制备可用于从气体流去除酸性气体的链烷醇胺的方法与流程
本发明涉及一种制备可用于从含有CO2和/或H2S的气体流去除CO2和/或H2S的链烷醇胺的方法。更具体地讲,本发明涉及这样一种方法,其中链烷醇胺的制备是使用专门选择的离子液体和使用专门选择的反应条件进行的。
背景技术:
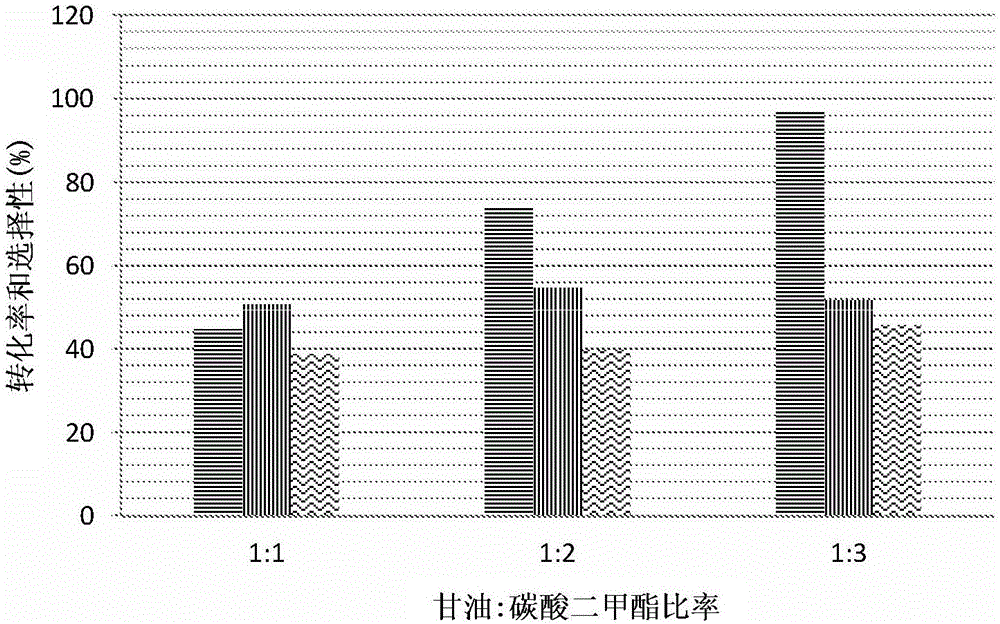
:气体流中CO2和H2S酸性气体(“酸气体”)的存在,是炼油厂及化工产品和气体加工厂的一个众所周知的问题。在大多数情况下,有必要增加额外的加工步骤以从气体流去除酸性气体,以便该气体流具有商业效用,例如作为燃料来源。已知有几种用于从气体流去除CO2和/或H2S的“净化(sweetening)”方法。一种这类方法涉及用基于链烷醇胺的溶剂吸收CO2和/或H2S。通常,链烷醇胺以水溶液的形式提供,其当与气体流中的酸性气体接触时发生放热且可逆的反应。含水的仲链烷醇胺和叔链烷醇胺与CO2和H2S的典型反应可由以下化学式表示:对于CO2吸收而言,仅伯链烷醇胺和仲链烷醇胺可直接从其与CO2的反应形成氨基甲酸,如以下反应所示。已知伯链烷醇胺和仲链烷醇胺形成氨基甲酸会限制链烷醇胺水溶液的摩尔CO2吸收能力。发生这种情况是因为,如以上的平衡反应(3)所示,对于每摩尔CO2,氨基甲酸的形成会消耗两摩尔链烷醇胺。因此,链烷醇胺的摩尔吸收能力受到限制,这取决于它是伯链烷醇胺、仲链烷醇胺还是叔链烷醇胺。此外,吸收能力还受到链烷醇胺的胺官能团的碱性的影响。一般而言,随着链烷醇胺的胺官能团的pKa提高,CO2吸收能力也提高。不可能直接从CO2与缺乏必要质子的叔链烷醇胺的反应形成氨基甲酸。相反,当在水的存在下与CO2反应时,叔链烷醇胺导致碳酸根阴离子的形成。在这种含水体系中,如以下反应所示,叔链烷醇胺充当CO2在水中水解时产生的氢离子的吸收剂(sink)。尽管过去已提议了多种链烷醇胺及其混合物用于气体净化,但仅有少数链烷醇胺被发现具有商业可行性。这些链烷醇胺包括单乙醇胺(MEA)、二乙醇胺(DEA)、甲基二乙醇胺(MDEA)、二异丙醇胺(DIPA)和二甘醇胺(DGA)。这很可能是使用它们时的经济效果的结果。例如,已知在CO2的捕集中使用含水的胺会消耗掉运行发电厂所需的能量的差不多30%(Fisheretal.,“Advancedaminesolventformulationsandprocessintegrationfornear-termCO2capturesuccess”,TrimericCorporation,Pennsylvania,2007)(Fisher等人,“用于近期CO2捕集成功的高级胺溶剂配方和工艺整合”,美国宾西法尼亚州TrimericCorporation公司,2007年)。尽管吸收能力是基于链烷醇胺的吸收剂的一个重要考虑因素,但CO2的吸收速率也是一个关键因素。CO2吸收速率慢会导致较长的气液接触时间,从而需要更大的吸收柱和更高的资金成本。因此,在高吸收能力的好处与较低吸收动力学的负面影响之间往往要折中。这对于使用MDEA作为酸性气体吸收剂是特别相关的。作为叔胺,MDEA具有比MEA更大的吸收能力。但是,它的吸收速率太慢,以至于它的吸收能力很少在商业上可行的系统中得到实现。正因为这个原因,MDEA通常与活化剂如哌嗪结合使用,如US4,336,233中所公开。WO95/03874也公开了使用MDEA和甲基单乙醇胺(MMEA)的含水混合物,会导致原位产生据称能改善酸性气体的去除的N,N’-双(二甲基)-N-羟乙基-乙二胺(DMHEED)。涉及链烷醇胺的循环利用即酸性气体的解吸的动力学,也是整个净化工艺中的一个重要经济考虑因素。例如,通常通过煮沸该水溶液(从而将CO2进行汽提)来进行从相应的氨基甲酸再生伯链烷醇胺和仲链烷醇胺,然后将该链烷醇胺冷却和回收,这是很耗能的。因此,期望高解吸速率以降低在工业规模上再生链烷醇胺时所耗费的能量。在一个相关方面,较低沸点的挥发性链烷醇胺的损失影响了产业界对链烷醇胺的选择,该损失往往是可在远超过100℃的温度下进行的解吸过程的结果。例如,正是因为这个原因,具有280℃的高得多的沸点的DEA取代了沸点为170℃的MEA,作为所选的链烷醇胺以便减少链烷醇胺损失。沸点为247℃的MDEA也因挥发性低于MEA而受人青睐。因而,通常优选沸点高而蒸气压低的吸收剂。又一个影响链烷醇胺吸收剂用于从气体流中去除CO2和/或H2S的经济可行性的问题,是制备期望的链烷醇胺所需的大宗材料的成本。例如,工业标准品MEA、DEA和MDEA是由环氧乙烷与氨制备的,环氧乙烷是特别昂贵的原材料。同时,由于MDEA最通常是以哌嗪活化的MDEA的形式使用,它的成本进一步被提升,这部分地是因为大规模制备哌嗪的成本昂贵。使用哌嗪活化的MDEA也不环保,对于这种MDEA的运输有限制。影响用链烷醇胺吸收剂进行气体净化的经济性的又一个问题是,链烷醇胺是按重量出售的,但就工业生产容量而言是按体积使用的。链烷醇胺的重量/体积比往往对它们的使用经济性有负面影响。仍需要替代的和经济上有利的用于捕集酸性气体的链烷醇胺吸收剂,特别是仍需要一种能够使用低成本材料有效制备有用的链烷醇胺的方法。技术实现要素:本发明基于出乎意料的发现,即从甘油(GL)、碳酸二甲酯(DMC)和各种胺的反应制备的链烷醇胺是特别有用的捕集酸性气体的吸收剂。具体地讲,已发现了一种用于制备有用的链烷醇胺的有利方法,该方法包括在专门选择的反应条件下使用离子液体。甘油在生物柴油的生产中作为副产物大量产生。随着人们日益关注使用生物柴油以至少部分替代石化燃料,甘油的产生已增加到了高出目前的需求的地步。因此,甘油是廉价和可现成获得的,特别是在盛行生产生物燃料的国家中。此外,由于甘油可源自生物质,它可被认为是绿色材料。碳酸二甲酯也被认为是绿色试剂(参见Kreutzberger.,CharlesB.(2001),"ChloroformatesandCarbonates",Kirk-OthmerEncyclopediaofChemicalTechnology,NewYork:JohnWiley(Kreutzberger.,CharlesB.,2001年,“氯甲酸酯和碳酸酯”,《Kirk-Othmer化学技术百科全书》,纽约:约翰·威立出版社)),并且也相对廉价,特别是与用于制备已知的链烷醇胺吸收剂(如工业标准品MEA、DEA和MDEA)的环氧乙烷相比。在一个方面,本发明提供一种用于制备式I的链烷醇胺化合物和/或式II的链烷醇胺化合物或者它们的盐的方法:其中:R1和R2独立地选自氢、C1-C8直链或支链烷基基团、C2-C8直链或支链烯基基团、C3-C8环烷基基团、C6-C10芳基基团、3-10元杂环基团、多糖基团、聚环氧乙烷基团,或者R1和R2与它们连接的氮原子一起形成杂环基团,其中所述烷基基团、烯基基团、环烷基基团、芳基基团和杂环基团未被取代或者可被一至三个选自以下的基团取代:C1-C6烷氧基、C2-C8烷氧基烷氧基、C3-C8环烷基、C6-C10芳基、3-10元杂环基、C7-C10芳烷基、3-10元杂环基-C1-C4烷基、-OH、-CH2CH(OH)CH2(OH)、-CH(CH2OH)2或-NR3R4,其中R3和R4独立地选自氢或C1-C6直链或支链烷基基团;所述方法包括以下步骤:i)通过甘油(glycerol)与碳酸二甲酯(dimethylcarbonate)的一锅反应制备缩水甘油(glycidol),该一锅反应在100℃至160℃的温度下,以1:4至1:10的甘油与碳酸二甲酯摩尔比,在具有下式的离子液体催化剂存在下进行:[Cat+][X-]其中:[Cat+]表示一种或更多种阳离子种类,[X-]表示一种或更多种阴离子种类;ii)使步骤i)的产物与式III的胺反应,其中:R1和R2如对式I和II所定义。本文所用的术语“芳基”,无论是单独使用还是作为另一基团的一部分使用,都是指芳族的环状或稠合双环的烃基。本文所指的芳基基团的例子包括苯基基团和萘基基团。优选的芳基基团是苯基。本文所用的术语“环烷基”指非芳族的环状的烃基。本文所指的环烷基基团的例子包括环丙基、环丁基、环戊基和环己基。优选的环烷基基团是环丙基。本文所用的术语“杂环基”指在环中包含O、N、NH和S中的一种或更多者的芳族或非芳族、环状或稠合双环的烃基。本文所指的杂环基基团的例子包括环氧乙烷(ethyleneoxide)、氮杂环丁烷基(azetidinyl)、吡咯烷基(pyrrolidinyl)、吡咯基(pyrrolyl)、吡唑基(pyrazolyl)、氧杂环丁烷(oxetanyl)、吡唑啉基(pyrazolinyl)、咪唑基(imidazolyl)、咪唑啉基(imidazolinyl)、咪唑烷基(imidazolidinyl)、噁唑基(oxazolyl)、噁唑烷基(oxazolidinyl)、异噁唑啉基(isoxazolinyl)、异噁唑基(isoxazolyl)、噻唑基(thiazolyl)、噻二唑基(thiadiazolyl)、噻唑烷基(thiazolidinyl)、异噻唑基(isothiazolyl)、异噻唑烷基(isothiazolidinyl)、呋喃基(furyl)、四氢呋喃基(tetrahydrofuryl)、噻吩基(thienyl)、噁二唑基(oxadiazolyl)、哌啶基(piperidinyl)、哌嗪基(piperazinyl)、氮杂卓基(azepinyl)、吡啶基(pyridyl)、吡嗪基(pyrazinyl)、嘧啶基(pyrimidinyl)、哒嗪基(pyridazinyl)、三嗪基(triazinyl)、三唑基(triazolyl)、四唑基(tetrazolyl)、四氢吡喃基(tetrahydropyranyl)、吗啉基(morpholinyl)、硫代吗啉基(thiamorpholinyl)。优选的杂环化合物包括吡咯基、吡咯烷基和哌啶基。本文所用的术语“催化剂”指能提高化学反应的速率而本身又不被该反应消耗的物质。具体地讲,所使用的离子液体催化剂能提高甘油和碳酸二甲酯之间酯交换形成甘油碳酸酯的速率和/或提高甘油碳酸酯脱羧形成缩水甘油的速率。本文所用的术语“离子液体”指能够通过熔化盐来产生,并且经如此产生时仅由离子组成的液体。离子液体可从包含一种阳离子和一种阴离子的均匀物质形成,或者它可由超过一种阳离子和/或超过一种阴离子组成。因此,离子液体可由超过一种阳离子和一种阴离子组成。离子液体还可由一种阳离子和一种或更多种阴离子组成。再另外,离子液体可由超过一种阳离子和超过一种阴离子组成。术语“离子液体”包括具有高熔点的化合物和具有低熔点(例如室温或低于室温)的化合物。因此,许多离子液体具有低于200℃、特别是低于100℃、室温左右(15至30℃)或甚至低于0℃的熔点。具有低于30℃左右的熔点的离子液体通常称为“室温离子液体”,并且往往源自具有含氮杂环阳离子的有机盐。在室温离子液体中,阳离子和阴离子的结构防止有序晶体结构的形成,因此该盐在室温下是液体。离子液体最广为人知的是作为溶剂。已证实许多离子液体具有可忽略不计的蒸气压、温度稳定性、低可燃性和可循环利用性。由于可供采用的阴离子/阳离子组合的数目众多,因此可以微调离子液体的物理性质(例如熔点、密度、粘度及与水或有机溶剂的可混溶性)以适合特定应用的要求。根据本发明,[Cat+]可包含选自以下的阳离子种类:铵(ammonium)、苯并咪唑鎓(benzimidazolium)、苯并呋喃鎓(benzofuranium)、苯并噻吩鎓(benzothiophenium)、苯并三唑鎓(benzotriazolium)、硼鎓(borolium)、噌啉鎓(cinnolinium)、二氮杂双环癸烯鎓(diazabicyclodecenium)、二氮杂双环壬烯鎓(diazabicyclononenium)、1,4-二氮杂双环[2.2.2]辛烷鎓(1,4-diazabicyclo[2.2.2]octanium)、二氮杂双环十一烯鎓(diazabicyclo-undecenium)、二噻唑鎓(dithiazolium)、呋喃鎓(furanium)、胍鎓(guanidinium)、咪唑鎓(imidazolium)、吲唑鎓(indazolium)、吲哚啉鎓(indolinium)、吲哚鎓(indolium)、吗啉鎓(morpholinium)、氧杂硼鎓(oxaborolium)、氧杂磷鎓(oxaphospholium)、噁嗪鎓(oxazinium)、噁唑鎓(oxazolium)、异噁唑鎓(iso-oxazolium)、氧代噻唑鎓(oxothiazolium)、磷鎓(phospholium)、鏻鎓(phosphonium)、二氮杂萘鎓(phthalazinium)、哌嗪鎓(piperazinium)、哌啶鎓(piperidinium)、吡喃鎓(pyranium)、吡嗪鎓(pyrazinium)、吡唑鎓(pyrazolium)、哒嗪鎓(pyridazinium)、吡啶鎓(pyridinium)、嘧啶鎓(pyrimidinium)、吡咯烷鎓(pyrrolidinium)、吡咯鎓(pyrrolium)、喹唑啉鎓(quinazolinium)、喹啉鎓(quinolinium)、异喹啉鎓(iso-quinolinium)、喹喔啉鎓(quinoxalinium)、奎宁环鎓(quinuclidinium)、硒唑鎓(selenazolium)、锍鎓(sulfonium)、四唑鎓(tetrazolium)、噻二唑鎓(thiadiazolium)、异噻二唑鎓(iso-thiadiazolium)、噻嗪鎓(thiazinium)、噻唑鎓(thiazolium)、异噻唑鎓(iso-thiazolium)、噻吩鎓(thiophenium)、硫脲鎓(thiuronium)、三嗪鎓(triazinium)、三唑鎓(triazolium)、异三唑鎓(iso-triazolium)和脲鎓(uranium)。在本发明的一个优选实施方案中,[Cat+]包含选自以下的无环阳离子:[N(Ra)(Rb)(Rc)(Rd)]+、[P(Ra)(Rb)(Rc)(Rd)]+和[S(Ra)(Rb)(Rc)]+,其中:Ra、Rb、Rc和Rd各自独立地选自C1-C30直链或支链烷基基团、C3-C8环烷基基团或C6-C10芳基基团;并且其中所述烷基基团、环烷基基团或芳基基团未被取代或者可被一个至三个选自以下的基团取代:C1-C6烷氧基、C2-C12烷氧基烷氧基、C3-C8环烷基、C6-C10芳基、C7-C10烷芳基、C7-C10芳烷基、-CN、-OH、-SH、-NO2、-CO-2Rx、-OC(O)Rx、-C(O)Rx、-C(S)Rx、-CS2Rx、-SC(S)Rx、-S(O)(C1-C6)烷基、-S(O)O(C1-C6)烷基、-OS(O)(C1-C6)烷基、-S(C1-C6)烷基、-S-S(C1-C6烷基)、-NRxC(O)NRyRz、-NRxC(O)ORy、-OC(O)NRyRz、-NRxC(S)ORy、-OC(S)NRyRz、-NRxC(S)SRy、-SC(S)NRyRz、-NRxC(S)NRyRz、-C(O)NRyRz、-C(S)NRyRz、-NRyRz或者杂环基团,其中Rx、Ry和Rz独立地选自氢或C1-C6烷基。更优选地,[Cat+]包含选自以下的阳离子:[N(Ra)(Rb)(Rc)(Rd)]+、[P(Ra)(Rb)(Rc)(Rd)]+和[S(Ra)(Rb)(Rc)]+,其中:Ra、Rb、Rc和Rd各自独立地选自C1-C15直链或支链烷基基团、C3-C6环烷基基团或C6芳基基团;并且其中所述烷基基团、环烷基基团或芳基基团未被取代或者可被一个至三个选自以下的基团取代:C1-C6烷氧基、C2-C12烷氧基烷氧基、C3-C8环烷基、C6-C10芳基、C7-C10烷芳基、C7-C10芳烷基、-CN、-OH、-SH、-NO2、-CO2Rx、-OC(O)Rx、-C(O)Rx、-C(S)Rx、-CS2Rx、-SC(S)Rx、-S(O)(C1-C6)烷基、-S(O)O(C1-C6)烷基、-OS(O)(C1-C6)烷基、-S(C1-C6)烷基、-S-S(C1-C6烷基)、-NRxC(O)NRyRz、-NRxC(O)ORy、-OC(O)NRyRz、-NRxC(S)ORy、-OC(S)NRyRz、-NRxC(S)SRy、-SC(S)NRyRz、-NRxC(S)NRyRz、-C(O)NRyRz、-C(S)NRyRz、-NRyRz或者杂环基团,其中Rx、Ry和Rz独立地选自氢或C1-C6烷基。更多的例子包括其中Ra、Rb、Rc和Rd独立地选自甲基、乙基、正丙基、正丁基、正戊基、正己基、正庚基、正辛基、正壬基、正癸基、正十一烷基、正十二烷基、正十三烷基、正十四烷基、正十五烷基、正十六烷基、正十七烷基和正十八烷基。更优选地,Ra、Rb、Rc和Rd中的两者或更多者,并且最优选地,Ra、Rb、Rc和Rd中的三者或更多者选自甲基、乙基、丙基和丁基。还更优选地,[Cat+]包含选自以下的阳离子:[N(Ra)(Rb)(Rc)(Rd)]+,其中:Ra、Rb、Rc和Rd如上所定义。在又一个优选的实施方案中,[Cat+]优选地包含选自以下的阳离子:[P(Ra)(Rb)(Rc)(Rd)]+,其中:Ra、Rb、Rc和Rd如上所定义。适合根据本发明使用的优选的铵阳离子和鏻鎓阳离子的具体例子包括:适合根据本发明使用的更优选的铵阳离子的具体例子包括:在本发明的又一个实施方案中,[Cat+]包含选自胍鎓、环状胍鎓、脲鎓、环状脲鎓、硫脲鎓和环状硫脲鎓的阳离子。更优选地,[Cat+]包含具有下式的阳离子:其中:Ra、Rb、Rc、Rd、Re和Rf各自独立地选自C1-C30直链或支链烷基基团、C3-C8环烷基基团或C6-C10芳基基团,或者Ra、Rb、Rc和Rd中的任何连接不同氮原子的两者形成亚甲基链-(CH2)q-,其中q为2-5;其中所述烷基基团、环烷基基团或芳基基团或者所述亚甲基链未被取代或者可被一个至三个选自以下的基团取代:C1-C6烷氧基、C2-C12烷氧基烷氧基、C3-C8环烷基、C6-C10芳基、C7-C10烷芳基、C7-C10芳烷基、-CN、-OH、-SH、-NO2、-CO2Rx、-OC(O)Rx、-C(O)Rx、-C(S)Rx、-CS-2Rx、-SC(S)Rx、-S(O)(C1-C6)烷基、-S(O)O(C1-C6)烷基、-OS(O)(C1-C6)烷基、-S(C1-C6)烷基、-S-S(C1-C6烷基)、-NRxC(O)NRyRz、-NRxC(O)ORy、-OC(O)NRyRz、-NRxC(S)ORy、-OC(S)NRyRz、-NRxC(S)SRy、-SC(S)NRyRz、-NRxC(S)NRyRz、-C(O)NRyRz、-C(S)NRyRz、-NRyRz或者杂环基团,其中Rx、Ry和Rz独立地选自氢或C1-C6烷基。适合根据本发明使用的胍鎓阳离子、脲鎓阳离子和硫脲鎓阳离子的具体例子包括:在又一个优选的实施方案中,[Cat+]包含具有富电子的硫部分或硒部分的阳离子。例子包括上文所定义的具有硫醇、硫醚或二硫化物侧取代基的阳离子。在本发明的另一个实施方案中,[Cat+]包含选自以下的芳族杂环阳离子种类:苯并咪唑鎓、苯并呋喃鎓、苯并噻吩鎓、苯并三唑鎓、噌啉鎓、二氮杂双环癸烯鎓、二氮杂双环壬烯鎓、二氮杂双环十一烯鎓、二噻唑鎓、咪唑鎓、吲唑鎓、吲哚啉鎓、吲哚鎓、噁嗪鎓、噁唑鎓、异噁唑鎓、氧杂噻唑鎓、二氮杂萘鎓、吡嗪鎓、吡唑鎓、哒嗪鎓、吡啶鎓、嘧啶鎓、喹唑啉鎓、喹啉鎓、异喹啉鎓、喹喔啉鎓、四唑鎓、噻二唑鎓、异噻二唑鎓、噻嗪鎓、噻唑鎓、异噻唑鎓、三嗪鎓、三唑鎓和异三唑鎓。更优选地,[Cat+]具有下式:其中:Ra、Rb、Rc、Rd、Re、Rf和Rg各自独立地选自氢、C1-C30直链或支链烷基基团、C3-C8环烷基基团或C6-C10芳基基团,或者Rb、Rc、Rd、Re和Rf中的任何连接相邻碳原子的两者形成亚甲基链-(CH2)q-,其中q为3-6;并且其中所述烷基基团、环烷基基团或芳基基团或者所述亚甲基链未被取代或者可被一个至三个选自以下的基团取代:C1-C6烷氧基、C2-C12烷氧基烷氧基、C3-C8环烷基、C6-C10芳基、C7-C10烷芳基、C7-C10芳烷基、-CN、-OH、-SH、-NO2、-CO2Rx、-OC(O)Rx、-C(O)Rx、-C(S)Rx、-CS-2Rx、-SC(S)Rx、-S(O)(C1-C6)烷基、-S(O)O(C1-C6)烷基、-OS(O)(C1-C6)烷基、-S(C1-C6)烷基、-S-S(C1-C6烷基)、-NRxC(O)NRyRz、-NRxC(O)ORy、-OC(O)NRyRz、-NRxC(S)ORy、-OC(S)NRyRz、-NRxC(S)SRy、-SC(S)NRyRz、-NRxC(S)NRyRz、-C(O)NRyRz、-C(S)NRyRz、-NRyRz或者杂环基团,其中Rx、Ry和Rz独立地选自氢或C1-C6烷基。Ra优选地选自C1-C30直链或支链烷基、更优选C2-C20直链或支链烷基、还更优选C2-C10直链或支链烷基、最优选C4-C8直链或支链烷基。另外的例子包括其中Ra选自甲基、乙基、正丙基、正丁基、正戊基、正己基、正庚基、正辛基、正壬基、正癸基、正十一烷基、正十二烷基、正十三烷基、正十四烷基、正十五烷基、正十六烷基、正十七烷基和正十八烷基。在包含Rg基团的阳离子中,Rg优选地选自C1-C10直链或支链烷基、更优选C1-C5直链或支链烷基,最优选Rg为甲基基团。在同时包含Ra基团和Rg基团的阳离子中,Ra和Rg各自优选地独立地选自C1-C30直链或支链烷基,并且Ra和Rg中的一个也可为氢。更优选地,Ra和Rg中的一个可选自C2-C20直链或支链烷基、还更优选C2-C10直链或支链烷基、最优选C4-C8直链或支链烷基,并且Ra和Rg中的另一个可选自C1-C10直链或支链烷基、更优选C1-C5直链或支链烷基、最优选甲基基团。在又一个优选的实施方案中,Ra和Rg在存在的情况下可各自独立地选自C1-C30直链或支链烷基和C1-C15烷氧基烷基。在又一个优选的实施方案中,Rb、Rc、Rd、Re和Rf独立地选自氢和C1-C5直链或支链烷基,更优选地,Rb、Rc、Rd、Re和Rf为氢。在本发明的这个实施方案中,[Cat+]优选地包含选自以下的阳离子:其中:Ra、Rb、Rc、Rd、Re、Rf和Rg如上所定义。更优选地,[Cat+]包含选自以下的阳离子:其中:Ra和Rg如上所定义。另外根据本发明的这个实施方案,[Cat+]可优选地包含选自以下的阳离子:其中:Ra、Rb、Rc、Rd、Re、Rf和Rg如上所定义。可根据本发明使用的优选的含氮芳族杂环阳离子的具体例子包括:在本发明的另一个优选实施方案中,[Cat+]包含选自环铵、1,4-二氮杂双环[2.2.2]辛烷鎓、吗啉鎓、环鏻鎓、哌嗪鎓、哌啶鎓、奎宁环鎓和环锍鎓的饱和杂环阳离子。更优选地,[Cat+]包含具有下式的饱和杂环阳离子:其中:Ra、Rb、Rc、Rd、Re、Rf和Rg如上所定义。还更优选地,[Cat+]包含具有下式的饱和杂环阳离子:并且最优选地为其中:Ra、Rb、Rc、Rd、Re、Rf和Rg如上所定义。适合根据本发明使用的优选的饱和杂环阳离子的一个具体例子是1-丁基-1-甲基吡咯烷鎓阳离子:另外根据本发明的这个实施方案,[Cat+]可优选地包含选自以下的饱和杂环阳离子:其中:Ra、Rb、Rc、Rd、Re、Rf和Rg如上所定义。在以上的饱和杂环阳离子中,Ra优选地选自C1-C30直链或支链烷基、更优选C2-C20直链或支链烷基、还更优选C2-C10直链或支链烷基、最优选C4-C8直链或支链烷基。另外的例子包括其中Ra选自甲基、乙基、正丙基、正丁基、正戊基、正己基、正庚基、正辛基、正壬基、正癸基、正十一烷基、正十二烷基、正十三烷基、正十四烷基、正十五烷基、正十六烷基、正十七烷基和正十八烷基。在包含Rg基团的饱和杂环阳离子中,Rg优选地选自C1-C10直链或支链烷基、更优选C1-C5直链或支链烷基,最优选Rg为甲基基团。在同时包含Ra基团和Rg基团的饱和杂环阳离子中,Ra和Rg各自优选地独立地选自C1-C30直链或支链烷基,并且Ra和Rg中的一个也可为氢。更优选地,Ra和Rg中的一个可选自C2-C20直链或支链烷基、还更优选C2-C10直链或支链烷基、最优选C4-C8直链或支链烷基,并且Ra和Rg中的另一个可选自C1-C10直链或支链烷基、更优选C1-C5直链或支链烷基、最优选甲基基团。在又一个优选的实施方案中,Ra和Rg在存在的情况下可各自独立地选自C1-C30直链或支链烷基和C1-C15烷氧基烷基。在又一个优选的实施方案中,Rb、Rc、Rd、Re和Rf独立地选自氢和C1-C5直链或支链烷基,更优选地,Rb、Rc、Rd、Re和Rf为氢。根据本发明,[X-]可包含一种或多种选自以下的阴离子:氢氧根(hydroxides)、卤根(halides)、过卤根(perhalides)、假卤根(pseudohalides)、硫酸根(sulphates)、亚硫酸根(sulphites)、磺酸根(sulfonates)、磺酰亚胺根(sulfonimides)、磷酸根(phosphates)、亚磷酸根(phosphates)、膦酸根(phosphonates)、甲基化物根(methide)、硼酸根(borates)、羧酸根(carboxylates)、唑根(azolate)、碳酸根(carbonates)、氨基甲酸根(carbamates)、硫代磷酸根(thiophosphates)、硫代羧酸根(thiocarboxylates)、硫代氨基甲酸根(thiocarbamates)、硫代碳酸根(thiocarbonates)、黄原酸根(xanthates)、硫代磺酸根(thiosulfonates)、硫代硫酸根(thiosulfates)、硝酸根(nitrate)、亚硝酸根(nitrite)、过氯酸根(perchlorate)、卤素金属酸根(halometallates)、氨基酸根(aminoacids)和硼酸根(borates)。因此,[X-]可表示一种或多种选自以下的阴离子:a)选自以下的卤根阴离子:F-、Cl-、Br-、I-;b)选自以下的过卤根阴离子:[I3]-、[I2Br]-、[IBr2]-、[Br3]-、[Br2C]-、[BrCl2]-、[ICl2]-、[I2Cl]-、[Cl3]-;c)选自以下的假卤根阴离子:[N3]-、[NCS]-、[NCSe]-、[NCO]-、[CN]-;d)选自以下的硫酸根阴离子:[HSO4]-、[SO4]2-、[RkOSO2O]-;e)选自以下的亚硫酸根阴离子:[HSO3]-、[SO3]2-、[RkOSO2]-;f)选自以下的磺酸根阴离子:[RjSO2O]-;g)选自以下的磺酰亚胺根阴离子:[(RjSO2)2N]-;h)选自以下的磷酸根阴离子:[H2PO4]-、[HPO4]2-、[PO4]3-、[RkOPO3]2-、[(RkO)2PO2]-、i)选自以下的亚磷酸根阴离子:[H2PO3]-、[HPO3]2-、[RkOPO2]2-、[(RkO)2PO]-;j)选自以下的膦酸根阴离子:[RjPO3]2-、[RjP(O)(ORk)O]-;k)选自以下的甲基化物根阴离子:[(RjSO2)3C]-;l)选自以下的硼酸根阴离子:[双草酸硼酸根]、[双丙二酸硼酸根];m)选自以下的羧酸根阴离子:[RkCO2]-;n)选自以下的唑根阴离子:[3,5-二硝基-1,2,4-三唑根]、[4-硝基-1,2,3-三唑根]、[2,4-二硝基咪唑根]、[4,5-二硝基咪唑根]、[4,5-二氰基-咪唑根]、[4-硝基咪唑根]、[四唑根];o)选自以下的含硫阴离子:硫代碳酸根(例如[RkOCS2]-);硫代氨基甲酸根(例如[Rk2NCS2]-);硫代羧酸根(例如[RjCS2]-);硫代磷酸根(例如[(RkO)2PS2]-);硫代磺酸根(例如[RS(O)2S]-);和硫代硫酸根(例如[ROS(O)2S]-);和p)硝酸根([NO3]-)或亚硝酸根([NO2]-)阴离子;q)选自以下的碳酸根阴离子:[CO3]2-、[HCO3]-、[RkCO3]-;优选[MeCO3]-;其中:Rj和Rk独立地选自C1-C10烷基、C6芳基、C1-C10烷基(C6)芳基和C6芳基(C1-C10)烷基,这些基团中的每一个都可被一个或更多个选自以下的基团取代:氟、氯、溴、碘、C1-C6烷氧基、C2-C12烷氧基烷氧基、C3-C8环烷基、C6-C10芳基、C7-C10烷芳基、C7-C10芳烷基、-CN、-OH、-SH、-NO2、-CO2Rx、-OC(O)Rx、-C(O)Rx、-C(S)Rx、-CS-2Rx、-SC(S)Rx、-S(O)(C1-C6)烷基、-S(O)O(C1-C6)烷基、-OS(O)(C1-C6)烷基、-S(C1-C6)烷基、-S-S(C1-C6烷基)、-NRxC(O)NRyRz、-NRxC(O)ORy、-OC(O)NRyRz、-NRxC(S)ORy、-OC(S)NRyRz、-NRxC(S)SRy、-SC(S)NRyRz、-NRxC(S)NRyRz、-C(O)NRyRz、-C(S)NRyRz、-NRyRz或者杂环基团,其中Rx、Ry和Rz独立地选自氢或C1-C6烷基,并且其中Rj还可为氟、氯、溴或碘。在一个优选的实施方案中,[X-]包含选自以下的卤根或过卤根阴离子:[F]-、[Cl]-、[Br]-、[I]-、[I3]-、[I2Br]-、[IBr2]-、[Br3]-、[Br2Cl]-、[BrCl2]-、[ICl2]-、[I2Cl]-、[Cl3]-。更优选地,[X-]包含选自以下的卤根或过卤根阴离子:[F]-、[Cl]-、[Br]-、[I]-、[I2Br]-、[IBr2]-、[Br2Cl]-、[BrCl2]-、[ICl2]-、[I2Cl]-。在又一个优选的实施方案中,[X-]包含选自以下的含氧阴离子:[NO3]-、[NO2]-、[H2PO4]-、[HPO4]2-、[PO4]3-、[RkOPO3]2-、[(RkO)2PO2]-、[H2PO3]-、[HPO3]2-、[RkOPO2]2-、[(RkO)2PO]-、[RjPO3]2-、[RjP(O)(ORk)O]-,其中Rj和Rk如上所定义。这个类别的阴离子的更多例子包括:[MeOPO3]2-、[EtOPO3]2-、[(MeO)2PO2]-、[(EtO)2PO2]-、[MePO3]2-、[EtPO3]2-、[MeP(O)(OMe)O]-、[EtP(O)(OEt)O]-。在又一个优选的实施方案中,[X-]包含选自[RkCO2]-的羧酸根阴离子;其中Rk如上所定义。这个类别的阴离子的更多例子包括:[HCO2]-、[MeCO2]-、[EtCO2]-、[CH2(OH)CO2]-、[CH3CH(OH)CH2CO2]-、[PhCO2]-、水杨酸根、丙氨酸根、精氨酸根、天冬酰胺根、天冬氨酸根、半胱氨酸根、谷氨酸根、谷氨酰胺根、甘氨酸根、组氨酸根、异亮氨酸根、亮氨酸根、赖氨酸根、甲硫氨酸根、苯丙氨酸根、脯氨酸根、丝氨酸根、苏氨酸根、色氨酸根、赖氨酸根、缬氨酸根、N-甲基甘氨酸根、2-氨基丁酸根、2-氨基异丁酸根、2-氨基-4-氨基氧基丁酸根、2-(甲基胍基)-乙酸根、2-吡咯烷酮-5-羧酸根、哌啶-2-羧酸根和1-哌啶丙酸根。在又一个优选的实施方案中,[X-]包含具有富电子的硫部分或硒部分的阴离子。例子包括:上文所定义的具有硫醇、硫醚或二硫化物侧取代基的阴离子、[NCS]-、[NCSe]-、[RkOCS2]-、[Rk2NCS2]-、[RkCS2]-、[(RkO)2PS2]-、[RjS(O)2S]-和[RjOS(O)2S]-,其中Rj和Rk如上所定义。这个类别的阴离子的更多例子包括:[CH2(SH)CO2]-、[CH3CH2(SH)CO2]-、[CH3CS2]-、[CH3CH2CS2]-、[PhCS2]-、[(MeO)2PS2]-、[(EtO)2PS2]-、[(PhO)2PS2]-、[(CH3)2NCS2]-、[(CH3CH2)2NCS2]-、[Ph2NCS2]-、[CH3OCS2]-、[CH3CH2OCS2]-、[PhOCS2]-、在又一个优选的实施方案中,[X-]包含选自以下的含硫阴离子:硫酸根阴离子([HSO4]-、[SO4]2-、[RkOSO2O]-)、亚硫酸根阴离子([HSO3]-、[SO3]2-、[RkOSO2]-)和磺酸根阴离子([RjSO2O]-)。这个类别的阴离子的更多例子包括:[FSO2O]-、[CF3SO2O]-、[MeSO2O]-、[PhSO2O]-、[4-MeC6H4SO2O]-、[二辛基磺基琥珀酸根]-、[MeOSO2O]-、[EtOSO2O]-、[C8H17OSO2O]-和[MeOSO2]-、[PhOSO2]-。在又一个优选的实施方案中,[X-]包含选自[RkCO3]-的碳酸根阴离子;其中Rk如上所定义。优选地,其中[X-]包含选自[RkCO3]-的碳酸根阴离子,Rk选自甲基、乙基、正丙基、正丁基、正戊基、正己基、正庚基、正辛基、正壬基、正癸基、正十一烷基、正十二烷基、正十三烷基、正十四烷基、正十五烷基、正十六烷基、正十七烷基和正十八烷基。更优选地,Rk选自甲基、乙基、正丙基、正丁基,最优选地,Rk为甲基。在又一个优选的实施方案中,[X-]可包含选自[OH]-和[SH]-的阴离子。在本发明的一个特别优选的实施方案中,[X-]可包含选自[CO3]2-、[HCO3]-、[MeCO3]-、[OH]-和[SH]-的阴离子,最优选选自[MeCO3]-和[OH]-的阴离子。在本发明的又一个实施方案中,[X-]可包含选自以下的氟化阴离子:[BF4]、[CF3BF3]-、[CF3CF2BF3]-、[PF6]-、[CF3PF5]-、[CF3CF2PF5]-、[(CF3CF2)2PF4]-和[(CF3CF2)3PF3]-。但是,这种类型的氟化阴离子与以上公开的阴离子类型相比通常较不优选。本发明并不限于包含仅具有单一电荷的阴离子和阳离子的离子液体。因此,式[Cat+][X-]旨在涵括包含例如具有双电荷、三电荷和四电荷的阴离子和/或阳离子的离子液体。所以,离子液体中的[Cat+]和[X-]的相对化学计量量并不固定,而是可进行改变以考虑具有多电荷的阳离子和阴离子。例如,式[Cat+][X-]应理解为包括具有式[Cat+]2[X2-]、[Cat2+][X-]2、[Cat2+][X2-]、[Cat+]3[X3-]、[Cat3+][X-]3等等的离子液体。还应认识到,本发明并不限于包含单种阳离子和单种阴离子的离子液体。因此,在某些实施方案中,[Cat+]可表示两种或更多种阳离子,如1,3-二甲基咪唑鎓、1-乙基-3-甲基咪唑鎓和1-3-二乙基咪唑鎓的统计学混合物。同样,在某些实施方案中,[X-]可表示两种或更多种阴离子,如三溴根([Br3]-)和双(三氟甲磺酰)亚胺根([N(SO2CF3)2]-)的混合物。在一个实施方案中,本发明的方法中使用的离子液体是三丁基甲基甲基碳酸铵(tributylmethylammoniummethylcarbonate)。在另一个实施方案中,本发明的方法中使用的离子液体是1-丁基-1-甲基甲基碳酸吡咯烷鎓(1-butyl-1-methylpyrrolidiniummethylcarbonate)。在又一个实施方案中,本发明的方法中使用的离子液体是四甲基氢氧化铵。根据本发明的式I和II的链烷醇胺的形成据信遵循以下反应途径:碳酸二甲酯(DMC)和甘油(GL)在离子液体催化剂的存在下反应形成甘油碳酸酯(GC)中间体,该中间体随后被转化为缩水甘油(GD)。随后缩水甘油与胺的反应产生期望的链烷醇胺。已出乎意料地发现,在离子液体催化的甘油与碳酸二甲酯的一锅合成反应中,通过在100℃至160℃的温度下并使用1:4至1:10的甘油与碳酸二甲酯摩尔比进行该反应,生成缩水甘油中间体的选择性可得到提高,同时维持高转化率。特别出乎意料的是,基于已知的使用离子液体催化剂从甘油和碳酸二甲酯合成缩水甘油的现有技术方法,这些反应条件既导致转化率高,也导致制备缩水甘油中间体的选择性更好。S.M.Gadeetal.,CatalysisCommunications,27,2012,pages184to188(S.M.Gade等人,《催化剂通讯》,第27期,2012年,第184至188页;下文简称“Gade等人”)报道了使用离子液体催化剂在温和条件下从甘油和碳酸二甲酯一锅合成缩水甘油。但是,根据该公开文献,选择性不受甘油:碳酸二甲酯摩尔比的变化的显著影响。根据Gade等人的那些研究,对于1:2的甘油:碳酸二甲酯摩尔比,观察到的最高缩水甘油选择性仅为55%。所报道的这个选择性仍十分低。这些结果在图2中示出;当甘油:碳酸二甲酯比率从2:1变为3:1时,观察到选择性下降。Gade等人还报道了在三种不同温度下(70℃、80℃和90℃)温度对转化率和选择性的影响。当温度从70℃提高到80℃时,发现甘油的转化率显著提高。但是,反应温度从80℃提高到90℃时,没有观察到转化率的进一步改进。此外,并且值得注意的是,当根据Gade等人报道的标准操作条件进行测试时,没有发现反应温度的变化显著影响缩水甘油选择性,该选择性始终显示在50%左右,并且实际上随着温度提高而下降。这些结果在图1中示出。因此,按照Gade等人提供的信息,根据本发明方法的步骤i)的甘油与碳酸二甲酯反应的条件会导致高转化率及高缩水甘油选择性,这是完全出乎意料的。缩水甘油制备的高选择性使得随后在本发明方法的步骤ii)中缩水甘油与式III的胺反应后,可提高式I和/或II的链烷醇胺的产率。根据本发明的一个实施方案,式I、II或III的R1和R2独立地选自氢、C1-C8直链或支链烷基基团、C2-C8直链或支链烯基基团、C3-C8环烷基基团、C6-C10芳基基团、3-10元杂环基团,或者式I、II或III的R1和R2与它们连接的氮原子一起形成杂环基团,其中所述烷基基团、烯基基团、环烷基基团、芳基基团和杂环基团未被取代或者可被一至三个选自以下的基团取代:C1-C6烷氧基、C2-C8烷氧基烷氧基、C3-C8环烷基、C6-C10芳基、3-10元杂环基、C7-C10芳烷基、3-10元杂环基-C1-C4烷基、-OH、-CH2CH(OH)CH2(OH)、-CH(CH2OH)2或-NR3R4,其中R3和R4独立地选自氢或C1-C6直链或支链烷基基团。在另一个实施方案中,式I、II或III的R1选自多糖基团或聚环氧乙烷基团,该多糖基团优选地源自壳聚糖多糖。在本发明的一个优选实施方案中,式I、II或III的R1和R2独立地选自氢、C1-C6直链或支链烷基基团、C3-C6环烷基基团、C6-C10芳基基团、3-8元杂环基团,或者式I、II或III的R1和R2与它们连接的氮原子一起形成杂环基团,其中所述烷基基团、环烷基基团、芳基基团和杂环基团未被取代或者可被一至三个选自以下的基团取代:C1-C6烷氧基、C2-C8烷氧基烷氧基、C3-C6环烷基、C6-C10芳基、3-8元杂环基、C7-C10芳烷基、3-10元杂环基-C1-C4烷基、-OH、-CH2CH(OH)CH2(OH)、-CH(CH2OH)2或-NR3R4,其中R3和R4独立地选自氢或C1-C6直链或支链烷基基团。在又一个优选实施方案中,式I、II或III的R1和R2独立地选自氢、C1-C6直链或支链烷基基团、C3-C6环烷基基团、C6-C10芳基基团、3-8元杂环基团,或者式I、II或III的R1和R2与它们连接的氮原子一起形成杂环基团,其中所述烷基基团、环烷基基团、芳基基团、杂环基团未被取代或者可被一至三个选自以下的基团取代:C1-C6烷氧基、C2-C8烷氧基烷氧基、C3-C6环烷基、C6-C10芳基、3-8元杂环基、-OH、-CH2CH(OH)CH2(OH)、-CH(CH2OH)2或-NR3R4,其中R3和R4独立地选自氢或C1-C6直链或支链烷基基团。在本发明的一个更优选实施方案中,式I、II或III的R1和R2独立地选自氢和C1-C6直链或支链烷基基团,或者式I、II或III的R1和R2与它们连接的氮原子一起形成杂环基团,其中所述烷基基团或者所述杂环基团未被取代或者可被一至三个选自以下的基团取代:C1-C6烷氧基、C2-C8烷氧基烷氧基、C3-C6环烷基、C6-C8芳基、3-8元杂环基、-OH、-CH2CH(OH)CH2(OH)、-CH(CH2OH)2或-NR3R4,其中R3和R4独立地选自氢或者C1-C6直链或支链烷基基团。在又一个更优选实施方案中,式I、II或III的R1和R2独立地选自氢和C1-C6直链或支链烷基基团,其中所述烷基基团未被取代或者可被一至三个选自以下的基团取代:C3-C6环烷基、C6-C8芳基、-OH和-NR3R4,其中R3和R4独立地选自氢或者C1-C6直链或支链烷基基团。在还又一个更优选实施方案中,式I、II或III的R1和R2独立地选自氢和C1-C6直链或支链烷基基团,其中所述烷基基团未被取代或者可被一至三个选自以下的基团取代:-OH和-NR3R4,其中R3和R4独立地选自氢或者C1-C6直链或支链烷基基团。根据本发明的直链和支链烷基基团的例子包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、正己基、异己基、正庚基和正辛基。优选的烷基基团包括甲基、乙基、正丙基、异丙基、正丁基、异丁基和叔丁基。可通过本发明方法制备的特别优选的式I和II的链烷醇胺是其中R1和R2为如下的基团的那些链烷醇胺:1)R1为H,R2为H;2)R1为H,R2为甲基;3)R1为H,R2为乙基;4)R1为H,R2为正丙基;5)R1为H,R2为异丙基6)R1为H,R2为正丁基;7)R1为H,R2为叔丁基;8)R1为甲基,R2为甲基;9)R1为乙基,R2为乙基;10)R1为正丙基,R2为正丙基;11)R1为异丙基,R2为异丙基;12)R1为正丁基,R2为正丁基;13)R1为叔丁基,R2为叔丁基;14)R1和R2如以上实施方案1)至12)中所定义,并且R2还被–OH基团取代;15)R1和R2如以上实施方案1)至13)中所定义,并且R2还被-NR3R4取代,其中R3和R4独立地选自氢或者C1-C6直链或支链烷基基团;及16)R1选自–CH2CH(OH)CH2(OH)或–CH(CH2OH)2并且R2选自甲基、乙基、正丙基、正丁基或叔丁基。根据本发明,在该方法的步骤i)中,甘油与碳酸二甲酯的摩尔比为1:4至1:10,优选1:5至1:8,例如1:6至1:7。因此,示例性的甘油与碳酸二甲酯摩尔比包括:1:5、1:6、1:7或1:8。该方法的步骤i)在100℃至160℃的温度下进行,优选地,该方法在110℃至140℃、更优选115℃至130℃的温度下进行。已发现115℃至125℃的温度,例如120℃,对于本发明的方法的步骤i)特别有利。该方法的步骤i)中的加热可使用任何合适的方法实现,本领域技术人员容易获知这些方法。例如,步骤i)中的反应可使用常规热力方法、微波加热或采用其他热源(如超声波或红外辐射)进行加热。在本发明的一个实施方案中,该方法的步骤i)中的加热通过常规热力加热方法实现。在本发明的另一个实施方案中,该方法的步骤i)中的加热通过在微波反应器中的微波加热来实现。离子液体由于微波吸收能力高,可容易地被结合到微波反应中,因此可支持快速且清洁的方法。在将微波反应器装置用于本发明的方法的情况中,通过由通常在2450MHz的频率下运行的磁控管产生的微波能量(即,频率为约108Hz至1012Hz的电磁辐射)来提供加热。反应混合物可在敞开的容器中加热,或者优选地在密闭容器中加热。优选地,微波反应器是自动控制的,使得在操作过程中可指定特定的温度、最大压力、最大功率输出和保持时间。适用于本发明的微波反应器包括CEMExplorer微波反应器和AntonPaarMonowave300微波反应器。该方法的步骤i)可在10,000至1,500,000帕(0.1至15巴)、更优选10,000至1,000,000帕(0.1至10巴)、最优选50,000至500,000帕(0.5至5巴)的压力下进行。本领域技术人员会理解,离子液体和甘油和碳酸二甲酯反应物可在步骤i)中通过连续工艺或分批工艺进行反应。任何常规的液-液或气-液接触装置都可根据本发明使用。例如,可使用逆流液-液接触装置、顺流液-液接触装置、逆流气-液接触装置、顺流气-液接触装置、液-液分批接触装置或气-液分批接触装置,来使离子液体和甘油和碳酸二甲酯反应物反应。如有需要,该方法的步骤i)可在与离子液体、甘油、二甲醚、甘油碳酸酯和缩水甘油相容的溶剂存在下进行。在期望改变离子液体的粘度的情况中,溶剂的使用可能是适当的。适用于这个目的的溶剂是非碱性非质子极性溶剂,如乙酸乙酯、丙酮、二乙醚、乙腈、二甲亚砜、二甲基甲酰胺和环丁砜(四氢噻吩1,1-二氧化物)。溶剂的存在还可有利于缩水甘油产物随后与离子液体分离。因此,在本发明的一个实施方案中,基于该方法的步骤i)中的反应混合物的总重量,溶剂以最多80重量%的量存在。例如,基于该方法的步骤i)中的反应混合物的总重量,溶剂可以以40重量%至80重量%、50重量%至80重量%或60重量%至80重量%的量存在。作为另一种选择,基于该方法的步骤i)中的反应混合物的总重量,溶剂以小于20重量%的量存在。在本发明的另一个实施方案中,基于该方法的步骤i)中的反应混合物的总重量,溶剂以小于10重量%的量存在。在又一个实施方案中,在该方法的步骤i)中,反应在基本上不存在溶剂(即,小于10重量%,优选小于5重量%,例如2重量%、1重量%或0重量%)的情况下进行。离子液体可被担载在与本发明的方法相容的固体上,优选多孔载体材料。适用于本发明的这个实施方案的固体载体包括二氧化硅、氧化铝、二氧化硅-氧化铝和活性炭。通常,供根据本发明的这个实施方案使用的担载离子液体包含50重量%至1重量%的离子液体,更优选20重量%至1重量%的离子液体。本发明方法的步骤i)中使用的离子液体催化剂的量没有具体限制,技术人员根据反应物的量能够容易确定合适的量。例如,离子液体催化剂可以以与基于甘油计至少2摩尔%相对应的量存在。但是,已出乎意料地发现,基于甘油计至少3摩尔%离子液体催化剂的量,对于本发明方法的步骤i)是特别有利的。因此,优选用于该方法的步骤i)的离子液体催化剂的量为基于甘油计至少3摩尔%,更优选至少5摩尔%,甚至更优选至少8摩尔%,例如10摩尔%。在本发明的一些实施方案中,离子液体在该方法的步骤i)中使用后进行循环利用。离子液体与产物/副产物材料的分离可容易地由技术人员使用已知的分离技术进行,所述已知的分离技术例如在不同液相(例如含水液相和有机液相)之间的分配。作为另一种选择,可利用离子液体的可忽略不计的蒸气压,将产物/副产物材料分离到气相中。该方法的步骤i)进行合适的时间,以获得甘油的定量或近乎定量(例如大于95%)的转化。应认识到,反应速率会根据所用的离子液体而不同。另外,其他反应参数如温度、压力和选择的溶剂(如果有的话)也可影响反应速率。甘油的定量或近乎定量的转化优选地在最多90分钟、更优选最多60分钟、还更优选最多30分钟、最优选最多15分钟的反应时间后获得。在反应的加热是在微波中完成的情况中,甘油的定量或近乎定量的转化优选地在最多90分钟、更优选最多60分钟、还更优选最多30分钟、最优选最多15分钟的微波反应保持时间后获得。本文所指的“保持时间”意指将反应混合物保持在处于预定温度下的微波反应器中的时间,而不是该反应混合物的总辐射时间。根据本发明方法的步骤ii),通过缩水甘油与式III的胺基团反应来制备式I和/或II的链烷醇胺化合物或其盐。通过环氧化物的开环来生产链烷醇胺的方法是本领域技术人员熟知的。这类反应例如在以下专利和文献中有描述:US4,863,563;US2,089,569;EP2295438;Org.Lett.,2005,7,第3649至3651页。根据本发明,该方法的步骤ii)可通过使步骤i)的缩水甘油(GD)产物与式III的胺反应来进行,而无须首先将缩水甘油从含有离子液体催化剂的反应混合物分离。作为另一种选择,可从反应混合物分离缩水甘油,然后将使其与式III的胺反应。在任何一种情况中,都是优选当缩水甘油在该方法的步骤ii)中与式III的胺反应时,缩水甘油以分批方式加入到胺,例如滴加。已发现这个方案可降低不期望的副反应(如碱催化的缩水甘油聚合)的可能性。在缩水甘油在步骤ii)中与式III的胺反应之前先进行分离的情况中,可通过技术人员知道的任何合适方式进行分离。例如,在一个实施方案中,使用液-液萃取从反应混合物中回收缩水甘油。缩水甘油可优先被分配到有机相如乙酸乙酯中,该有机相然后可例如通过重力分离(例如在沉淀装置中)进行分离。不同的相还可使用例如倾析器、旋液分离器、静电凝聚过滤器或离心机进行分离。另一种用于从反应混合物分离缩水甘油的合适方法是例如使用枯烯(cumene)进行共沸蒸馏,这例如在US3,374,153中有描述。因此,遵循这种方法的典型共沸蒸馏包括首先在室温下在真空下去除任何多余的碳酸二甲酯和所产生的甲醇。然后加入大量过剩的枯烯,例如每当量缩水甘油加入50当量枯烯,接着将所得混合物在减压下蒸馏。在US3,374,153中描述的方法中,随后通过将缩水甘油萃取到水中使其与枯烯分离,然后使其进行进一步的分馏。但是,在本发明的情形中,这最后的步骤不太优选,因为缩水甘油也会与水形成共沸混合物,并且在这样的步骤中缩水甘油的水解可能会降低缩水甘油的总产率。有利地,已发现枯烯是本发明方法的步骤ii)的合适溶剂。因此,优选的是,在本发明方法的步骤ii)中与式III的胺反应之前,缩水甘油不与枯烯分离。这样,所形成的具有比枯烯高得多的沸点的链烷醇胺,可容易地通过在真空下去除枯烯而从溶剂中分离出来。因此,在本发明的一个实施方案中,在该方法的步骤ii)中进行反应之前,通过共沸蒸馏(优选地使用枯烯)使缩水甘油与反应混合物分离。更优选地,从使用枯烯进行的共沸蒸馏获得的缩水甘油/枯烯混合物直接被用于该方法的步骤ii)中与式III的胺的反应。该方法的步骤ii)在导致可接受的反应速率而又不导致任何反应物的热力分解的合适温度下进行。优选地,该方法的步骤ii)在10℃至100℃的温度下进行,更优选地在30℃至70℃、还更优选地40℃至60℃、例如50℃的温度下进行。技术人员会认识到,在式III的胺为氨的情况下,可能要求较高的温度以获得可接受的反应速率并由此补偿与氨的氮原子相关的较低亲核性。因此,在式III的胺为氨的情况下,步骤ii)的温度可例如高达225℃。优选地,在式III的胺为氨的情况下,步骤ii)的温度为200℃或更低,更优选地,在式III的胺为氨的情况下,步骤ii)的温度为150至200℃。该方法的步骤ii)中的加热可使用任何合适的方法实现,这些方法是本领域技术人员容易获知的,并且可合适地包括前文所述的常规热力方法或微波方法。该方法的步骤ii)可在10,000至1,500,000帕(0.1至15巴)、更优选10,000至1,000,000帕(0.1至10巴)、最优选50,000至500,000帕(0.5至5巴)的压力下进行。本领域技术人员会理解,缩水甘油和式III的胺可在步骤ii)中通过连续工艺或分批工艺反应。任何常规的液-液或气-液接触装置都可根据本发明使用。例如,可使用逆流液-液接触装置、顺流液-液接触装置、逆流气-液接触装置、顺流气-液接触装置、液-液分批接触装置或气-液分批接触装置,来使缩水甘油与式III的胺的反应。如有需要,该方法的步骤ii)可在与反应物相容的溶剂存在下进行。适用于这个目的的溶剂是非碱性非质子的极性溶剂和非极性溶剂,如乙酸乙酯、丙酮、二乙醚、乙腈、二甲亚砜、二甲基甲酰胺、环丁砜(四氢噻吩1,1-二氧化物)、二氯甲烷、氯仿和枯烯。在本发明的一个实施方案中,基于该方法的步骤ii)中的反应混合物的总重量,溶剂以小于80重量%的量存在。在本发明的另一个实施方案中,基于该方法的步骤ii)中的反应混合物的总重量,溶剂以小于50重量%的量存在。在本发明的一个优选实施方案中,基于该方法的步骤ii)中的反应混合物的总重量,溶剂以小于30重量%的量存在。在本发明的一个更优选实施方案中,基于该方法的步骤ii)中的反应混合物的总重量,溶剂以小于10重量%的量存在。在又一个实施方案中,在该方法的步骤ii)中,反应在实质上不存在溶剂(即,小于10重量%,优选小于5重量%,例如2重量%、1重量%或0重量%)的情况下进行。技术人员会认识到,在式III的胺为伯胺的情况下,换句话说在R1和R2中的一者为H的情况下,或者在式III的胺为氨的情况下,单个胺化合物可能会与多个缩水甘油分子反应,从而导致形成相应的二聚体或三聚体链烷醇胺化合物。技术人员会知道,通过使用相比于缩水甘油大量过量的式III的胺化合物,可实质上消除与单个胺分子的多重开环反应。通过将缩水甘油分批加入到该胺中也可实现类似的效果;这代表了该方案相比于将缩水甘油与式III的胺混合的又一个优点。因此,本发明方法的步骤ii)中使用的缩水甘油的量没有具体限制,技术人员根据要反应的式III的胺的量能够容易确定合适的量。如上提到,在式III的胺为伯胺或氨的情况下,可使用大量过量的胺(例如5至10当量的该胺)以减少单个胺化合物与多个缩水甘油分子反应的可能性。因此,在一个优选的实施方案中,在式III的胺为伯胺或氨的情况下,将至少5当量的该胺用于步骤ii)中的反应。更优选地,使用至少10当量。但是,也已发现在该胺为仲胺的情况下,较小量过量的胺(例如1.1当量至1.3当量之间)是合适的,特别是将缩水甘油滴加到该胺中时。步骤ii)中获得的链烷醇胺可进行纯化或者作为粗产物混合物的一部分直接使用。可按需使用技术人员知道的任何合适的纯化方法。例如,可通过蒸馏(例如在减压下蒸馏)从粗产物混合物分离式I和/或II的链烷醇胺。已发现,当通过本发明的方法获得的式I和/或II的链烷醇胺用作酸性气体净化剂时,使用纯化的链烷醇胺样品比使用粗产物混合物得到更好的结果。因此,优选的是,将式I和/或II的链烷醇胺与粗产物混合物分离,例如通过蒸馏分离,然后再用作酸性气体净化剂。缩水甘油与式III的胺的反应通常导致形成对应于本文式I和II的化合物的结构异构体的混合物;根据式I的异构体通常为主要组分。由于对应于本文式I和II的链烷醇胺化合物的不同几何异构体都适合用作酸性气体净化剂,因此在将这些异构体用于酸性气体吸收之前不必将它们分离。但是,如果需要,可通过已知的方法(如色谱方法)分离结构异构体。已发现,式I和II的链烷醇胺当用作酸性气体吸收剂时是特别有利的,其吸收速率与已知的链烷醇胺吸收剂(如MEA和DEA)相当。根据式I和II的链烷醇胺的特征性二醇官能团据认为赋予合乎需要的特性,如高吸收速率和相对高的沸点(约200℃以上),这使得式I和II的链烷醇胺尤其可用作酸性气体吸收剂。不想受任何具体理论的约束,但据信式I和/或II的链烷醇胺化合物对CO2和/或H2S的吸收既通过化学吸收过程(如前所讨论)进行,也通过物理吸收过程进行。这通过实验进行了说明(参见图8),其中当CO2分压增加时,CO2的吸收超过0.5摩尔CO2/摩尔链烷醇胺的理论最大值。因此,在另一个实施方案中,本发明的方法还包括以下步骤:iii)使含有CO2和/或H2S的气体流与步骤ii)中制备的式I的链烷醇胺化合物和/或式II的链烷醇胺化合物或者它们的盐接触;以及iv)获得与步骤iii)的含有CO2和/或H2S的气体流相比CO2和/或H2S含量降低的经处理的气体流。应认识到,该方法的接触步骤iii)要求存在至少一种根据式I或式II的链烷醇胺化合物。因此,在本发明的一个实施方案中,将一种式I化合物用于该方法的接触步骤iii)。在另一个实施方案中,将一种式II化合物用于该方法的接触步骤iii)。也可将链烷醇胺化合物的混合物用于该方法的接触步骤iii)。因此,可将两种或更多种式I的链烷醇胺化合物的混合物用于该方法的接触步骤iii)。同样,可将两种或更多种式II的链烷醇胺化合物的混合物用于该方法的接触步骤iii)。此外,也可将至少一种式I的化合物和至少一种式II的化合物的混合物用于该方法的接触步骤iii)。优选地,式I和/或II的链烷醇胺以溶液的形式用于接触步骤iii)。根据式I或II的链烷醇胺化合物在该溶液中的总量优选地为20重量%至70重量%,更优选30重量%至60重量%,最优选30重量%至50重量%,例如40重量%。适用于本发明方法的接触步骤iii)的溶剂包括极性质子溶剂,这类溶剂包括水、醇,例如甲醇、乙醇、正丁醇和异丙醇。优选地,溶剂为水。因此,在本发明的一个实施方案中,式I和/或式II的化合物以水溶液的形式提供用于接触步骤iii)。作为另一种选择,接触步骤iii)中采用的链烷醇胺可以以被载体担载的形式提供。适用于本发明的载体包括膜载体。合适的聚合物膜载体(包括聚醚砜(PES)载体在内)的例子列举于Nasir等人关于胺聚合物膜(APM)的综述中(Nasiretal.,WorldAcademyofScience,EngineeringandTechnology81,2013,pp465to468(Nashir等人,《世界科学、工程与技术学会》,第81卷,2013年,第465至468页))在一个实施方案中,式I和/或式II的链烷醇胺化合物以被载体担载的形式用于接触步骤iii),其中式I和/或II的R1和R2不形成杂环基团,R1选自多糖基团(优选壳聚糖)或聚环氧乙烷基团。尽管不必要用式I和/或II的链烷醇胺实现酸性气体的充分吸收,但接触步骤iii)可在可增强式I和/或II的链烷醇胺的吸收特性的活化剂化合物的存在下进行。合适的活化剂化合物为例如哌嗪。接触步骤iii)可在吸收器或接触柱中进行,优选地在10至80℃、更优选20至60℃、最优选30至50℃的温度下进行。例如,可使气体流与式I和/或II的链烷醇胺在30℃或30℃左右的温度下接触。优选地使气体流与式I和/或II的链烷醇胺在100至2000kPa、更优选200至1000kPa的压力下接触。例如,可使气体流与该二氧化碳吸收剂在500kPa或500kPa左右的压力下接触。用式I或II的链烷醇胺从气体流去除CO2和/或H2S可构成分批或连续工艺的一部分。在一些实施方案中,该链烷醇胺以溶液的形式提供,该溶液接触气体流后被送到解吸器或汽提柱。可通过降低该溶液上方的酸性气体的分压和/或用蒸汽将该溶液进行汽提,使酸性气体从该溶液中分离出来。在一些实施方案中,在链烷醇胺是以水溶液的形式提供的情况中,可通过简单地在再沸器中进行加热来产生汽提蒸汽。然后可将废链烷醇胺溶液循环回到吸收器。接触步骤iii)可用于从多种不同类型的气体流中去除CO2和/或H2S,并任选地去除一种或更多种另外的物质。例如,式I和/或II的链烷醇胺可用于从来自燃烧过程的废气(如来自炉子和发电厂的烟道气)中去除CO2和/或H2S。式I和/或II的链烷醇胺还可用于从含烃气体流特别是含甲烷气体流中去除CO2和/或H2S,并任选地去除一种或更多种另外的物质。因此,式I和/或II的链烷醇胺可有利地用于从天然气和/或生物气中去除CO2和/或H2S,并任选地去除一种或更多种另外的物质。式I和/或II的链烷醇胺可有利地用于从生命支持系统中的呼吸气体混合物中去除CO2和/或H2S。应认识到,式I和/或II的链烷醇胺作为酸性吸收剂的用途可被整合到加工厂中,作为气体流的多阶段加工的一个阶段。例如,通过本发明方法产生的式I和/或II的链烷醇胺可被用于天然气精炼厂中,作为商业天然气产品的生产中的一个阶段,其中其他阶段可包括氮气的去除和重质烃的去除。作为另一种选择,通过本发明方法产生的式I和/或II的链烷醇胺可被用于烟道气处理厂中,作为烟道气的多阶段加工的一个阶段,其中其他阶段可例如包括颗粒物的去除和NOx的催化转化。在另一个方面,本发明还提供通过本文描述的方法制备的式I和/或II的链烷醇胺化合物用于从含CO2和/或H2S的气体流中去除CO2和/或H2S的用途。因此,在一个实施方案中,根据本发明方法制备的式I和/或式II的链烷醇胺化合物被用于从含有甲烷的气体流(如天然气和/或生物气)中去除CO2和/或H2S以及任选地去除一种或更多种另外的物质。在另一个实施方案中,根据本发明方法制备的式I和/或式II的链烷醇胺化合物被用于从烟道气流中去除CO2和/或H2S。在还又一个实施方案中,根据本发明方法制备的式I和/或式II的链烷醇胺化合物被用于从生命支持系统中的呼吸气体混合物中去除CO2和/或H2S。现将通过以下实施例并参考以下附图对本发明进行举例说明。附图说明图1图示显示了Gade等人所报道的温度对转化率和选择性的影响;图2图示显示了Gade等人所报道的甘油:碳酸二甲酯比率对转化率和选择性的影响;图3图示显示了对于其中甘油:碳酸二甲酯比率为1:5、使用三丁基甲基甲基碳酸铵、1-丁基-1-甲基甲基碳酸吡咯烷鎓或四甲基氢氧化铵作为离子液体催化剂的缩水甘油合成而言,温度(微波加热,15分钟保持时间)对缩水甘油选择性的影响;图4图示显示了工业标准品MEA、DEA和MDEA的蒸气压-温度曲线;图5图示显示了工业标准品DEA和MDEA的蒸气压-温度曲线及根据本发明方法制备的链烷醇胺A和B的蒸气压-温度曲线的对比;图6图示比较了根据本发明方法制备的链烷醇胺A和B及工业标准品MEA、DEA和MDEA的CO2吸收能力;图7图示说明了根据本发明方法制备的链烷醇胺A和B及工业标准品MEA、DEA和MDEA的反复CO2吸收实验的结果的再现性;图8图示说明了根据本发明方法制备的链烷醇胺如何同时表现出化学吸收特性和物理吸收特性。具体实施方式离子液体的制备从市售的25%四甲基铵水溶液制备四甲基氢氧化铵。使用旋转蒸发仪从该溶液去除水。根据Holbrey等人,《绿色化学》(GreenChem.),2010年,第12卷,第407至413页中报道的微波辅助的甲基碳酸盐合成,制备三丁基甲基甲基碳酸铵和1-丁基-1-甲基甲基碳酸吡咯烷鎓。将三丁胺(1.854g,10mmol)、DMC(0.90g,10mmol)和甲醇(2ml)连同磁力搅拌棒加入到10ml微波处理玻璃小瓶中,然后将小瓶密封并放入CEMExplorer微波反应器内。将所得溶液在160℃和磁力搅拌下加热保持1小时时间。在减压下去除挥发性溶剂和过量DMC后,分离三丁基甲基甲基碳酸铵。将1-丁基吡咯烷(1.272g,10mmol)、DMC(0.90g,10mmol)和甲醇(2ml)连同磁力搅拌棒加入到10ml微波处理玻璃小瓶中,然后将小瓶密封并放入CEMExplorer微波反应器内。将所得溶液在140℃和磁力搅拌下加热保持1小时时间。在减压下去除挥发性溶剂和过量DMC后,分离1-丁基-1-甲基甲基碳酸吡咯烷鎓。微波反应使用CEMExplorer微波反应器进行微波反应,操作频率为2450MHz,最大功率输出为80W。将各成分连同磁力搅拌棒加入到10ml微波处理玻璃小瓶中,然后将小瓶密封并放入该反应器内。然后将样品在指定的保持温度下运行预定的时间。除非另有指明,否则下文所指的运行时间是指样品被保持在特定温度下的时间,而不是总的辐射时间。产物样品的分析反应后,使用Agilent6890N气相色谱仪对样品进行气相色谱(GC)分析,该气相色谱仪具有HP-Innowax毛细管柱,采用氦载气,根据以下条件操作:i)流速0.7cm3min-1,50℃,1分钟;ii)以25℃min-1线性梯度升至200℃;iii)以3℃min-1线性梯度从200℃升至230℃;iv)230℃下保持18分钟。从碳酸二甲酯和甘油一锅法制备缩水甘油实施例1将1-丁基-1-甲基甲基碳酸吡咯烷鎓(0.02173g,0.1mmol)与甘油(0.093g,1mmol)和碳酸二甲酯(0.45g,5mmol)连同磁力搅拌棒加到压力定额为1000kPa(10巴)的20ml密封玻璃管中。将密封玻璃管放入预热至120℃的油浴中,剧烈磁力搅拌15分钟。然后从油浴中取出玻璃管,让其冷却至室温后取样品进行气相色谱(GC)分析。实施例2重复实施例1的过程,例外的是在120℃下加热反应30分钟。催化剂装载量基于甘油计保持恒定在10摩尔%,并且采用同样的甘油:碳酸二甲酯摩尔比(1:5)。实施例3将1-丁基-1-甲基甲基碳酸吡咯烷鎓(0.02173g,0.1mmol)与甘油(0.093g,1mmol)和碳酸二甲酯(0.45g,5mmol)连同磁力搅拌棒加到10ml微波处理玻璃小瓶中,然后将小瓶密封。将样品放入CEMExplorer微波反应器内,在120℃、550kPa(5.5巴)压力和磁力搅拌下加热15分钟的保持时间,然后直接通过气相色谱法(GC)分析反应混合物。实施例4重复实施例3的过程,例外的是使用三丁基甲基甲基碳酸铵代替1-丁基-1-甲基甲基碳酸吡咯烷鎓。催化剂装载量基于甘油计保持恒定在10摩尔%,并且采用同样的甘油:碳酸二甲酯摩尔比(1:5)。实施例5重复实施例3的过程,例外的是使用四甲基氢氧化铵代替1-丁基-1-甲基甲基碳酸吡咯烷鎓。催化剂装载量基于甘油计保持恒定在10摩尔%,并且采用同样的甘油:碳酸二甲酯摩尔比(1:5)。实施例6重复实施例3的过程,例外的是使用1:8的甘油:碳酸二甲酯摩尔比。催化剂装载量基于甘油计保持恒定在10摩尔%。实施例7针对一系列的不同保持温度(100℃、140℃和160℃),重复实施例3至5的过程。在每种情况中,催化剂装载量基于甘油计保持恒定在10摩尔%,并且采用同样的甘油:碳酸二甲酯摩尔比(1:5)。实施例8重复实施例3的过程,例外的是使用1:15的甘油:碳酸二甲酯摩尔比。催化剂装载量基于甘油计保持恒定在10摩尔%。下表1显示了实施例1至8的结果。表1中的结果(对应于项目3、4和7至16)也被用于生成图示(图3)。表1:1反应时间=15分钟;加热=油浴2反应时间=30分钟;加热=油浴3微波加热GL=甘油;DMC=碳酸二甲酯,GD=缩水甘油,GC=甘油碳酸酯表1的结果显示根据本发明的方法的步骤i)实现了出乎意料地高的转化率和缩水甘油选择性,并且可在短的反应时间内获得。例如,当GL和DMC以1:5的GL:DMC摩尔比,在1-丁基-1-甲基甲基碳酸吡咯烷鎓催化剂存在下,使用来自油浴的热量在120℃下反应15分钟时,获得100%的GL转化率和85%的GD选择性(项目1)。当GL和DMC以1:5的GL:DMC摩尔比,在1-丁基-1-甲基甲基碳酸吡咯烷鎓催化剂存在下,在微波中反应15分钟的保持时间时,获得100%的GL转化率和90%的GD选择性(项目4)。当GL和DMC以1:5的GL:DMC摩尔比,在四甲基氢氧化铵催化剂存在下,在微波中反应15分钟的保持时间时,获得97%的GL转化率和82%的GD选择性(项目10)。缩水甘油的分离分离离子液体催化的反应中形成的缩水甘油的一般方法是通过液-液萃取进行。具体地讲,在反应完成后,将基于反应混合物的体积计两体积当量的乙酸乙酯与基于反应混合物的体积计一当量的水加入到反应容器。缩水甘油优先分配到有机相中,接着通过倾析分离有机相,然后在减压下去除有机溶剂,得到缩水甘油粗产物。缩水甘油与式III的胺的反应将从碳酸二甲酯与甘油在离子液体存在下反应制备的缩水甘油加入到含有胺的容器,并将所得的混合物在高温下搅拌以形成相应的链烷醇胺。在以下实施例中,缩水甘油以纯形式使用。但是,可以将该方法的步骤ii)的包含缩水甘油和离子液体催化剂的产物混合物直接转移到胺,而不需首先分离缩水甘油。也可以将胺转移到步骤ii)的包含缩水甘油和离子液体催化剂的产物混合物,尽管这不太优选。不过,这个方法可能会增加不期望的副反应的可能性,如缩水甘油聚合,从而会降低链烷醇胺产率。实施例9链烷醇胺混合物A将缩水甘油(4.96g,64mmol)在5℃下滴加到正丙胺(39.9g,675mmol,10.5当量),并将所得混合物搅拌16小时。到时间后,在真空下去除过量的正丙胺。回收到7.49g(88.1%收率)的无色、轻微粘稠的液体。由1HNMR得知,所形成的产物为如上所示的开环结构异构体的混合物,并且相对于缩水甘油而言转化是完全的。由于信号重叠的缘故,所形成的每种结构异构体的量不能从1HNMR确定。实施例10链烷醇胺混合物B将缩水甘油(5.143g,69mmol)在50℃下滴加到二乙胺(5.608g,77mmol,1.12当量),并将所得混合物搅拌16小时。到时间后,在真空下去除过量的二乙胺。回收到7.61g(89.7%收率)的无色、轻微粘稠的液体。由1HNMR得知,所形成的产物为如上所示的开环结构异构体的混合物,并且相对于缩水甘油而言转化是完全的。由于信号重叠的缘故,所形成的每种结构异构体的量不能从1HNMR确定。评估链烷醇胺的物理性质实施例11在减压下测量链烷醇胺混合物A和混合物B以及工业标准品MEA、DEA和MDEA的沸点。随后使用列线图解法通过外推确定大气压下的沸点。结果在下表2中给出。结果显示,链烷醇胺混合物A和混合物B的沸点均显著高于MEA,并且均与DEA和MDEA的沸点相当。如前文所讨论,高沸点对于降低链烷醇胺循环利用时酸性气体解吸过程中的损失是有利的。链烷醇胺混合物A的沸点比链烷醇胺混合物B的沸点高30℃。这据信是由于仲链烷醇胺相对于叔链烷醇胺而言氢键键合加强的缘故。表2化合物沸点(℃)MEA170.8DEA268.8MDEA246-248链烷醇胺A262-263链烷醇胺B232-233实施例12使用装备有爱德华兹(Edwards)压力传感器的液体蒸气压力计,测定链烷醇胺混合物A和混合物B以及MEA、DEA和MDEA的蒸气压。图4显示工业标准品MEA、DEA和MDEA的蒸气压-温度曲线,从中明显看出MEA的蒸气压显著高于DEA和MDEA。图5显示工业标准DEA和MDEA的蒸气压-温度曲线与链烷醇胺A和B的蒸气压-温度曲线的对比。图5证明了链烷醇胺A和B的蒸气压与DEA和MDEA相当。CO2吸收研究——实验程序在一个典型的实验中,首先通过使压力容器(Parr压力系统)在减压下排空并接着将处于一定温度和压力下的已知量的气体泵入该容器中,来测定该容器的体积。气体的量的测量值从质量流量控制器(BROOKSSmartMassflow)作为标准条件下的气体体积读取。使用理想气体定律计算该压力容器的实际体积。将已知质量和体积的链烷醇胺-水混合物放入压力容器中,并将该容器排空至10kPa。然后在20.0℃通过质量流量控制器将二氧化碳泵入处于搅拌下的压力容器(500rpm),直到500kPa。让该系统平衡1小时,或者直到根据质量流量控制器得知不再有气体加入。使用质量流量控制器中的读数,计算引入到压力容器中的气体的总量。通过理想气体定律计算气相中的实际气体量,其中气相的体积等于压力容器的体积减去液相的体积。通过从引入到压力容器中的气体的总量减去气相中的实际气体量,计算溶解在液相中的气体的量。实施例13制备链烷醇胺混合物A的40重量%水溶液。将4.858ml的该混合物转移到高压锅(500rpm),随后平衡至20℃。施加瞬间真空,然后通过质量流量控制器引入CO2。将该反应器加压到500kPa,记录加入的CO2的总体积。结果在(错误!未找到引用源。)中作为每升吸收剂吸收的CO2摩尔数、每公斤吸收剂吸收的CO2摩尔数和每摩尔链烷醇胺吸收的CO2摩尔数示出。实施例14制备链烷醇胺混合物B的40重量%水溶液。将4.923ml的该混合物转移到高压锅,随后平衡至20℃。施加瞬间真空,然后通过质量流量控制器引入CO2。将该反应器加压到500kPa,记录加入的CO2的总体积。结果在(错误!未找到引用源。)中作为每升吸收剂吸收的CO2摩尔数、每公斤吸收剂吸收的CO2摩尔数和每摩尔链烷醇胺吸收的CO2摩尔数示出。比较例1制备二乙醇胺(DEA)的40重量%水溶液。将4.984ml的该混合物转移到高压锅,随后平衡至20℃。施加瞬间真空,然后通过质量流量控制器引入CO2。将该反应器加压到500kPa,记录加入的CO2的总体积。结果在(错误!未找到引用源。)中作为每升吸收剂吸收的CO2摩尔数、每公斤吸收剂吸收的CO2摩尔数和每摩尔链烷醇胺吸收的CO2摩尔数示出。比较例2制备甲基二乙醇胺(MDEA)的40重量%水溶液。将4.887ml的该混合物转移到高压锅,随后平衡至20℃。施加瞬间真空,然后通过质量流量控制器引入CO2。将该反应器加压到500kPa,记录加入的CO2的总体积。结果在(错误!未找到引用源。)中作为每升吸收剂吸收的CO2摩尔数、每公斤吸收剂吸收的CO2摩尔数和每摩尔链烷醇胺吸收的CO2摩尔数示出。表3表3中给出的结果证明,根据本发明的式I和II的链烷醇胺表现出与工业标准链烷醇胺DEA和MDEA相当的CO2吸收。根据本发明的链烷醇胺(A和B)的显著优点是,它们从廉价和容易获得的甘油前体制备,从而成本效益性更高。实施例15使用不同的反应器压力(在100kPa和700kPaCO2分压之间)重复实施例13和14的方法。对40重量%的MEA水溶液,在相同的CO2分压范围内也采用相同的程序。对于每个所测试的CO2分压,测定链烷醇胺吸收剂的相应吸收能力‘α’(molCO2/mol链烷醇胺)。这些实验的结果连同比较例1和2的结果(500kPaCO2分压)在图6中示出。结果表明,可通过本发明方法制备的链烷醇胺混合物A和混合物B的CO2吸收能力与工业标准MEA、DEA和MDEA相当。图7中显示针对链烷醇胺混合物A和混合物B及工业标准MEA、DEA和MDEA,基本上如实施例13和14及比较例1和2中所描述进行并且在500kPa(5巴)的CO2分压下进行的CO2反复吸收实验的结果。图7证明了根据本发明制备的链烷醇胺的结果具有高水平的再现性,与在工业标准MEA、DEA和MDEA上观察到的再现性相当。化学和物理吸收实施例16为观察根据本发明方法制备的链烷醇胺的化学和物理吸收行为,针对不同的CO2分压,测量链烷醇胺混合物A的40重量%含水混合物的CO2吸收。链烷醇胺混合物A:将该链烷醇胺溶液加到经过清洁/干燥的容器,并将该容器排空至10kPa(0.1巴)。然后将CO2压力调整至100kPa(1巴),通过质量流量控制器引入CO2。2小时后,使压力平衡在100kPa(1巴),记录加入的CO2的体积。随后,使压力提高到150kPa(1.5巴)并且平衡,之后以50kPa(0.5巴)为单位提高。CO2在该链烷醇胺混合物中的溶解度结果在图8中示出。图8清楚地显示,随着CO2分压提高,CO2的吸收超过0.5molCO2/mol链烷醇胺,这表明了除了化学吸收以外还有物理吸收。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1