工合成的真核生物的染色体,标志着人类向人工合成生命又迈进了一步。尽管DNA的合成成本随着技术的进步而不断降低,然而超大片段的DNA甚至是完整基因组的组装由于受限于大片段DNA的操作困难,效率仍然是比较低的。技术实现要素:本发明第一方面提供一种核酸构建物,该核酸构建物含有与cas9基因操作性连接的酵母Tef1启动子,且该核酸构建物能在酵母中组成型表达Cas9。在一个具体实施例中,该核酸构建物是质粒。在一个具体实施例中,该核酸构建物还含有来自pBR322的复制起始位点及其筛选标记,如氨苄青霉素抗性基因。在一个具体实施例中,所述核酸构建物还含有来源于CEN6ARS4的复制区及其筛选标记,如MET14。在一个具体实施例中,所述核酸构建物在酵母中为单拷贝复制,在大肠杆菌中为高拷贝复制。在一个具体实施例中,所述核酸构建物含有依次操作性相连的来自pBR322的复制起点、启动子Tef1、cas9基因、终止序列CYC1、lacZα、f1起点、筛选标记Met14、来自CEN6ARS4的复制区以及筛选标记氨苄青霉素抗性基因。在一个具体实施例中,所述核酸构建物含有SEQIDNO:1第65-319位碱基所示的来自CEN6ARS4的复制区和SEQIDNO:1第3610-7749位碱基所示的cas9基因。在一个具体实施例中,所述核酸构建物如SEQIDNO:1所示。本发明第二方面提供一种核酸构建物,所述核酸构建物在酵母中组成型表达sgRNA,所述sgRNA含有操作性连接的酵母启动子SNR52、20bp识别位点和HandleCas9序列。在一个具体实施例中,所述核酸构建物是质粒。在一个具体实施例中,所述核酸构建物还含有来源于2μ的载体复制区及其筛选标记,如TRP1。在一个具体实施例中,所述核酸构建物还含有来自pBR322的复制起始位点及其筛选标记,如氨卞青霉素抗性基因。在一个具体实施例中,所述核酸构建物含有依次操作性相连的来自pBR322的复制起点、SNR52启动子、20bp识别位点1、HandleCas9序列、SNR52启动子、20bp识别位点2、HandleCas9序列、CYC1引物、CYC1终止序列、lacZα、f1起点、TRP1、来源于2μ的载体复制区和氨苄抗性基因。在一个具体实施例中,所述核酸构建物含有SEQIDNO:2第4624-4998和第5012-5386位所示的序列。在一个具体实施例中,所述核酸构建物如SEQIDNO:2所示。本发明第三方面提供一种核酸构建物,该核酸构建物含有来源于CEN6ARS4的载体复制区及其筛选标记如ADE2,和来源于pBeloBAC11的载体复制起点及分配区及其筛 选标记如氯霉素抗性基因。在一个具体实施例中,所述核酸构建物是质粒。在一个具体实施例中,所述核酸构建物含有操作性相连的来源于CEN6ARS4的载体复制区、筛选标记ADE2、氯霉素抗性基因、分配调控基因rctB、弧菌二号染色体复制区oriCII、以及分配调控基因inc、rctA、parA和parB。在一个具体实施例中,所述核酸构建物如SEQIDNO:3所示。本发明第四方面提供一种细胞,所述细胞含有本发明任意一种、两种或全部三种核酸构建物。在一个具体实施例中,所述细胞为大肠杆菌或酵母细胞。在一个具体实施例中,所述核酸构建物以游离的质粒形式存在于所述细胞中。在一个具体实施例中,所述核酸构建物在所述细胞中组成型表达。本发明第五方面提供一种拼接DNA的方法,所述方法包括:(1)将含有待拼接的DNA序列的供体质粒、能在酵母中组成型表达Cas9的核酸构建物、用于拼接的线性化的受体载体和能够引导Cas9切割供体质粒的含sgRNA的核酸构建物转入步骤同一酵母细胞中;和(2)孵育步骤(1)所述的酵母细胞;从而实现DNA拼接。在一个具体实施例中,通过酵母原生质体融合技术将含有待拼接的DNA序列的供体质粒和能在酵母中组成型表达Cas9的核酸构建物转移到同一酵母细胞中。在一个具体实施例中,采用醋酸锂法将用于拼接的线性化的受体载体和能够引导Cas9切割供体质粒的含sgRNA的核酸构建物转入酵母细胞中。在一个具体实施例中,先采用原生质体融合技术将含有待拼接的DNA序列的供体质粒和能在酵母中组成型表达Cas9的核酸构建物转入酵母细胞中,然后采用LiAc法将用于拼接的线性化的受体载体和能够引导Cas9切割供体质粒的含sgRNA的核酸构建物转入上述酵母细胞中。在一个具体实施例中,供体质粒包含酵母中的单拷贝复制区、筛选标记、能被sgRNA识别的序列以及能被Cas9识别切割的序列。在一个具体实施例中,待拼接的DNA两端含有与其拼接的DNA末端同源的序列。在一个具体实施例中,所述同源序列长度为300~800bp,例如约500bp。在一个具体实施例中,除在DNA两端外,在DNA的其它区域不存在与所述同源序列同源的序列。在一个具体实施例中,受体载体两端含有与待拼接的DNA的两端同源的的序列。在一个具体实施例中,所述同源序列的长度为60~500bp,例如约60~100bp。在一个具体实施例中,所述能在酵母中组成型表达Cas9的质粒为本发明第一方面所述的核酸构建物。在一个具体实施例中,所述受体载体为本发明第三方面所述的核酸构建物。在一个具体实施例中,所述含sgRNA的质粒为本发明第二方面所述的核酸构建物。在一个具体实施例中,步骤(2)之后,通过受体载体、能表达Cas9的质粒和以及含sgRNA的质粒上的标记筛选出需要的转化子。在一个具体实施例中,本发明方法包括:(i)将本发明上述第一、二和三方面所述的核酸构建物和含有待拼接的DNA序列的供体质粒转入同一酵母细胞中;和(ii)孵育步骤(i)所述的酵母细胞;从而实现DNA拼接。本发明还提供一种试剂盒,所述试剂盒含有本发明前述第一、二和三方面所述的任意一种、任意两种或全部三种核酸构建物;和任选的本发明所述的供体质粒。试剂盒中还可任选得含有用于通过酵母原生质体融合技术或LiAc法将所述核酸构建物或供体质粒转入酵母细胞所需的各种试剂。附图说明图1显示CRISPR/Cas9介导的大片段DNA拼接方法原理图。三个含有不同供体质粒的酵母菌(其中一个酵母菌含有Cas9表达质粒)通过原生质体融合,使三个质粒融合到同一个酵母细胞中;LiAc转化线性化受体载体和psgRNA。sgRNA在酵母体内表达后与Cas9结合,将后者定位到特定识别位点使其可以切割供体质粒上大片段DNA两端附近的位点(箭头所指位置),切割下的片段与线性受体载体在酵母体内通过同源重组拼接成环形质粒。图2显示CRISPR/Cas9介导的大片段DNA拼接方法流程图。通过原生质体融合方法可以将Cas9表达质粒和所有供体质粒融合到同一个酵母细胞中,该步骤需要7天时间。在此期间可以准备线性化的受体载体以及进行psgRNA的构建。通过LiAc法将线性化受体载体和psgRNA转化进入已包含了Cas9表达质粒和所有供体质粒的酵母细胞中。CRISPR/Cas9在酵母体内的表达可以切割供体质粒释放大片段DNA,利用酵母的同源重组系统,这些含同源臂的大片段DNA与线性受体载体可以正确拼接成环形质粒,该步骤需要5天时间。图3显示Cas9表达质粒pMetcas9示意图。质粒pMetcas9含有启动子Tef1使Cas9可在酵母中组成型表达。该质粒含酵母复制区(CEN6ARS4)和筛选标记(MET14),在酵母中为单拷贝复制;同时含大肠杆菌复制区(来源于pBR322)和筛选标记氨苄青霉 素(Ampicillin),在大肠杆菌中为高拷贝复制。序列见SEGIDNO:1。图4显示sgRNA表达质粒psgRNA示意图。质粒psgRNA含有的启动子SNR52使sgRNATarget1和Target2可在酵母中组成型表达。该质粒含酵母复制区(来源于2μ)和筛选标记(TRP1),在酵母中为多拷贝复制;同时含大肠杆菌复制区(来源于pBR322)和筛选标记氨苄青霉素(Ampicillin),在大肠杆菌中为高拷贝复制。序列见SEGIDNO:2。图5显示线性化载体模板pcriv0质粒示意图。质粒pcriv0含酵母复制区(CEN6ARS4)和筛选标记(ADE2),在酵母中为单拷贝复制;同时含弧菌二号染色体复制区oriCII及分配调控相关基因rctA、rctB、inc、parA、parB和氯霉素筛选标记,在大肠杆菌中为单拷贝复制。序列见SEGIDNO:3。图6显示pCriv6酶切验证图及示意图。A显示pCriv6酶切验证图,其中M为LambdaLadderPFGMarker;1为pCriv6NotI酶切pCriv6;2为SpeI酶切pCriv6。pCriv6经NotI酶切得到311kb大小的一条带,而经SpeI酶切可以得到98kb和213kb大小的两条带。PFGE条件:1%琼脂糖凝胶,6V/cm,14℃,16小时,转换时间1-25秒;B显示pCriv6示意图。图7显示pCriv7酶切结果及示意图。A为pCriv7酶切验证图,其中M为LambdaLadderPFGMarker;1为SpeI酶切pCriv7。pCriv7经SpeI酶切得到片段200kb、182kb、157kb、98kb、32kb大小的五条带。其中32kb条带与酵母染色体碎片大小相近,因此在胶上无法分开。PFGE条件:1%琼脂糖凝胶,6V/cm,14℃,16小时,转换时间1-25秒;B为pCriv7示意图。具体实施方式本发明旨在发展一种高效拼接大片段DNA的方法。利用该发明减少了困难且低效率的大片段DNA的体外操作,使大片段DNA的拼接变得简单易行。本发明提供了一种利用CRISPR/Cas9系统,在酵母中高效拼接DNA的方法(原理见图1)。通过酵母原生质体融合技术,将含有待拼接的两端带有同源序列的大片段DNA的供体质粒转移到同一个酵母细胞中,同时该酵母细胞中含有一个组成型表达Cas9的质粒;LiAc法共同转化一个用于拼接的线性化受体载体和一个能够切割供体载体的sgRNA质粒;供体载体被切割后,大片段DNA通过酵母体内同源重组拼接到受体载体上,通过受体载体、Cas9载体和sgRNA载体上的标记筛选出需要的转化子(见图2)。与前人已建立的拼接方法相比,本方法的优点在于实验操作过程中避免了繁琐低效的体外抽取、酶切、回收及转化大片段DNA的步骤,使实验过程变得简单、易操作。本发明对待拼接DNA的大小没有严格限制,有应用于超大片段DNA的拼接的潜力,比如用于拼接几兆 碱基对的基因组的组装和拼接。例如,适用于本发明方法拼接的DNA片段至少为100kbp,例如至少500kbp,可达几兆碱基对。本文中,高拷贝复制指拷贝数大于1,一般指5以上,例如10以上。本发明具有下列详细特征:Cas9表达载体的构建:构建在大肠杆菌中为高拷贝复制(复制起始位点来源于pBR322,筛选标记为氨苄青霉素(Ampicillin)抗性基因),在酵母中单拷贝复制(载体复制区来源于CEN6ARS4,筛选标记MET14)且能组成型表达Cas9的质粒pMetcas9。CEN6ARS4可获自pSH47(Guldener,U.等,Anewefficientgenedisruptioncassetteforrepeateduseinbuddingyeast,NucleicAcidsResearch24(13):2519-2524)。该复制区序列可如SEQIDNO:1第65-319位碱基所示。Cas9的序列可如SEQIDNO:1第3610-7749位碱基所示。HpaI、SpeI双酶切质粒p415-GalL-hCAS9〔DiCarlo,J.E.等,GenomeengineeringinSaccharomycescerevisiaeusingCRISPR-Cassystems,NucleicAcidsResearch,2013,41(7):p.4336-4343〕,回收两个5.4kbp的片段。PCR扩增MET14标记(以酿酒酵母S.cerevisiaes288c基因组为模板)和Tef1启动子(以质粒pAG36〔Goldstein,A.L.等,ThreenewdominantdrugresistancecassettesforgenedisruptioninSaccharomycescerevisiae.Yeast.1999Oct;15(14):1541-53〕为模板),通过引物3’端带上40bp与待拼接的相邻片段的同源序列,PCR产物纯化后转入酵母体内进行拼接,拼接的质粒经PCR及测序验证正确后转化大肠杆菌DH10B,抽取质粒备用。图3显示了本发明pMetcas9的质粒图谱,其核苷酸序列如SEQIDNO:1所示。sgRNA表达载体构建:构建在大肠杆菌中为高拷贝复制(复制起始位点来源于pBR322,筛选标记为氨苄青霉素抗性基因),在酵母中多拷贝复制(载体复制区来源于2μ,筛选标记TRP1)且能组成型表达sgRNA的质粒psgRNA。sgRNA包括SNR52启动子、20bp识别位点和HandleCas9序列。所述20bp识别位点负责识别供体质粒上的对应的识别位点,而HandleCas9负责招募Cas9至切割位点,从而实现对供体质粒的切割。所述20bp的识别位点可以根据供体质粒载体的骨架序列随意设计。在一个具体实施例中,SEQIDNO:2的第4624-4998位碱基显示了sgRNA的序列,第5012-5386位碱基显示另一sgRNA的序列,其包括了SNR5启动子、20bp识别位点和HandleCas9序列。在本发明的例子中由于各供体质粒所用的骨架都相同,所以只需要两个识别位点就可以完成对所有供体质粒的切割。20bp识别位点的序列可根据本领域常规的技术进行设计。表一显示了本发明具体实施例中所用的两个20bp的识别位点的序列。在一个具体实施例中,人工合成两个DNA片段Target1(SEQIDNO:4)和Target2(SEQIDNO:5),其中Target1用BglII和SpeI酶切,Target2用SpeI和NcoI酶切,分 别回收400bp大小的酶切片段,与BglII和NcoI酶切的载体pTRPgRNA(由质粒p426-SNR52p-gRNA.Y-SUP4t〔DiCarlo,J.E.等,同上〕质粒改造,将URA3筛选标记改为TRP1筛选标记)连接,转化大肠杆菌DH10B,得到含有两个靶向识别位点的sgRNA的质粒。拼接的质粒经PCR及测序验证正确。表一:sgRNA识别序列图4显示了本发明psgRNA的质粒图谱,其核苷酸序列如SEQIDNO:2所示。受体载体pcriv0的构建和线性化:PCR扩增pcriv0载体的复制区(弧菌二号染色体复制区〔Egan,E.S.和M.K.Waldor,DistinctReplicationRequirementsfortheTwoVibriocholeraeChromosomes,Cell,2003,114(4):p.521-530〕)和氯霉素筛选标记(以商业化质粒pBeloBAC11(NEB公司)为模板),以及酵母复制区(来源于CEN6ARS4)和ADE2筛选标记(来源于酿酒酵母基因组),通过引物3’端带上40bp与待拼接的相邻片段的同源序列。PCR产物纯化后转入酵母体内进行拼接。阳性克隆经PCR验证及测序正确后转化大肠杆菌DH10B。抽质粒酶切验证正确后,经测序确认供体载体pcriv0的序列正确性。线性化的载体可以经PCR扩增得到,通过引物带上与待拼接大片段DNA同源的60~500bp、优选60~200bp(例如约60bp)的序列。图5显示了本发明受体载体pcriv0的质粒图谱,其核苷酸序列如SEQIDNO:3所示。含大片段DNA的供体质粒的准备:含有待拼接大片段DNA的供体质粒需要包含酵母中单拷贝复制区(来源于CEN6ARS4)及不同的选择标记。此外,大片段DNA两端需要加入与待拼接的相邻大片段DNA同源的300~800bp(例如500bp左右)的序列。所有同源序列在这些大片段DNA的其他区域无显著同源序列。此外,应理解,供体质粒应含有能被sgRNA识别的序列。通常,此序列位于供体载体骨架上大片段DNA插入位置的外侧,并与sgRNA中的20bp识别位点相同。供体质粒中还应含有能被Cas9切割的位点,例如PAM位点。可根据本领域已知的技术设计这样的序列。作为一个例子,可在供体质粒上设计如下识别/切割位点:ATAGTGTCACCTAAATAGCTTGG和CGTAGCAACCAGGCGTTTAAGGG(即表一中的20bp序列加PAM位点)。应理解,本发明方法可拼接2个以上大片段DNA。因此,本发明可使用两个以上供体质粒,例如3个、4个甚至更多个,只要这些供体质粒的结构满足上述要求即可。转化/原生质体融合:对于小于30kb的多个质粒,可以用LiAc方法〔Gietz,R.D.和R.H.Schiestl,High-efficiencyyeasttransformationusingtheLiAc/SScarrierDNA/PEGmethod,NatureProtocols,2007,2(1):p.31-34〕转化进入酵母中。大于30kb的质粒由于用LiAc方法难以转化进入酵母中,则需要采用酵母原生质体融合方法〔Curran,B.P.和V.C.Bugeja,ProtoplastfusioninSaccharomycescerevisiae,Methodsinmolecularbiology(Clifton,NJ),1996,53:p.45-9〕。两个或三个含不同供体质粒的酵母细胞可以一次融合,用全部质粒的标记去筛选,会得到所有质粒在同一个细胞中的酵母菌。大片段DNA在酵母体内的拼接:将psgRNA和线性化受体载体转化已包含所有待拼接大片段DNA质粒的酵母细胞。在sgRNA和Cas9的共同作用下,切割所有供体质粒,释放出全部待拼接的大片段DNA。通过酵母体内高效的同源重组系统,这些末端含有同源序列大片段DNA以及线性化的受体载体可以拼接在一起。通过共同筛选pMetcas9、psgRNA和受体载体所带的营养缺陷标记,得到需要的转化重组子。阳性克隆需要经PCR、测序以及脉冲场凝胶电泳验证。下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件如Sambrook&Russell所著的MolecularCloning:ALaboratoryManual(分子克隆实验指南第三版)中所述的条件,或按照制造厂商所建议的条件。除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明中。文中所述的较佳实施方法与材料仅作示范之用。实施例1:两个大片段DNA的拼接本例中选用本实验室在酿酒酵母VL6-48中已分别构建好的pSP5和pTP3-U质粒(表二)。pSP5含有116643bp的供体DNA,命名为SP5;pTP3-U含有184475bp的供体DNA,命名为TP3。SP5和TP3序列均来源于大肠杆菌,它们是通过PCR扩增大肠杆菌某些必需基因,通过酵母菌内两级或三级拼接得到的较大质粒,SP5右末端及TP3左末端有491bp的同源序列。表二:质粒pSP5、pTP3-U信息供体质粒筛选标记供体DNA名称插入片段大小(bp)pSP5HIS3SP5116643pTP3-UURA3TP3184475将Cas9表达质粒pMetcas9(图3,SEQIDNO:1)转入已含pSP5质粒的酵母细胞VL6-48中。将VL6-48/pSP5/pMetcas9和VL6-48/pTP3-U进行原生质体融合,酵母原生质体融合方法和试剂配制参考文献〔Curran,B.P.和V.C.Bugeja,ProtoplastfusioninSaccharomycescerevisiae,Methodsinmolecularbiology(Clifton,NJ),1996.53:p.45-9〕和〔Kouprina,N.和V.Larionov,Selectiveisolationofgenomiclocifromcomplexgenomesbytransformation-associatedrecombinationcloningintheyeastSaccharomycescerevisiae,NatureProtocols,2008,3(3):p.371-377〕,在此基础做了一定改进,其步骤为:1.VL6-48/pSP5/pMetcas9和VL6-48/pTP3-U分别在SC-Met,Ura和SC-His平板上划线,30℃培养3天。2.挑酵母菌单菌落接至5ml相对应的SC液体培养基中,30℃,200rpm过夜培养;3.过夜酵母菌液稀释10倍测OD600,此时OD应为0.3-0.5(表示生长良好)。1:20到1:25转接到50ml相对应的SC液体培养基,使起始OD600小于或等于0.2。30℃,200rpm培养至OD600为1.0-1.5,约5-6个小时。4.20℃,5000rpm离心5分钟收菌,用30ml无菌水重悬,离心收菌;用20ml1M的无菌山梨醇溶液重悬细胞,5000rpm离心5分钟收菌。5.用20mlSPE溶液(1M山梨糖醇,0.01M磷酸钠,0.01MNa2EDTA,pH7.5)重悬细胞,加入20μl酵母裂解酶(zymolyase)溶液(20mg/mlZymolyase-20T,50%甘油,2.5%葡萄糖,50mMTris-HCl,pH8.0)和40μlβ-巯基乙醇,混匀,30℃处理30min。6.5℃,570g离心5分钟收菌,用50ml1M山梨醇洗一次,300-600g离心5min收菌。7.用20mlMP缓冲液(1M山梨糖醇,0.1MNaCl,0.01MHAc,用10M的氢氧化钠溶液调节pH值为5.5)重悬细胞,600g离心5min收菌。8.沉淀用3mlMP缓冲液重悬,稀释1/80倍,用血小板计数器计数。三个菌株分别取1*107个细胞,混匀,300-600g离心5min,沉淀用2ml60%的PEG4000和0.2ml1MCaCl2溶液重悬。9.室温放置3min,加入6mlMP缓冲液,混匀,室温放置6min。10.按上述离心条件收菌,用10mlMP缓冲液洗一次,离心收菌后,重悬于1mlMP缓冲液中。11.分别取100μl和900μl加入到事先融化的上层培养基(SC-Met,Ura,His)中,涂布平板,30℃培养5-7天。原生质体融合后,在SC三缺培养基上长出30个融合子,这些融合子应是含有pMetcas9、pSP5和pTP3-U三个质粒的酵母菌,命名为ZJ1。设计两条分别与SP55’端和TP3-U3’端有60bp左右同源序列的引物pCriv6-VR和pCriv6-VF(见表三),以pcriv0为模板,用KOD-plus-neo(购自Toyobo公司)PCR扩增出线性供体载体,PCR扩增条件为:95℃预变性5min;98℃变性10s,52℃退火20s,68℃延伸10min,30个循环。表三:实施例一和实施例二中所用引物醋酸锂(LiAc)转化1ug线性受体载体(图5,SEQIDNO:3)和1ugpsgRNA(图4,SEQIDNO:2)到菌株ZJ1中。LiAc转化方法及试剂、培养基的配制见参考文献〔Gietz,R.D.和R.H.schiestl,2007,同前〕,在该文献的基础上做了一些改进,其基本步骤为:1.酵母菌ZJ1在SC-Met,Ura,His板上划线,30℃培养2天;2.挑酵母菌单菌落接至5mlYPAD液体培养基中,30℃,200rpm过夜培养;3.过夜酵母菌液稀释10倍测OD600,此时OD应为0.4-0.5(表示生长良好)。1:20到1:25转接到2*YPAD液体培养基,使起始OD600小于或等于0.2。30℃,200rpm培养至OD600为0.8-1.0,约4-5个小时;4.20℃,5000rpm离心5分钟收菌,用1/2体积无菌水重悬(如50ml菌液用25ml无菌水重悬)离心收菌;5.1/2体积无菌水再洗一次,离心收菌,用1ml无菌水重悬后转至1.5mlEP管中,13000g,30秒离心收菌;6.用1/50体积无菌水重悬(如50ml菌液最后用1ml无菌水重悬),100μL每管分装备用;7.取出-20℃冻存的ssDNA(鲑精DNA)100℃变性5分钟后,立刻插入冰里;8.将备用的酵母菌13000g离心30秒,弃上清。依次加入(可先准备除DNA片段外的混合物,再分别加DNA片段):注:正对照:环形的酵母-大肠杆菌穿梭质粒pXX11。负对照:只加ddH2O34μL。9.用上述混合液重悬酵母菌(可涡旋);10.42℃热击25分钟(每5分钟混匀一次);11.13000g离心1分钟去上清,用200uLddH2O重悬酵母菌,涂SC-Met,Trp,Ade三缺板。用SC-Met,Trp,Ade三缺板筛选,30℃培养3-4天,得到28个转化子,菌落PCR验证的阳性率为5/7,含有正确的拼接质粒pCriv6。菌落PCR验证的步骤为:在片段之间的接头处设计三对验证引物(见表三),扩增片段大小为0.5-1.5kbp之间,挑取7个酵母转化子悬于20ul无菌水中,混匀后取0.2uL为模板,用KOD-FX酶进行PCR验证,PCR扩增条件为:95℃预变性5min;98℃变性10s,52℃退火20s,68℃延伸1.5min,35个循环。挑选一个酵母菌准备制作胶块,进行脉冲场凝胶电泳检测。酵母菌制作胶块方法为:1.接单克隆到50mlSC-Met,Trp,Ade培养基中,30℃培养过夜。2.OD600=1.0-2.0时,4000rpm离心5min收菌。3.用50mlddH2O,重悬菌体,4000rpm离心5min收菌,4.用10ml50mMEDTApH8.0重复步骤3,洗一次细胞。5.用750μl细胞重悬缓冲液(10mMTris-HCl,pH7.2)重悬,4000rpm离心5min,细胞沉淀用150μl细胞重悬缓冲液重悬,放50℃平衡。6.加入85μlZymolyase-20T溶液(20mg/mlZymolyase-20T,50%甘油,2.5%葡萄糖,50mMTris-HCl,pH8.0)和225μl2%的TE25S溶解的低熔点琼脂糖(事先配好,50℃平衡)。混匀后,倒入模具中,放冰上或4℃冰箱冷却。7.约30min后,把胶块取出,加入5ml酵母裂解酶(lyticase)缓冲液(10mMTris-HClpH7.5,50mMEDTApH8.0)和500μlZymolyase-20T,37℃孵育2h.8.胶块用25ml水洗一次,然后用洗涤缓冲液洗涤缓冲液(20mMTris-HClpH8.0,50mMEDTA)洗涤一次。9.每1ml胶块加入5ml蛋白酶k反应液(100mMEDTApH8.0,0.2%脱氧胆酸钠,1%月桂酰肌氨酸钠,1mg/ml蛋白酶K),50℃消化过夜(细胞特性不同,有些样品需要延长消化时间到4d,但这并不会损伤DNA)。10.用50ml洗涤缓冲液洗涤胶块4次,于室温轻柔震荡,每次30-60min。11.脉冲场凝胶电泳将酵母线性基因组跑出胶块,环形质粒仍然留在胶块中,电泳条件为,0.5XTBE缓冲液,电压6v/cm,转换时间为60-120秒,12h。12.第二天取出胶块,如步骤10,用50ml洗涤缓冲液洗涤胶块4次,于室温轻柔震荡,每次30-60min。第2、3次洗涤时加入1mM的PMSF,使蛋白酶K失活。13.在限制性内切酶消化前,先用10倍稀释的洗涤缓冲液或TE洗涤胶块30min,再用1Xcutsmart缓冲液(购自NEB公司)浸泡1h。14.NotI、SpeI(购自NEB公司)分别酶切1/3胶块。脉冲场电泳检测,电泳条件为:0.5XTBE缓冲液,电压6v/cm,转换时间为1-25秒,24h。脉冲场凝胶电泳结果显示,pCriv6经NotI酶切得到一条311kb大小的条带,而经SpeI酶切可以得到98kb和213kb大小的两条带,与预测条带大小相同(见图6)。实施例2:三个大片段DNA的拼接本例中选用本实验室在酿酒酵母VL6-48中已分别构建好的pTP1、pTP2和pTP3-L质粒(表四)。表四:质粒pTP1、pTP2、pTP3-L信息筛选标记插入片段名称插入片段大小(bp)pTP1HIS3TP1177147pTP2URA3TP2297952pTP3-LLYS2TP3184475pTP1含有177147bp的供体DNA,命名为TP1;pTP2含有297952bp的供体DNA,命名为TP2;pTP3-L含有184475bp的供体DNA,命名为TP3。TP1、TP2和TP3序列均来源于大肠杆菌,它们的构建方法和实例一中pSP5和pTP3-U的构建方法类似。TP1右末端与TP2左末端有385bp的同源序列,而TP2右末端及TP3左末端有491bp的同源序列。将Cas9表达质粒pMetcas9(见图3和SEQIDNO:1)转入已含pTP2质粒的酵母细胞中。将酵母菌VL6-48/pTP1、VL6-48/pTP2/pMetcas9、VL6-48/pTP3-L用实施例一中所述原生质体融合的方法将三个质粒融合在同一酵母菌细胞中得到菌株ZJ2。设计两条分别与片段TP15’端和TP33’端60bp左右同源的引物pCriv7-VR和pCriv7-VF(见表二),以pCriv0为模板,用KOD-plus-neoPCR扩增出线性供体载体,PCR扩增条件为:95℃预变性5min;98℃变性10s,52℃退火20s,68℃延伸10min,30个循环。1μg供体线性载体(图5,SEQIDNO:3)和1μgpsgRNA(图4,SEQIDNO:2)用LiAc法共同转化ZJ2,用SC-Met,Trp,Ade三缺板筛选,30℃培养3-4天,得到7个转化子,菌落PCR验证的阳性率为2/3,正确的拼接质粒命名为pCriv7。PCR验证的引物见表三。挑选一个酵母菌准备制作胶块,进行脉冲场凝胶电泳检测拼接质粒pCriv7。pCriv7经SpeI酶切得到片段200kb、182kb、157kb、98kb、32kb(被酵母染色体基因组覆盖)大小的5条带,PFGE结果请见附图7。上文所列的各相关文献均以全篇引入本申请作为参考。本发明所涉及的多个方面已做如上阐述。然而,应理解的是,在不偏离本发明之精神与范围的前提下,对上述描述的任何修饰都是允许的。同样,类似的情况也包括在权利要求中。当前第1页1 2 3
相关技术
网友询问留言
已有1条留言
-
 0访客 来自[中国] 2024年05月22日 23:14参考文献有哪些呀
0访客 来自[中国] 2024年05月22日 23:14参考文献有哪些呀
1