用于聚(ADP‑核糖)聚合酶‑1(PARP‑1)的放射性标记示踪物,其方法和用途与流程
本申请根据35USC§119(e)要求于2014年1月5日提交的美国临时申请序列号61/923,759的优先权权益,其全部内容通过引用并入。
关于联邦资助研究或开发的声明
本发明是在美国国立卫生研究院的资助NIH P01 HL13851和NIH R01 HL116389的支持下进行的。美利坚合众国政府可能在这项研究中具有某些权利。
技术领域
本教导属于用于成像PARP-1分布的放射性标记示踪物的领域。
背景技术:
聚(ADP-核糖)聚合酶-1(PARP-1)是核酶的PARP家族的最丰富的成员之一。1其最特征的功能是检测DNA损伤和促进DNA修复,但PARP-1也参与许多其它的DNA相关的细胞过程,如细胞凋亡调控、细胞分裂、分化、转录调控和染色体稳定化。2-5PARP-1也可以在调节炎症反应方面发挥中心作用。PARP-1,113kD的蛋白质具有三个独特结构域:具有特异性结合受损的DNA链断裂的两个锌指的N-末端DNA结合结构域;1,6中央自我修饰结构域;和循序地将ADP-核糖亚基从烟酰胺腺嘌呤二核苷酸(NAD+)转移到蛋白质受体的C末端催化结构域。7由于其在DNA修复方面的关键作用,在过去的20年里,已经积极地追求PARP-1作为药物靶标,投入了巨大努力,开发了几代PARP-1抑制剂(图1)用于治疗目的,特别是在缺血再灌注损伤和癌症领域。5,8最近,已经证明PARP1抑制是一种用于在依赖于PARP1活性而生存的癌症中诱导合成致死性的有效方法。9另外,PARP抑制剂或转基因小鼠中缺乏PARP表达降低所存在的炎症的程度,因此保护各种器官,包括肺,免受持续性炎症的不利影响。因此,一些PARP抑制剂,包括奥拉帕尼(olaparib)(AZD2281KU-59436),维利帕尼(veliparib)(ABT-888),鲁卡帕尼(rucaparib)(PF-01367338,AG014699)和尼拉帕尼(niraparib)(MK4827),现作为抗癌药物正处于临床试验评价中10-14。这些PARP抑制剂有效地抑制PARP1活性以及其它的PARP样酶如PARP2和端锚聚合酶(tankyrase1)的活性15。其它PARP抑制剂包括苯并咪唑甲酰胺(NU1085)21及其衍生物(AG014361)。22
尽管具有与无进展生存期相关的来自临床试验的有前景的结果,然而,通过目前的测定不能准确地确定不同的PARP抑制剂抑制肿瘤PARP活性的能力的差异。此外,最近发现最初作为PARP抑制剂开发的尼帕尼(iniparib)的作用机制最有可能与PARP活性的抑制无关16。因此,需要用于体内非侵入性定量肿瘤的PARP活性的方法以便用于证实肿瘤特异性PARP抑制以及评估有效的PARP抑制的持续时间以指导给药决定。
用正电子发射断层扫描(PET)成像可以是用于非侵入性地确定PARP活性水平的有效方法。[11C]PJ-34,一种PARP-1靶向示踪物,显示在成像糖尿病的动物模型中的PARP-1表达的一些潜质。17最近,通过使用反式-环辛烯和四嗪之间的[4+2]环加成的两步标记策略合成了18F标记的奥拉帕尼/AZD2281衍生物并用于体外细胞研究和微型PET肿瘤成像。18-20
技术实现要素:
在各种实施方案中,本教导包括用于测量PARP-1表达和PET成像PARP-1体内分布的放射性标记的PARP-1抑制剂。在各种实施方案中,本教导包括结合PARP-1并标记有发射正电子的放射性核素如18F的各种化合物。在各种实施方案中,也公开了这些化合物的合成方法。还公开了本发明教导的化合物对PARP-1的抑制效力。
在一些配置中,2-[对-(2-氟乙氧基)苯基]-1.3.10-三氮杂三环[6.4.1.04,13]十三烷基-2,4(13),5,7-壬四烯-9-酮(IC50=6.3±1.3nM)可以用18F标记。在各种配置中,18F标记可以是在一个步骤中,且可以提供高的化学和放射化学纯度。在一些配置中,微型PET成像可以用来证明MDA-MB-436肿瘤中的[18F]12摄取增加。在一些配置中,这种增加的摄取可以由12和奥拉帕尼/AZD2281阻止。
在各种不同的实施方案中,已开发了PARP-1抑制剂12(IC50=6.3nM)。可使用常规标记方法来合成具有高纯度和比活性的[18F]12。携带MBA-MD-436肿瘤的小鼠中的[18F]12的微型PET研究证实这些肿瘤中的放射性蓄积,已知这些肿瘤会过表达PARP-1。肿瘤摄取可由PARP-1抑制剂奥拉帕尼和12阻止,支持[18F]12摄取对PARP-1活性的特异性。因此,[18F]12可用作用于体内成像PARP-1表达的PET示踪物。
在各种不同的实施方案中,可以通过用[18F]氟化物亲核取代28的氟乙氧基或使用2-[18F]氟乙基叠氮化物的Cu(I)催化的点击反应实现放射性核素掺入。29,30可以根据方案1和2以仅仅几个步骤分别合成NU1085和AG014361的相应的2-氟乙氧基和2-氟乙基三唑类似物。虽然炔丙基和三唑基略微降低了类似物的抑制效力,这些中最有效的是12(IC50=6.3nM),其具有2-氟乙氧基基团,其次是(IC50=10.8)。
在一些配置中,可以自动化[18F]12在典型条件下的一步标记以便临床生产这种示踪物。在一些配置中,用于注射的最终剂量可具有高的化学和放射化学纯度和具有优异的比活性。在各种配置中,[18F]12在微型PET研究中的注射质量可以是0.0037-0.012μg/剂量(0.2mCi/剂量),其可以比阻断剂量和治疗剂量低得多。质量的这个量可以是不太可能有药理作用。因此,[18F]12可用作PET示踪物用于成像PARP-1表达和肿瘤。在一些配置中,PARP-1示踪物如[18F]12也可以用于确定患有慢性炎性疾病的患者,在这些患者中PARP抑制剂治疗可能延缓炎症疾病的进展。
因此,本教导包括具有如下结构的化合物及其药学上可接受的盐:
其中R可以选自由以下组成的组:其中代表键。在一些配置中,R可以是在一些配置中,F可以是18F。在一些配置中,R可以是在一些配置中,R可以是
在一些实施方案中,本教导包括成像受试者中的肿瘤的方法,以及成像受试者中的炎症的方法。在各种配置中,这些方法可以包括对受试者施用具有如下结构的化合物:
其中R可以选自由以下组成的组:其中代表键。在一些配置中,R可以是在一些配置中,F可以是18F。在一些配置中,R可以是在一些配置中,R可以是其中该化合物包括发射正电子的放射性核素如18F。该方法可以进一步包括对受试者进行PET扫描。
在本教导的各种配置中,受试者可以是患有或怀疑患有肿瘤或炎症的人。受试者也可以是患有或怀疑患有肿瘤或炎症的动物。该动物可以是哺乳动物,例如伴侣动物如,例如猫或狗,实验室动物诸如,例如,小鼠、豚鼠、兔或大鼠,或大型动物,诸如,例如,马、绵羊、牛或山羊。
附图说明
图1示出PARP-1抑制剂的实施例(现有技术)。
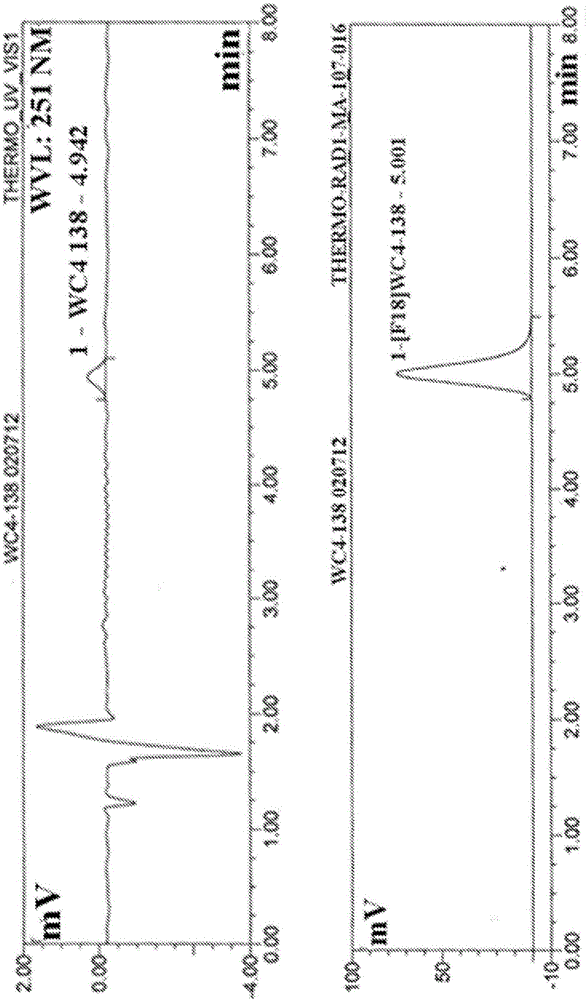
图2示出[18F]12(WC-4-138)的HPLC分析,其表现出高的化学和放射化学纯度。(顶部图:UV,底部图:放射性;比活性=11500mCi/μmol)。
图3A-D示出在60分钟时在基线条件下使用[18F]12并用奥拉帕尼(i.p.50mg/kg 20min预处理)和12(i.p.1mg/kg 20min预处理)阻断的小鼠中的MDA-MB-231和MDA-MB-436肿瘤的微型PET图象。上排:用奥拉帕尼(i.p.50mg/kg 20min预处理)治疗前和后的MDA-MB-231肿瘤。下排:12(i.p.1mg/kg20min预处理)前和后的MDA-MB-436(右)和MDA-MB-231(左)肿瘤。图3A,图3B和图3C分别表示红色、绿色和蓝色RGB通道,各自以黑色和白色渲染。图3D表示以黑色和白色渲染的RGB通道的复合。
图4示出在基线条件下并用奥拉帕尼(i.p.50mg/kg 20min预处理)和12(i.p.1mg/kg 20min预处理,顶部图)阻断的小鼠中的MDA-MB-436肿瘤中的[18F]12的时间放射性曲线。经奥拉帕尼治疗的MDA-MB-231肿瘤还显示[18F]12摄取下降(底部图)。
图5示出癌细胞系中的[18F]WC-4-138(12)摄取和PARP活性的分析。
图6示出如在成年小鼠中评价的[18F]WC-4-138示踪物的代谢稳定性。
图7示出在[18F]WC-4-138示踪物注射后进行60分钟动态成像的携带MDA/MB-231(乳腺癌)肿瘤的小鼠。
图8A-B示出在[18F]WC-4-138示踪物注射后进行60分钟动态成像的携带MDA/MB-231(乳腺癌)或SCC1(HNSCC)肿瘤的小鼠。
具体实施方式
在各种实施方案中,本教导包括可用于通过PET扫描成像肿瘤或炎症的示踪物。在各个方面,所述示踪物可以高亲和性结合PARP-1。在各种实施方案中,本教导还包括示踪物的合成方法。本教导的示踪物,当用正电子发射放射性同位素如18F标记时,可通过静脉内或其它合适的方式施用到受试者。
本文所描述的方法使用本领域技术人员公知的实验室技术,并且可在实验室手册和教科书中找到指导,如Sambrook,J.,et al.,Molecular Cloning:A Laboratory Manual,3rd ed.Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY,2001;Spector,D.L.et al.,Cells:A Laboratory Manual,Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY,1998;Hedrickson et al.,Organic Chemistry 3rd edition,McGraw Hill,New York,1970;Carruthers,W.,and Coldham,I.,Modern Methods of Organic Synthesis(4th Edition),Cambridge University Press,Cambridge,U.K.,2004;Curati,W.L.,Imaging in Oncology,Cambridge University Press,Cambridge,U.K.,1998;Welch,M.J.,and Redvanly,C.S.,eds.Handbook of Radiopharmaceuticals:Radiochemistry and Applications,J.Wiley,New York,2003。可以根据本领域技术人员熟知的药理学标准原则确定药物的施用方法和给药方案,使用标准参考文本如Remington:the Science and Practice of Pharmacy(Alfonso R.Gennaro ed.19th ed.1995);Hardman,J.G.,et al.,Goodman&Gilman’s The Pharmacological Basis of Therapeutics,Ninth Edition,McGraw-Hill,1996;和Rowe,R.C.,et al.,Handbook of Pharmaceutical Excipients,Fourth Edition,Pharmaceutical Press,2003提供的方法。
在本文所述的实验中,试剂和材料从商业供应商购买,除非另有说明不经进一步纯化即可使用。化学品购自Sigma-Aldrich化学公司(圣路易斯,密苏里州美国),除非另有指明。所有反应均通过标准无空气和无湿气技术用干燥溶剂在惰性氮气氛下进行,除非另有说明。
快速柱色谱法可以使用各种方法和仪器进行,包括Scientific Adsorbents公司的硅胶,60A,“40μm Micron Flash”(32-63μm)。可以使用本领域中公知的多种方法和仪器,包括MEL-TEMP 3.0装置来测定熔点。在一些配置中,熔点数据未经校正。可以通过多种常规方法在各种仪器,包括Varian Mercury-VX分光计上记录300MHz的1H和13C NMR谱。在一些配置中,可以将化学位移报告为从四甲基硅烷(TMS)往低场的百万分率(ppm)。以赫兹(Hz)给出所有的耦合常数(J)。通常将分裂模式描述为如下:s,单峰;d,双重峰;t,三重峰;m,多重峰。
可以通过各种商业合同组织,如GA诺克罗斯的Atlantic Microlab公司,来确定元素分析(C,H,N)。可用紫外线检测器和良好闪烁NaI(Tl)检测器以及相关的用于放射性检测的电子器件实施高效液相色谱(HPLC)。Grace Altima C18 250×10mm 10μ半制备柱(A)和Altima C18 250×4.6mm×10μ分析柱(B)可分别用于制备和分析。可以通过RDS111回旋加速器中的富集的(95%)[18O]水的质子辐照由18O(p,n)18F反应产生[18F]氟化物。可以使用Bioscan AR-2000成像扫描仪(Bioscan公司,华盛顿,DC)实现放射性-TLC。已公布的方法用于化合物527和1122的合成。按照国家研究理事会的“护理和使用实验动物的指南”的建议,在华盛顿大学动物研究委员会IACUC批准的协议下进行所有动物实验。
实施例
本教导包括在实施例中提供的不打算限制任何权利要求的范围的说明。除非以过去时明确提出,一个实施例可以是预示性或实际的实施例。提供下面的非限制性实施例以进一步说明本发明的教导。本领域的技术人员,考虑到本公开内容,将认识到可以在公开的具体实施方案中进行许多变化并仍然获得相同或类似的结果而不背离本教导的精神和范围。
实施例1
这个实施例说明了PARP-1抑制剂的合成。化合物编号参见方案1和2,见下文。
本教导的PARP-1抑制剂的合成示于方案1和2中。2,3-二氨基苯甲酸甲酯(5)在吡啶和二氯甲烷中与4-(2-氟乙氧基)苯甲酰氯反应,得到中间体6a和苯并咪唑化合物6的混合物。在溶剂蒸发后,在甲醇中用甲磺酸回流该混合物,得到6。然后在甲醇中使用铵将6的甲酯转化为酰胺化合物8。类似地,从5和4-(丙-2-炔基氧基)苯甲酰氯开始合成炔烃类似物9。在DMF中使用CuSO4·5H2O和抗坏血酸钠通过2-氟乙基叠氮化物和9的铜(I)催化的点击反应制备三唑化合物10。
从二胺中间体11合成三环化合物。化合物11在吡啶和二氯甲烷中与4-(2-氟乙氧基)苯甲酰氯反应,得到中间体12a和苯并咪唑12的混合物。在溶剂蒸发后,在甲醇中用甲磺酸回流该混合物,得到12。类似地,13和14分别从相应的苄基氯制备。使用2-氟乙基叠氮化物和13在与用于10时相同的条件下通过点击反应制备化合物15。通过在乙腈中回流14和甲磺酸银合成用于用18F标记12的甲磺酸酯前体16。
方案1.NU1085的衍生物(2)的合成
试剂和条件:(a)ROC6H4COCl(R=CH2CH2F用于6a和6,R=CH2C≡CH用于7a和7)吡啶,CH2Cl2;(b)CH3SO3H,MeOH;(c)NH3,MeOH;(d)9,FCH2CH2N3,CuSO4,抗坏血酸钠,DMF。
方案2.AG014361的衍生物(3)的合成
试剂和条件:(a)ROC6H4COCl(R=CH2CH2F用于12a和12,R=CH2C≡CH用于13a和13,R=CH2CH2Br用于14a和14),吡啶,CH2Cl2;(b)CH3SO3H,MeOH;(c)13,FCH2CH2N3,CuSO4,抗坏血酸钠,DMF;(d)14,AgOMs,乙腈。
实施例2
本实施例说明2-(4-(2-氟乙氧基)苯基)-1H-苯并[d]咪唑-4-羧酸甲酯(6)的合成。化合物编号参见方案1和2,见上文。
在23℃下,2,3-二氨基苯甲酸甲酯5(500mg,3mmol)和4-(2-氟乙氧基)苯甲酰氯(638mg,3.15mmol)在CH2Cl2(10ml)和吡啶(10ml)中的混合物搅拌过夜。减压除去溶剂后,将残余物溶于甲醇(50mL)中,并随后加入CH3SO3H(1mL)。将混合物回流3小时后,减压下除去甲醇,将残余物溶解在乙酸乙酯(75ml)中。将溶液用饱和Na2CO3(50ml),水(50ml)和饱和NaCl(50mL)洗涤,并经Na2SO4干燥。在蒸发溶剂后,将粗产物通过硅胶柱色谱纯化,用己烷-乙酸乙酯(1:1)洗脱,得到6,为白色固体(686mg,73%),mp 134.2-134.6℃。1H NMR(300MHz,CDCl3)δ10.58(s,1H),7.98(d,J=8.7Hz,2H),7.95(d,J=8.7Hz,1H),7.83(d,J=7.2Hz,1H),7.26(t,J=7.8Hz,1H),6.98(d,J=9.0Hz,2H),4.75(dt,J=47.1Hz,4.2Hz,2H),4.22(dt,J=27.6Hz,4.2Hz,2H),3.96(s,3H).13C NMR(75MHz,CDCl3)δ167.0,160.2,152.3,144.8,135.0,128.2,124.4,124.2,122.3,121.7,114.9,113.0,81.6(d,J=169.7Hz),67.1(d,J=20.6Hz),52.0。
实施例3
本实施例说明2-(4-(丙-2-炔基氧基)苯基)-1H-苯并[d]咪唑-4-羧酸甲酯(7)的合成。化合物编号参见方案1和2,见上文。
按照用于化合物6(实施例2)的相同程序制备化合物7,除了使用化合物5(500mg,3mmol)和4-(丙-2-炔基氧基)苯甲酰氯(613mg,3.15mmol)作为起始原料。将粗产物通过硅胶柱色谱纯化,用己烷-乙酸乙酯(1:1)洗脱,得到7,为白色固体(724mg,79%),mp 176.0-176.8℃。1H NMR(300MHz,CDCl3)δ10.58,8.03(d,J=9.0Hz,2H),7.98(d,J=8.1Hz,1H),7.86(d,J=7.8Hz,1H),7.29(t,J=8.1Hz,1H),7.10(d,J=9.0Hz,2H),4.76(d,J=2.4Hz,2H),4.00(s,3H),2.57(t,J=2.4Hz,1H).13C NMR(75MHz,CDCl3)δ167.1,159.4,152.4,144.9,135.1,128.2,124.6,124.3,122.8,121.8,115.4,113.0,77.9,76.0,55.8,52.1。
实施例4
本实施例说明2-(4-(2-氟乙氧基)苯基)-1H-苯并[d]咪唑-4-甲酰胺(8)的合成。化合物编号参见方案1和2,见上文。
在23℃下搅拌化合物6(315mg,1mmol)在甲醇(10ml)中在7N NH3中的溶液3天。在蒸发溶剂后,将粗产物通过硅胶柱色谱纯化,用己烷-乙酸乙酯(1:2)洗脱,得到8,为白色固体(245mg,82%),mp 195.8-197.4℃。1H NMR(300MHz,DMSO-d6)δ9.44(s,1H),8.23(d,J=9.0Hz,2H),7.89(d,J=7.5Hz,1H),7.80(s,2H),7.74(d,J=7.8Hz,1H),7.34(t,J=7.5Hz,1H),7.20(d,J=8.4Hz,2H),4.81(dt,J=47.7Hz,3.6Hz,2H),4.37(dt,J=30.0Hz,3.9Hz,2H).13C NMR(75MHz,DMSO-d6)δ166.4,160.1,152.0,135.5,128.6,122.7,122.0,115.1,82.1(d,J=166.2Hz),67.3(d,J=19.4Hz)。C16H14FN3O2.0.5H2O分析计算:C,62.33;H,4.90;N,13.63。实测:C,62.54;H,4.87;N,13.67。
实施例5
本实施例说明2-(4-(丙-2-炔基氧基)苯基)-1H-苯并[d]咪唑-4-甲酰胺(9)的合成。化合物编号参见方案1和2,见上文。
按照用于化合物8的相同程序制备化合物9,不同的是使用化合物7(460mg,1.5mmol)作为起始原料。将粗产物通过硅胶柱色谱纯化,用己烷-乙酸乙酯(1:2)洗脱,得到9,为白色固体(378mg,86%),mp 183.4-183.9℃。1H NMR(300MHz,DMSO-d6)δ9.39(s,1H),8.21(d,J=8.4Hz,2H),7.87(d,J=7.8Hz,1H),7.78(s,2H),7.72(d,J=7.8Hz,1H),7.32(t,J=7.8Hz,1H),7.20(d,J=9.0Hz,2H),4.92(d,J=1.8Hz,2H),3.63(s,1H).13C NMR(75MHz,DMSO-d6)δ166.4,159.0,152.0,128.5,122.7,122.3,121.9,115.4,78.9,78.6,55.7。C17H18N3O2.0.5H2O分析计算:C,67.99;H,4.70;N,13.99。实测:C,67.97;H,4.72;N,13.71。
实施例6
本实施例说明2-(4–((1-(2-氟乙基)-1H-1,2,3-三唑-4-基)甲氧基)苯基)-1H-苯并[d]咪唑-4-甲酰胺(10)的合成。化合物编号参见方案1和2,见上文。
在23℃下搅拌9(291mg,1.0mmol),1-叠氮基-2-氟乙烷(1.68mmol),抗坏血酸钠(990mg,5.0mmol)和CuSO4.5H2O(125mg,0.5mmol)在DMF(10ml)中的混合物过夜。用乙酸乙酯(75ml)稀释反应混合物,并用水(2×50ml)和饱和NaCl(50mL)洗涤,经Na2SO4干燥。蒸发溶剂后,将粗产物通过硅胶柱色谱纯化,用乙酸乙酯洗脱,得到10,为白色固体(255mg,67%),mp256.4-257.3℃。1H NMR(300MHz,DMSO-d6)δ9.42(s,1H),8.33(s,1H),8.21(d,J=8.7Hz,2H),7.87(d,J=7.5Hz,1H),7.78(s,2H),7.71(d,J=7.8Hz,1H),7.32(t,J=7.8Hz,1H),7.27(d,J=8.7Hz,2H),5.28(s,2H),4.85(dt,J=46.8Hz,4.5Hz,2H),4.76(dt,J=27.6Hz,4.2Hz,2H).13C NMR(75MHz,DMSO-d6)δ166.3,159.9,152.0,142.5,141.6,135.3,128.6,125.1,122.7,122.1,121.9,115.3,114.7,99.5,81.9(d,J=167.3Hz),61.3,50.1(d,J=20.5Hz)。C19H17FN6O2:C分析计算,59.99;H,4.50;N,22.09。实测:C,60.10;H,4.67;N,21.49。
实施例7
本实施例说明5,6-二氢-2-(4-(2-氟乙氧基)苯基)-咪唑并[4,5,1-jk][1,4]苯并二氮杂卓-7(4H)-酮(12)的合成。化合物编号参见方案1和2,见上文。
按照用于化合物6的相同程序制备化合物12(WC-4-138),除了使用化合物11(177mg,1mmol)和4-(2-氟乙氧基)苯甲酰氯(213mg,1.05mmol)作为起始材料。将粗产物通过硅胶柱色谱纯化,用乙酸乙酯-甲醇(10:1)洗脱,得到12,为白色固体(247mg,76%),mp 236.0-237.5℃。1H NMR(300MHz,DMSO-d6)δ8.44(t,J=5.1Hz,1H),7.89-7.80(m,4H),7.34(t,J=7.8Hz,1H),7.17(d,J=8.7Hz,2H),4.79(dt,J=48.9Hz,3.6Hz,2H),4.44(m,2H),4.35(dt,J=31.2Hz,3.9Hz,2H),3.53(m,2H).13C NMR(75MHz,DMSO-d6)δ167.8,159.9,154.1,143.7,132.9,131.7,125.5,123.1,122.5,121.9,118.1,115.1,82.5(d,J=165.0Hz),67.7(d,J=19.3Hz),50.9,40.8。C18H16FN3O2分析计算:C,66.45;H,4.96;N,12.92。实测:C,66.43;H,5.03;N,12.92。
实施例8
本实施例说明5,6-二氢-2-(4-(丙-2-炔基氧基)苯基)-咪唑并[4,5,1-jk][1,4]苯并二氮杂卓-7(4H)-酮(13)的合成。化合物编号参见方案1和2,见上文。
根据用于化合物6的相同程序制备化合物13,除了使用化合物11(177mg,1mmol)和4-(丙-2-炔基氧基)苯甲酰氯(204mg,1.05mmol)作为起始原料。将粗产物通过硅胶柱色谱纯化,用乙酸乙酯-甲醇(10:1)洗脱,得到13,为白色固体(268mg,84%),mp 258.0-259.1℃。1H NMR(300MHz,DMSO-d6)δ8.47(s,1H),7.85(m,4H),7.34(t,J=7.8Hz,1H),7.18(d,J=6.9Hz,2H),4.93(s,2H),4.46(m,2H),3.64(s,1H),3.54(m,2H).13C NMR(75MHz,DMSO-d6)δ167.4,158.5,153.6,143.3,132.5,131.1,125.1,122.7,122.4,121.5,117.7,115.0,79.0,78.5,55.6,50.5。C19H15N3O2分析计算:C,71.91;H,4.76;N,13.24。实测:C,71.71;H,4.82;N,12.98。
实施例9
本实施例说明2-(4-(2-溴乙氧基)苯基)-5,6-二氢-咪唑并[4,5,1-jk][1,4]苯并二氮杂卓-7(4H)-酮(14)的合成。化合物编号参见方案1和2,见上文。
根据用于化合物6的相同程序制备化合物14,除了使用化合物11(177mg,1mmol)和4-(2-溴乙氧基)苯甲酰氯(277mg,1.05mmol)作为起始原料。将粗产物通过硅胶柱色谱纯化,用乙酸乙酯-甲醇(10:1)洗脱,得到14,为白色固体(255mg,66%),分解熔点280℃。1H NMR(400MHz,DMSO-d6)δ8.44(t,J=5.6Hz,1H),7.87(d,J=8.0Hz,1H),7.85(d,J=8.0Hz,1H),7.82(d,J=8.4Hz,2H),7.34(t,J=7.6Hz,1H),7.16(d,J=8.4Hz,2H),4.44(m,4H),3.86(t,J=5.2Hz,2H),3.53(m,2H).13C NMR(100MHz,DMSO-d6)δ167.8,159.7,154.1,143.7,132.9,131.7,125.5,123.1,122.6,122.0,118.1,115.2,68.4,50.5,40.8。
实施例10
本实施例说明5,6-二氢-2-(4-((1-(2-氟乙基)-1H-1,2,3-三氮唑-4-基)甲氧基)苯基)-咪唑并[4,5,1-jk][1,4]苯并二氮杂卓-7(4H)-酮(15)的合成。化合物编号参见方案1和2,见上文。
按照用于化合物10的相同程序制备化合物15,不同的是使用化合物13(159mg,0.5mmol)作为起始原料。将粗产物通过硅胶柱色谱纯化,用乙酸乙酯-甲醇(10:1)洗脱,得到15,为白色固体(147mg,72%),mp 226.5-227.6℃。1H NMR(300MHz,DMSO-d6)δ8.46(t,J=5.7Hz,1H),8.33(s,1H),7.89-7.81(m,4H),7.34(t,J=7.8Hz,1H),7.25(d,J=8.4Hz,2H),5.28(s,2H),4.85(dt,J=47.1Hz,4.2Hz,2H),4.76(dt,J=27.6Hz,4.2Hz,2H),4.45(m,2H),3.54(m,2H).13C NMR(75MHz,DMSO-d6)δ167.4,159.4,143.3,142.6,131.2,125.1,125.0,122.7,122.0,121.5,117.7,114.8,81.9(d,J=167.3Hz),61.2,50.5,50.1(d,J=20.5Hz),40.4。C21H19FN6O2.1.5H2O分析计算:C,58.19;H,5.12;N,19.39。实测:C,57.89;H,4.54;N,18.92。
实施例11
本实施例说明5,6-二氢-2-(4-(2-(甲磺酰氧基)乙氧基)苯基)-咪唑并[4,5,1-jk][1,4]苯并二氮杂卓-7-(4H)-酮(16)的合成。化合物编号参见方案1和2,见上文。
回流14(193mg,0.5mmol)和AgOMs(508mg,2.5mmol)的混合物8小时。在蒸发溶剂后,将粗产物通过硅胶柱色谱纯化,用乙酸乙酯-甲醇(10:1)洗脱,得到16,为白色固体(129mg,64%),mp 253.2-254.1℃。1H NMR(400MHz,CD3OD)δ7.97(d,J=8.0Hz,1H),7.88(d,J=8.0Hz,1H),7.76(d,J=8.8Hz,2H),7.40(t,J=8.0Hz,1H),7.17(d,J=8.8Hz,2H),4.59(t,J=4.0Hz,2H),4.49(t,J=4.0Hz,2H),4.35(t,J=4.0Hz,2H),3.65(m,2H),3.12(s,3H).13C NMR(100MHz,CD3OD)δ160.2,148.1,142.7,135.7,132.2,131.1,131.0,125.8,122.6,122.1,121.4,116.9,114.6,68.3,66.0,50.5,40.6,36.0。
实施例12
该实施例说明PARP-1活性测定。
使用Putt和Hergenrother描述的方法评估新合成的PARP-1抑制剂抑制活性PARP-1的能力。23结果显示在表1中。三环苯并咪唑化合物比它们各自的苯并咪唑类似物具有更高的抑制效力(例如,12对比8,15对比10)。在苯并咪唑和三环苯并咪唑类似物中,具有氟乙氧基取代基的类似物比各自的具有氟乙基三唑基的类似物具有三倍更高的抑制效力(例如,8对比10,12对比15)。因此,选择最有效的抑制剂12用于18F标记。
表1.PARP-1抑制剂的抑制效率
a.报告值:IC50=20nM,EC50=35nm;3,24
实施例13
这个实施例示出了PARP-1酶活性测定。
该测定基于NAD+的化学定量,即当活性PARP-1C-末端催化结构域循序地将ADP-核糖亚基从烟酰胺腺嘌呤二核苷酸(NAD+)转移到蛋白质受体时所消耗的NAD+的量。7
高比活性的PARP-1和活化的DNA购自Trevigen(马里兰州盖瑟斯堡)。该测定法所需的所有其它试剂包括NAD+均购自Sigma-Aldrich(圣路易斯,密苏里州)。在这些实验中用作对照的已知的PARP-1抑制剂PJ-34在内部合成。为了测试用于PARP-1抑制的化合物,首先在pH 8.0,50mM Tris-HCl,2mM MgCl2(PARP测定缓冲液)中制备250nM NAD+的溶液并且将20μL转移至96孔黑色荧光板的每个孔。在PARP测定缓冲液中制备活化DNA的50μg/ml溶液并将10μL加至每个孔中。首先在DMSO中制备测试化合物的储备溶液,在PARP测定缓冲液中稀释至不同浓度,并将10μL加至每个孔中。为了引发反应,将10μL的在PARP测定缓冲液中的10μg/ml PARP-1酶加入到每个孔中。总体积为50μL,使每孔终浓度为2μg/ml PARP-1酶,10μg/ml活化DNA,以及100nM NAD+。然后将板在室温下孵育20分钟。然后通过首先加入20μL 2M KOH,随后加入20μL的20%苯乙酮(在乙醇中)至每孔来确定所存在的NAD+的量。将板在4℃下孵育10分钟。然后加入90μL的88%甲酸,导致222mM KOH,2.2%苯乙酮,44%甲酸的终浓度,和不同的NAD+浓度。将板在100℃下孵育5分钟,使之冷却,然后使用360nM激发和450nM发射滤光片在Perkin Elmer Victor微孔板荧光计(沃尔瑟姆,马萨诸塞州)上读数。使用用于Windows的GraphPad Prism版本5.04(圣地亚哥,CA)生成剂量-响应曲线,其中仅含有NAD+的对照孔设定为0%PARP活性且仅包含PARP-1的对照孔设定为100%PARP活性。从由至少三个独立的实验生成的剂量-响应曲线计算IC50值,并在表1中报告为平均值±标准偏差(SD)。
实施例14
本实施例说明放射性标记的PARP-1抑制剂的合成。
在常规条件(K222/K2CO3)下在DMF中在105℃下,通过甲磺酸酯前体16的亲核取代合成[18F]12(方案3),在反相HPLC纯化及固相萃取之后以40-50%产率(经衰变校正的)得到[18F]12(图2)。总合成时间为90分钟。在10%乙醇/盐水中的最后剂量的比活性为5500-18000mCi/μmol。
方案3.[18F]12的放射合成
试剂和条件:[18F]KF,K222,K2CO3,DMF,105℃,10min。
实施例15
这个实施例进一步说明了合成[18F]化合物12(WC-4-138)。
将[18F]氟化物(在100-500μL[18O]水中可达50mCi)转移到含K222(2.2mg,5.8μmol)和K2CO3(0.3mg,2.2μmol)的BD真空采血管(13×75mm,5ml,玻璃,无添加剂)中。然后用乙腈(3×1mL)在N2气体的平缓流动下将混合物通过105℃下的共沸蒸馏干燥。当干燥接近完成时,从油浴移出真空采血管,并在室温下通过N2流除去溶剂残余物(<100μL)。将16(0.65mg,1.6μmol)在DMF(300μL)中的溶液加入到该真空采血管中,然后摇动,并在105℃下加热10分钟。在室温下,用水(2ml)稀释该反应混合物,然后装载到用于纯化的半制备柱(A)(18%乙腈/82%水/0.1%TFA,4ml/分钟,251nm)上。收集含有[18F]12的HPLC流分,并使用标准的固相萃取方法在乙醇中获得[18F]12。将剂量稀释至10%乙醇在盐水中。分析柱(B)用于分析所述剂量(32%乙腈/68%0.1M甲酸铵缓冲液pH=4.5,2ml/分钟,251nM)。总合成时间为90分钟,衰变校正收率40-50%,放射化学纯度为100%,而比活性在合成结束时在5500至18000mCi/μmol的范围内。
实施例16
本实施例说明小鼠中的肿瘤组织在微型PET中的可视化。
使用[18F]12通过PET可视化免疫缺陷小鼠中的MDA-MB-436人乳腺癌异种移植肿瘤。在示踪物注射后的60分钟,这些肿瘤显示有别于背景的摄取增加(图3)。示踪物注射之前的20分钟用奥拉帕尼(50mg/kg i.p.)或12(1mg/kg i.p.)治疗的相同小鼠减少肿瘤中的[18F]12摄取到背景组织活性水平(图3)。来自0-60分钟的微型PET研究的肿瘤中的[18F]12的时间-放射性曲线证实微型PET图像的视觉评估(图4),显示作为用奥拉帕尼或12治疗的结果的[18F]12摄取显著减少。
MDA-MB-436是具有天生高水平的PARP-1活性19的人乳腺癌细胞系,并已用在小鼠模型中用于评估18F标记的奥拉帕尼衍生物用微型PET成像PARP-1活性的功效。在1小时微型PET获取过程中,[18F]12在肿瘤中逐步积累,并且[18F]12摄取在用奥拉帕尼或12预处理的动物中被阻断。奥拉帕尼和12是具有高的抑制效力(IC50=5nM31和6.3nM,分别)的有竞争力的PARP-1抑制剂。因此,我们的数据表明,小鼠模型中的[18F]12摄取是由于PARP-1表达,而且特别地[18F]12是用于PARP-1表达的体内成像的有效PET示踪物。
实施例17
这个实施例说明了使用PET可视化小鼠中的肿瘤组织。
使用补充有2mM L-谷氨酰胺,1mM丙酮酸钠,0.1mM非必需氨基酸2%用于MEM的维生素,和5%胎牛血清的Eagle’s最低必需培养基(含Earle’s平衡盐溶液和2mM L-谷氨酰胺),在标准条件下使用5%CO2气氛,将MDA-MB-436和MDA-MB-231人乳腺癌细胞保持在细胞培养物中。胰蛋白酶消化并收获指数生长的细胞用于肿瘤植入。计数后,将细胞重新悬浮在冰冷的1:1Matrigel和PBS中,以得到所需的浓度,并保持在冰上。
在肿瘤植入用于这些串行成像研究前,使来自Charles River实验室的成熟的雌性无胸腺nu/nu小鼠在AALAC认可的畜舍设施中适应至少1周。将1:1Matrigel:PBS中的MDA-MB-436乳腺癌细胞植入雌性nu/nu小鼠的乳腺脂肪垫(腋淋巴结附近)中。植入后2-3周进行成像研究。
将荷瘤小鼠置于含有异氟烷/氧气的吸气室中,然后固定到用于安置尾静脉导管的定制双人床;通过鼻锥在异氟烷/氧气下维持麻醉用于动态成像程序。用150-200μCi的[18F]12注入小鼠并使用Focus 220和Inveon PET/CT扫描仪扫描0-60分钟。从肿瘤和周围的“背景”组织的关注的手工绘制区域生成标准的摄取值(SUVs)。通过比较基线扫描和在用阻断剂奥拉帕尼(50mg/kg,IP)或12(1mg/Kg,IP)预处理之后的20分钟通过示踪物注射获得的图像确定具体摄取的可视化。
实施例18
本实施例说明细胞培养测定中的[18F]WC-4-138(化合物12)的摄取。
在这些实验中,在补充有中10%胎牛血清(FBS,Gibco),1%青霉素-链霉素(P/S,Gibco),和100ng/ml氢化可的松(Sigma-Aldrich)的Dulbecco’s改良Eagle’s培养基(DMEM,Gibco)中繁殖头和颈部鳞状细胞癌系SCC1,SCC15和SCC25(ATCC)。在补充有10%FBS和1%P/S的RPMI培养基(Gibco)中繁殖小细胞肺癌细胞系NCI-H69和NCI-H82(ATCC)。在补充有5%FBS,2%用于MEM的维生素(Gibco),1%200mM L-谷氨酰胺(Gibco),1%10mM非必需氨基酸(NEAA,Gibco)的Eagle’s最低必需培养基(Gibco)中繁殖人类乳腺癌细胞系MDA-MB-231(ATCC)。
对于每个实验重复,将大约1μCi[18F]WC-4-138稀释在1ml细胞培养基中,并加入到106个细胞。在5,30或60分钟后,收集培养基并在0.7ml磷酸盐缓冲盐水(PBS,Gibco)中洗涤细胞两次。通过刮擦细胞培养皿收集粘附细胞并转移到微量离心管。在收集的培养基,PBS或细胞沉淀中测定放射性。使用标准化学发光PARP ELISA试剂盒(Trevigen#4520-096-K)定量来自细胞沉淀的蛋白质。所有数据都经衰变校正和归一化为细胞沉淀中的总蛋白量。对于药物治疗研究,在与[18F]WC-4-138孵育前二十小时,用10μM奥拉帕尼或尼帕尼孵育细胞。
头部和颈部细胞系SCC1和SCC25在组成上占用少量的示踪物,显示为细胞沉淀中的0.001-0.002μCi/μg(图5A)。肺细胞系NCI-H69和NCI-H82占用在细胞沉淀中显示为0.004-0.006μCi/μg的[18F]WC-4-138(图5A)。在用[18F]WC-4-138孵育5,30或60分钟(在每个时间点n=3)后,在头部和颈部鳞状细胞癌(HNSCC)系(SCC1和SCC25)或小细胞肺癌(SCLC)系(NCI-H69和NCI-H82)中测定放射性。这些附图代表对于SCC细胞1-2单位的PARP活性和对于NCI细胞大约3.5单位的PARP活性(图5B)。在HNSCC或肺SCLC细胞中测定PARP活性。(图5C-D)在用奥拉帕尼或尼帕尼治疗的HNSCC细胞系中测定[18F]WC-4-138摄取。在用示踪物孵育30或60分钟(*p<0.05)之后,用于摄取的数据是平均值±标准差。所有数据表示为平均值±SD,n=3,除非另有说明。
该活性由奥拉帕尼(图5C)而不是尼帕尼(图5D)消除。
实施例19
这个实施例说明化合物12([18F]WC-4-138)的代谢稳定性。
在这些实验中,在将400μCi注入到成年雄性C57BL/6J小鼠(Jackson实验室)的尾静脉后评估[18F]-WC-4-138的代谢稳定性。注射后5或30分钟通过颈脱位法处死小鼠。撕裂下腔静脉并从腹腔采集血液。通过以14000rpm离心从红血细胞分离血浆。血浆(100μL)与乙腈以1:1.5比例混合并以14000rpm离心。测定与红血细胞沉淀、全血浆和乙腈-可溶性和不溶性流份关联的放射性。也在指定的时间收集每只动物的肝脏并在干冰上冷冻。肝脏在2ml乙腈中匀浆并且1ml的匀浆的肝脏以14000rpm离心。测定与上清液和沉淀相关的放射性。
用水以1:1比例混合乙腈可溶性血浆或肝脏上清液(100μL)并通过反相HPLC分离。还通过HPLC分离母体化合物作为参考。测定与每个HPLC流份相关联的放射性。将各样品的母体化合物百分比计算为与预期包含母体化合物的HPLC流份相关联的放射性部分。
化合物12([18F]WC-4-138)在血液中快速代谢,在注射后5分钟相对于参考化合物存在50%并且在注射后30分钟观察到相对于参考化合物只有约10%的化合物(图6)。与此相反,化合物在肝脏中具有长得多的半衰期,在注射后5分钟相对于参考化合物存在约80%并且在注射后30分钟相对于参考化合物存在约70%(图6)。
实施例20
这个实施例说明了化合物12([18F]WC-4-138)在小鼠中的生物分布。
经由尾静脉用[18F]WC-4-138注射八周大,雌性,无胸腺裸鼠(Harlan)。在IV注射示踪物30μCi后5或30分钟,注射45μCi后1或2小时或注射60μCi的[18F]WC-4-138后4小时,通过颈脱位法处死小鼠。通过反相高效液相色谱法(HPLC)分离血浆或肝脏的乙腈可溶流份和对照母体化合物并且定量各个流份的放射性。将各样品的母体化合物百分比计算为与预期包含母体化合物的HPLC流份相关联的放射性部分。数据表示为平均值±SD,n=3。
在每个时间点处死四只小鼠,除了2小时时间点(n=3)。从每只动物收集血液、心脏、肺、肌肉、肝、脾、脂肪、肾上腺、肾、子宫、卵巢、骨、骨髓、胰腺、胃、小肠以及大肠。所有器官弄干以除去过量的血液,称重,并在贝克曼6000γ计数器中计数。对每个器官测定组织的每克注射剂量百分比(%ID/g)。结果在0.5%至15%每克组织注射剂量之间变化,并且在表1和2中报告。
表1无胸腺裸鼠中的[18F]WC-4-138的器官生物分布
%ID/g
%ID/g的=每克组织注射剂量百分比。
所有数据都是平均值±标准差。对于所有组n=4,除了在120分钟n=3。
表2.从小鼠生物分布研究估算的人类剂量
使用标准MIRD方法,从根据表1中示出的小鼠生物分布数据计算出的器官滞留时间获得估算。
实施例21
这个实施例说明了化合物12([18F]WC-4-138)在小鼠中的摄取。
将106个SCC1细胞或107个MDA-MB-231细胞植入到成熟的雌性无胸腺裸小鼠(Charles River实验室)的乳腺脂肪垫中。在植入后5周成像携带SCC1肿瘤的小鼠以及在植入后2.5周成像携带MDA-MB-231细胞的小鼠。用2%异氟烷/氧麻醉小鼠,并在整个成像过程中通过鼻锥使小鼠保持在1%异氟烷/氧气中。在Inveon PET/CT扫描仪上获得整个动物微型CT图像。小鼠经尾静脉注射11.36±0.5MBq(307±13μCi)的[18F]WC(4)-138,并使用Focus 220或Inveon PET/CT扫描仪使小鼠经历60分钟动态扫描。用Integrated Research Workflow软件(西门子)配准微型PET和微型CT图像。在肿瘤上画出关注的区域,以确定时间活性曲线。对于药物治疗研究,成像前30分钟,动物接受50mg/Kg奥拉帕尼或尼帕尼腹膜内注射。在基线,或IP注射奥拉帕尼或尼帕尼后30分钟成像小鼠。MDA-MB-231荷瘤小鼠的横向视图显示于图7。箭头指示CT上所确定的肿瘤位置。肿瘤中和基线(图7,每个通道视图的顶部,箭头)中的小鼠的其它部分大量摄取示踪物,但通过用奥拉帕尼治疗,消除了[18F]12的肿瘤摄取(图7,每个通道面板左下,箭头),但示踪物仍然存在于该部分的其它区域。与此相反,尼帕尼治疗不影响肿瘤细胞中的[18F]WC(4)-138的摄取,但废除在其它类型的细胞中的所述摄取(图7,每个通道视图的右下,箭头)。
实施例22
这个实施例示出了定量分析,其显示基线和MDA-MB-231肿瘤(图8A)和SCC肿瘤(图8B)药物治疗之间的每ml注射剂量显著降低。在图8中,在基线或IP注射奥拉帕尼或尼帕尼后的30分钟成像小鼠。(A)在基线或药物治疗后为每个MDA/MB-231肿瘤计算每ml注射剂量的百分比。奥拉帕尼处理的小鼠中的示踪物摄取显著降低(通过配对t检验确定*p=0.0038,n=6肿瘤)。尼帕尼处理的小鼠中的示踪物摄取不变(通过配对t检验确定p=0.1098,n=6肿瘤)。(B)在基线或药物治疗后为每个SCC1肿瘤计算每ml注射剂量的百分比。奥拉帕尼处理的小鼠中的示踪物摄取显著降低(通过配对t检验确定*p=0.001,n=8肿瘤)。尼帕尼处理的小鼠中的示踪物摄取不变(通过配对t检验确定p=0.7216,n=5肿瘤)。
实施例23
受试者呈现潜在PARP-1相关的乳腺癌症状。医师要求进行PET扫描,并且技术人员施用有效量的[18F]WC(4)-138和执行PET扫描。相比于受试者的周围组织,肿瘤显示出大量的示踪物摄取。医师诊断PARP-1相关的癌症并开具PARP-1抑制剂作为治疗方案的一部分。
实施例24
受试者呈现肺的异常炎症。医师要求进行PET扫描,并且技术人员施用有效量的[18F]WC(4)-138和执行PET扫描。相比于对照肺组织,肿瘤组织显示出大量的示踪物摄取,并且医师诊断PARP-1相关的炎症并开具PARP-1抑制剂作为治疗方案的一部分。
参考文献
1.Hassa,P.O.;Hottiger,M.O.Frontiers in Bioscience:a Journal and Virtual Library 2008,13,3046.
2.d'Adda di Fagagna,F.;Hande,M.P.;Tong,W.M.;Lansdorp,P.M.;Wang,Z.Q.;Jackson,S.P.Nature Genetics 1999,23,76.
3.Jagtap,P.;Szabo,C.Nature Reviews.Drug Discovery 2005,4,421.
4.Schreiber,V.;Dantzer,F.;Ame,J.C.;de Murcia,G.Nature Reviews.Molecular Cell Biology 2006,7,517.
5.Virag,L.;Szabo,C.Pharmacological Reviews 2002,54,375.
6.Gradwohl,G.;Menissier de Murcia,J.M.;Molinete,M.;Simonin,F.;Koken,M.;Hoeijmakers,J.H.;de Murcia,G.Proceedings of the National Academy of Sciences of the United States of America 1990,87,2990.
7.Kameshita,I.;Matsuda,Z.;Taniguchi,T.;Shizuta,Y.The Journal of Biological Chemistry 1984,259,4770.
8.Ferraris,D.V.Journal of Medicinal Chemistry 2010,53,4561.
9.Basu,B.;Sandhu,S.K.;de Bono,J.S.药物2012,72,1579.
10.Sandhu,S.K.;Schelman,W.R.;Wilding,G.;Moreno,V.;Baird,R.D.;Miranda,S.;Hylands,L.;Riisnaes,R.;Forster,M.;Omlin,A.;Kreischer,N.;Thway,K.;Gevensleben,H.;Sun,L.;Loughney,J.;Chatterjee,M.;Toniatti,C.;Carpenter,C.L.;Iannone,R.;Kaye,S.B.;de Bono,J.S.;Wenham,R.M.The Lancet Oncology2013,14,882.
11.Plummer,R.;Lorigan,P.;Steven,N.;Scott,L.;Middleton,M.R.;Wilson,R.H.;Mulligan,E.;Curtin,N.;Wang,D.;Dewji,R.;Abbattista,A.;Gallo,J.;Calvert,H.Cancer Chemotherapy and Pharmacology 2013,71,1191.
12.Gelmon,K.A.;Tischkowitz,M.;Mackay,H.;Swenerton,K.;Robidoux,A.;Tonkin,K.;Hirte,H.;Huntsman,D.;Clemons,M.;Gilks,B.;Yerushalmi,R.;Macpherson,E.;Carmichael,J.;Oza,A.The Lancet Oncology 2011,12,852.
13.Kummar,S.;Chen,A.;Ji,J.;Zhang,Y.;Reid,J.M.;Ames,M.;Jia,L.;Weil,M.;Speranza,G.;Murgo,A.J.;Kinders,R.;Wang,L.;Parchment,R.E.;Carter,J.;Stotler,H.;Rubinstein,L.;Hollingshead,M.;Melillo,G.;Pommier,Y.;Bonner,W.;Tomaszewski,J.E.;Doroshow,J.H.Cancer Research 2011,71,5626.
14.Javle,M.;Curtin,N.J.Therapeutic Advances in Medical Oncology 2011,3,257.
15.Wahlberg,E.;Karlberg,T.;Kouznetsova,E.;Markova,N.;Macchiarulo,A.;Thorsell,A.G.;Pol,E.;Frostell,A.;Ekblad,T.;Oncu,D.;Kull,B.;Robertson,G.M.;Pellicciari,R.;Schuler,H.;Weigelt,J.Nature Biotechnology 2012,30,283.
16.Liu,X.;Shi,Y.;Maag,D.X.;Palma,J.P.;Patterson,M.J.;Ellis,P.A.;Surber,B.W.;Ready,D.B.;Soni,N.B.;Ladror,U.S.;Xu,A.J.;Iyer,R.;Harlan,J.E.;Solomon,L.R.;Donawho,C.K.;Penning,T.D.;Johnson,E.F.;Shoemaker,A.R.Clinical Cancer Research:an Official Journal of the American Association for Cancer Research 2012,18,510.
17.Tu,Z.;Chu,W.;Zhang,J.;Dence,C.S.;Welch,M.J.;Mach,R.H.Nuclear Medicine and Biology 2005,32,437.
18.Keliher,E.J.;Reiner,T.;Turetsky,A.;Hilderbrand,S.A.;Weissleder,R.ChemMedChem 2011,6,424.
19.Reiner,T.;Keliher,E.J.;Earley,S.;Marinelli,B.;Weissleder,R.Angew Chem Int Ed Engl 2011,50,1922.
20.Reiner,T.;Lacy,J.;Keliher,E.J.;Yang,K.S.;Ullal,A.;Kohler,R.H.;Vinegoni,C.;Weissleder,R.Neoplasia 2012,14,169.
21.Delaney,C.A.;Wang,L.Z.;Kyle,S.;White,A.W.;Calvert,A.H.;Curtin,N.J.;Durkacz,B.W.;Hostomsky,Z.;Newell,D.R.Clinical cancer research:an official journal of the American Association for Cancer Research 2000,6,2860.
22.Skalitzky,D.J.;Marakovits,J.T.;Maegley,K.A.;Ekker,A.;Yu,X.H.;Hostomsky,Z.;Webber,S.E.;Eastman,B.W.;Almassy,R.;Li,J.;Curtin,N.J.;Newell,D.R.;Calvert,A.H.;Griffin,R.J.;Golding,B.T.Journal of Medicinal Chemistry 2003,46,210.
23.Putt,K.S.;Hergenrother,P.J.Analytical Biochemistry 2004,326,78.
24.Jagtap,P.;Soriano,F.G.;Virag,L.;Liaudet,L.;Mabley,J.;Szabo,E.;Hasko,G.;Marton,A.;Lorigados,C.B.;Gallyas,F.,Jr.;Sumegi,B.;Hoyt,D.G.;Baloglu,E.;VanDuzer,J.;Salzman,A.L.;Southan,G.J.;Szabo,C.Critical Care Medicine 2002,30,1071.
25.Ruf,A.;de Murcia,G.;Schulz,G.E.Biochemistry 1998,37,3893.
26.Kinoshita,T.;Nakanishi,I.;Warizaya,M.;Iwashita,A.;Kido,Y.;Hattori,K.;Fujii,T.FEBS letters 2004,556,43.
27.White,A.W.;Almassy,R.;Calvert,A.H.;Curtin,N.J.;Griffin,R.J.;Hostomsky,Z.;Maegley,K.;Newell,D.R.;Srinivasan,S.;Golding,B.T.Journal of Medicinal Chemistry 2000,43,4084.
28.Lasne,M.C.;Perrio,C.;Rouden,J.;Barre,L.;Roeda,D.;Dolle,F.;Crouzel,C.Top Curr Chem 2002,222,201.
29.Glaser,M.;Arstad,E.Bioconjugate chemistry 2007,18,989.
30.Zhou,D.;Chu,W.;Dence,C.S.;Mach,R.H.;Welch,M.J.Nuclear Medicine and Biology 2012.
31.Menear,K.A.;Adcock,C.;Boulter,R.;Cockcroft,X.L.;Copsey,L.;Cranston,A.;Dillon,K.J.;Drzewiecki,J.;Garman,S.;Gomez,S.;Javaid,H.;Kerrigan,F.;Knights,C.;Lau,A.;Loh,V.M.,Jr.;Matthews,I.T.;Moore,S.;O'Connor,M.J.;Smith,G.C.;Martin,N.M.Journal of Medicinal Chemistry 2008,51,6581.
本文引用的所有出版物都通过引用并入,每个都以其整体。如本文中所使用的,单数形式“一”和“该”也意图包括复数形式,除非上下文另有说明。
- 还没有人留言评论。精彩留言会获得点赞!
- 抑制人pard6a基因表达的小干扰核糖核酸序列及其抗卵巢癌增殖和转移的用图
- 光交联的形成抑制方法、及自交联体形成抑制型光响应性核酸的制作方法
- 糖修饰的核酸分子的制作方法
- 一种用于信息存储和加密的单链脱氧核糖核酸的制作方法
- Rnase4作为药物靶点在脑胶质瘤抑制药物中的应用
- (2S,3R,4R)-3,5-二取代-2-脱氧-2-羟基-2-甲基-D-核糖-γ-内酯的制备方法及其中间体的制作方法
- Rnase4作为药物靶点在脑胶质瘤抑制药物中的应用
- (2S,3R,4R)-3,5-二取代-2-脱氧-2-羟基-2-甲基-D-核糖-γ-内酯的制备方法及其中间体的制作方法
- 脱氧核糖核酸酶在制备用于治疗男性低生育力的药物中的应用
- 基于适体的靶向激活循环放大spr检测蛋白质的方法
- 抑制人pard6a基因表达的小干扰核糖核酸序列及其抗卵巢癌增殖和转移的用图
- 光交联的形成抑制方法、及自交联体形成抑制型光响应性核酸的制作方法
- 糖修饰的核酸分子的制作方法
- 一种用于信息存储和加密的单链脱氧核糖核酸的制作方法
- Rnase4作为药物靶点在脑胶质瘤抑制药物中的应用
- (2S,3R,4R)-3,5-二取代-2-脱氧-2-羟基-2-甲基-D-核糖-γ-内酯的制备方法及其中间体的制作方法
- Rnase4作为药物靶点在脑胶质瘤抑制药物中的应用
- (2S,3R,4R)-3,5-二取代-2-脱氧-2-羟基-2-甲基-D-核糖-γ-内酯的制备方法及其中间体的制作方法
- 脱氧核糖核酸酶在制备用于治疗男性低生育力的药物中的应用
- 基于适体的靶向激活循环放大spr检测蛋白质的方法