一种用于恶臭假单胞菌NBRC14164的基因敲除方法与流程
本发明涉及一种用于恶臭假单胞菌NBRC 14164的基因敲除方法,其属分子生物学研究领域。
背景技术:
恶臭假单胞菌(Pseudomonas putida)是一类革兰氏阴性菌,属γ-变形菌,广泛分布于自然界的生态位,由于其强大的代谢能力而对环境有较强适应性,如可生存于高浓度重金属污染或有机污染物等环境中[1,2,3]。随着测序技术的发展,许多典型P.putida菌株的基因组序列得以查询,为菌株的广泛应用、遗传背景解析、分子改造等提供了强有力的支撑。如极具代表性的P.putida KT2440在PHA生产[4]方面有良好表现,而其基因组尺度的代谢网络模型的构建更让其大大提高了PHA产量[5]。
目前,基因敲除方法有很多,其中利用同源重组原理发展出来的技术有着广泛的应用。同源重组是指基因工程中利用活细胞DNA可与外源DNA序列发生重组的性质,来进行定点修饰改造基因组上某一目的基因的技术[6],可分为诱导性基因敲除法、基因插入突变技术、自杀载体构建技术等。其中,自杀载体的原理为:将需缺失的目的基因克隆到自杀载体上,通过接合等使其进入宿主,由于在宿主菌中不存在复制基因启始所需的复制蛋白而无法复制,在外界选择性压力的作用下,自杀载体所携带的突变基因就与宿主染色体上的野生型基因发生二次同源重组,产生了带有突变位点的突变株,而载体自身由于自杀性特性连同染色体上原有的野生型基因一起随着细菌的传代从菌体内消失[7]。
P.putida NBRC 14164的全基因组序列于2014年测出[3],在DDBJ/EMBL/GenBank等多个数据库中的编号为AP013070。在此之前,该菌株仅作为研究中的对比分析,如用于分类方法研究[8]、有毒污染物降解[9]等;时至今日,仍鲜有针对该菌株的专门研究的报道。从分子层面探索P.putida NBRC 14164的可操作性,提供一种行之可效的操作方法,将为其深入研究打开新的通道,开辟广阔的空间。
[1]Wu X,Monchy S,Taghavi S,et al.,Comparative genomics and functional analysis of niche-specific adaptation in Pseudomonas putida.FEMS Microbiol.,35:299-323(2011).
[2]Poblete-Castro I,Becker J,Dohnt K,et al.,Industrial biotechnology of Pseudomonas putida and related species.Appl.Microbiol.Biotechnol.,93:2279-2290(2012).
[3]Shoko O,Atsushi Y,Akira H,et al.,The complete genome sequence of Pseudomonas putida NBRC 14164T confirms high intraspecies variation.Genome Announcements,2(1):e00029-14(2014).
[4]Wang Q,Nomura CT,Monitoring differences in gene expression levels and polyhydroxyalkanoate(PHA)production in Pseudomonas putida KT2440grown on different carbon sources.Journal of Bioscience&Bioengineering,110(6):653-659(2010).
[5]Nogales J,PalssonThiele I,A genome-scale metabolic reconstruction of Pseudomonas putida KT2440:iJN746as a cell factory.BMC Systems Biology,2(37):1-20(2008).
[6]贺淹才,简明基因工程原理[M].北哀科学出版社,2005:389.
[7]卢福芝,孙靓,黄靖华,等,质粒pUC19-CM-D的构建及应用.安徽农业科学,38(19):9953-9954(2010).
[8]Tamura H,Hotta Y,Sato H,Novel accurate bacterial discrimination by MALDI-time-of-flight MS based on ribosomal proteins coding in S10-spc-alpha operon at strain level S10-GERMS.Journal of the American Society for Mass Spectrometry,24(8):1185-93(2013).
[9]Nonaka K,Ohta H,Sato Y,et al.,Utilization of phenylpropanoids by Pseudomonas putida soil isolates and its probable taxonomic significance.Microbes&Environments,23(4):360-364(2008).
技术实现要素:
鉴于上述,本发提出一种用于恶臭假单胞菌NBRC 14164的基因敲除方法,并验证其可行性,具体技术如下:
首先构建基因敲除质粒,然后制备接合转化的辅助菌株E.coli S17-1感受态细胞,再将基因敲除质粒转化E.coli S17-1感受态细胞,最后利用接合转化法导入质粒至P.putida NBRC 14164中,电泳验证即可。
具体说明如下:
恶臭假单胞菌Pseudomonas putida NBRC 14164,购买自中国工业微生物菌种保藏管理中心。埃希氏大肠杆菌Escherichia coli S17-1在接合转化中起辅助作用,与载体质粒pK18mobSacB均为实验室保藏(原始菌株购自武汉淼灵生物科技有限公司)。
一种用于恶臭假单胞菌NBRC 14164的基因敲除方法,其步骤如下:
1)基因敲除质粒构建;
2)E.coli S17-1感受态细胞的制备;
3)基因敲除质粒转化E.coli S17-1感受态细胞;
4)接合转化法导入质粒至P.putida NBRC 14164中。
所述的步骤1)是:查找拟敲除基因序列,利用软件Primer Premier 5设计基因敲除引物与敲除后验证引物,两对引物扩增产物相差300-400bp,敲除引物扩增产物经酶切连接质粒即构建成功。
所述的步骤2)是:用于接合转化的辅助菌株E.coli按照热激转化法制备感受态细胞,用Mg2+溶液、Ca2+溶液的洗涤,全程无菌操作。
所述的步骤3)是:用热激转化法将基因敲除质粒转化进入E.coli感受态细胞。
所述的步骤4)是:接合转化利用两次同源重组将基因敲除,第一次同源重组质粒从E.coli进入P.putida中并整合到染色体上,第二次同源重组质粒与敲除引物扩增的基因从染色体上脱离,即基因敲除成功。
第一次同源重组条件为恶臭假单胞菌与含有重组自杀载体质粒的大肠杆菌于30℃,200rpm培养12h,再静置12h。
第二次同源重组条件为筛选后的第一次同源重组成功的单菌落接种到无抗性LB液体培养基中,30℃,200rpm培养24h。
本发明的优点是:采用接合转化方法,利用同源重组原理,借助辅助菌株E.coli S17-1、自杀载体质粒pK18mobSacB共同完成基因敲除。首次从分子层面探索P.putida NBRC 14164的可操作性,提供一种行之可效的操作方法;此方法不仅适用于基因敲除,亦可将外源基因整合至P.putida染色体,相比于外源质粒导入具有更好的稳定性。此方法将为P.putida NBRC 14164的深入研究打开新的通道,开辟广阔的空间。
附图说明
图1酶切连接示意图
待敲除基因先从P.putida基因组上扩增下来,其中用于扩增的上下游引物含有酶切位点。将扩增后的基因与自杀载体质粒用相同的限制性内切酶切割后,经连接即可成功构建基因敲除质粒。
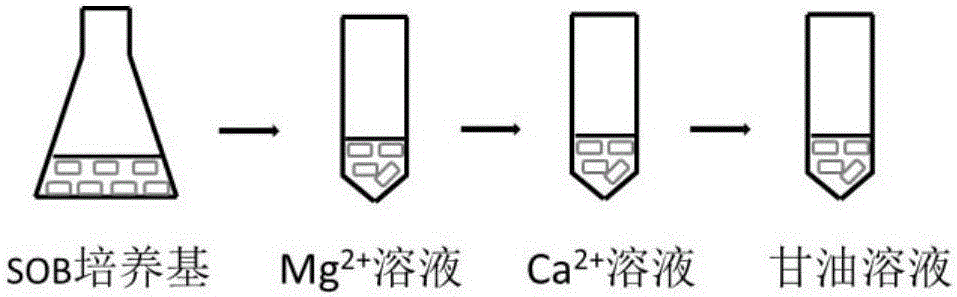
图2感受态细胞制备示意图
用于接合转化的辅助菌株E.coli经Mg2+溶液、Ca2+溶液的洗涤,最后用甘油溶液重悬即可成为适用于热激转化的感受态细胞。
图3热激转化示意图
空心圆点代表自杀载体质粒,经热激与冰浴,自杀载体质粒穿过E.coli的细胞壁与细胞膜进入到细胞中。
图4基因敲除示意图
载体上灰色片段为拟敲除基因片段,基因组上灰黑相间片段为完整基因;第一次同源重组后,自杀载体质粒整合至基因组;第二次同源重组后,自杀载体质粒脱离基因组,剩下黑色片段为敲除后基因片段。
图5 prs和rpiA基因敲除前后电泳图
泳道3为DNA分子量标准,泳道1和4分别为敲除前的prs和rpiA基因,泳道2和5分别为敲除后的prs和rpiA基因。
具体实施方式
下面结合附图和具体实施例,对本发明做进一步的详细说明:
1.培养基配方与溶液配方
SOB培养基:胰蛋白胨20g/L、酵母粉5g/L、氯化钠0.5g/L、250mM氯化钾10mL/L、2M氯化镁5mL/L;
LB培养基:蛋白胨10g/L、酵母粉5g/L、氯化钠10g/L;
LBS培养基:蛋白胨10g/L、酵母粉5g/L、氯化钠10g/L、蔗糖60g/L;
配制固体培养基时加入琼脂粉15g/L;
氨苄青霉素(Amp)工作浓度:100μg/mL;
卡那霉素(Kan)工作浓度:50μg/mL;
Mg2+溶液:100mM氯化镁、10mM TriS,pH7.5;
Ca2+溶液:100mM氯化钙、10mM TriS,pH7.5;
甘油溶液:8.5mL Ca2+溶液、1.5mL甘油。
2.基因敲除质粒构建
(1)在NCBI数据库中查找P.putida基因的全序列,以prs和rpiA为例设计基因敲除引物与敲除后验证引物如表1所示。其中验证引物扩增产物约为基因全长,敲除引物扩增产物约为基因全长去掉首尾100-200bp,即两对引物扩增产物相差200-400bp。
表1prs和rpiA基因敲除引物与敲除后验证引物
(2)以P.putida甘油菌为模板用敲除引物扩增基因,将电泳验证正确的扩增产物与pK18mobSacB的序列两端用相同的限制性内切酶切割1h、连接2h,即得基因敲除质粒。
酶切连接过程如图1所示。
3.E.coli S17-1感受态细胞的制备
(1)活化E.coli,按1%(v/v)的接种量将种子液接种到200mL SOB培养基中,37℃,200rpm,培养至OD600为0.6左右。
(2)将培养物在冰水中放置30min后,用15mL的预冷的Mg2+溶液、Ca2+溶液各洗涤菌体一次。
(3)用5mL甘油溶液重悬菌体,悬浮菌液分装每管40μL,保存于-80℃。
感受态细胞制备过程如图2所示。
4.基因敲除质粒转化E.coli S17-1感受态细胞
(1)取出E.coli置于冰上融化后加入10μL质粒,轻轻混匀,冰浴30min。
(2)42℃热击90s后迅速置于冰上3min,再加入890μL的LB培养基,30℃,200rpm培养1h。
(3)取100μL转化液涂在含有Kan的LB平板上,30℃培养18h。
热激转化过程如图3所示。
5.接合转化法导入质粒至P.putida NBRC 14164中
(1)各取活化后的E.coli和P.putida菌液500μL,混合在同一无菌离心管中,8000rpm离心3min,用1mL LB培养基洗涤一次,加入1mL LB培养基重悬菌体,30℃,200rpm培养12h,再静置12h,第一次同源重组完成。
(2)100μL混合液涂在LB-Amp-Kan的平板上,30℃培养24h。
(3)挑取单菌落,接种到无抗性LB液体培养基中,30℃培养24h,第二次同源重组完成。
(4)将培养液用无菌水稀释100倍,取100μL涂布在LBS-Amp的平板上,30℃培养24h。
(5)LBS-Amp平板上的菌挑取单菌落,按编号依次点在LBS-Amp和LB-Kan平板上,30℃培养24h,标出LBS Amp平板上有生长而LB-Kan平板上无生长的菌落即为阳性菌落。
(6)菌落PCR、电泳验证。
两次同源重组过程如图4所示,prs和rpiA基因敲除前后电泳如图5所示。
- 还没有人留言评论。精彩留言会获得点赞!