一种新型作物特异启动子分离方法与流程
本发明涉及到生物技术和植物基因工程技术领域。具体而言,本发明涉及大规模分离鉴定作物启动子的方法。
背景技术:
启动子是转基因的重要元件,直接决定了转入基因的作用效果和利用效率。在基因工程中,为了同时达到有效性状改良和减少能量损失的目的,使用合适强度且具有时空或环境特异性启动子是最具可行性的技术路线。而该技术的前提是建立包含较多不同表达强度和表达特性的内源启动子。在作物中,特别是水稻中,启动子的研究远落后于基因的研究。有限的启动子研究更多的着眼于解释其驱动基因的功能,而对于特异性和强度这两个基因工程应用重要指标的鉴定却相对较少。因此大规模筛选克隆系列不同表达强度同时具有严谨特异型的植物启动子在作物转基因品种改良的研究和实践中具有重要意义。
启动子的批量分离鉴定手段十分有限。传统方法中,利用功能基因组学研究所构建的启动子陷阱或增强子陷阱类水稻t-dna插入突变文库,即构建一个无启动子的报告基因表达载体,只有该载体在插入到染色体基因启动子下游并临近转录位点且方向正确时,才能在染色体基因启动子的指导下表达,才会有报告基因和被捕获的内源基因上游编码区的融合转录,从而发现、分离和研究启动子及其相关基因。该方法能获得一些启动子,但也存在一些缺点,如易出现重复捕获现象,成功率低,而且这种方法前期投入巨大,所得启动子表达特征随机性较大,序列结构不明确,需要繁 杂的分离和验证工作;且受转基因插入频率的制约,无法充分的挖掘天然资源。通过生物信息学手段挖掘转录组数据是提高特异性启动子和调控元件研究能力的思路,2008年后,在水稻和拟南芥中已有报道通过分析基因芯片表达数据,定向预测并分离鉴定到一组具有组织和诱导特异性的启动子。但这样的方法在研究环境诱导启动子时,所使用的转录数据多是在极端的处理条件下收集,筛选基因的诱导强度可能并不与水稻耐逆性直接相关。虽然分离的启动子活性在极端环境下常常达到甚至高于组成型启动子35s或actin的强度,但在耐逆生理过程中的表达强度却并不确定。因此,在基因工程中,这些启动子可能并不能最大程度发挥所驱动耐逆基因的效率。此外,目前所分离启动子序列均来自于已测序品种,比如水稻中大部分启动子来源于日本晴,日本晴与实际应用的水稻品种存在一定的遗传差异,因此日本晴启动子在需进行遗传改良的背景材料中是否具有高度保守的表达能力还存在疑问。
因此,虽然现有技术中给出了少量获取启动子的方法,但是现有方法均存在其局限性或难以弥补的缺陷,目前还没有一种能够高效地批量进行启动子分离的技术。
技术实现要素:
本发明希望提供一种高效的、大规模高效分离作物中各种未知启动子的方法。
本发明主要是基于这样的思路:数千年的驯化历史,使作物具有丰富的与农业性状相关的遗传多样性。这些遗传多样性主要是由其基因组上碱基的多态性(单个碱基变异、片段重复、缺失、插入)等决定的。这些多态性既会出现在基因序列上,也会出现在启动子上。大量的研究已经表明,基因序列上的碱基多态性影响了其功能从而产生了表型的变化。对于启动子序列的多态性,虽然研究较少,但普遍认可这些snp(单核苷酸多态性) 或片段插入缺失可能通过改变启动子活性影响基因表达,从而产生性状的差异。以此为理论基础,本发明拟以不同表型品种为材料,通过综合转录组和基因组深度测序数据,以生物信息学手段批量分析分离与表型差异相关的、具有核酸多态性的特异型启动子,再通过连接报告基因的实验验证,鉴定获得一批与关键性状调控相关且具有生理表达强度的新型启动子。
具体而言,本发明提供一种新型作物特异启动子分离方法,其特征在于,所述方法利用启动子上碱基的多态性可能通过改变启动子活性影响基因表达,从而产生性状差异的原理,以不同表型的品种为材料,综合转录组和基因组深度测序数据,以生物信息学手段批量分析并分离与表型差异相关的、具有核酸多态性的特异型启动子,再通过连接报告基因的实验进行验证,获得一批与关键性状调控相关且具有生理表达强度的新型启动子。
进一步地,所述方法包括下述步骤:
(1)、确定待分离启动子的特异性;
(2)、基于所述特异性选定具有所述特异性的作物品种作为待分离作物;
(3)、选定不具有所述特异性或者特异性弱于所述待分离作物的相同作物的另一品种作为对照作物;
(4)、分别取预定数目的待分离作物植株和对照作物植株,在两种作物植株中诱导所述特异性表达,并进行转录组测序;
(5)、将待分离作物植株的转录组测序结果与对照作物植株的转录组测序结果进行比对,找到与所述特异性相关的差异表达基因;
(6)、分别提取所述差异表达基因上游及下游的启动子序列;
(7)、判断所述启动子序列上是否存在碱基多态性;
(8)、如果所述启动子序列上存在碱基多态性或者碱基多态性超过目标值,并该碱基多态性与所述特异性相关,则将其作为候选启动子;
(9)、克隆分离所筛选的候选启动子,转入无所述特异性的作物品种 中鉴定其表达特性,若所转入的作物品种表现出所述特异性,则判定所述候选启动子为具有所述特异性的目标启动子。
进一步地,所述步骤(5)还包括:对所述差异表达基因进行实时荧光定量pcr分析,以获取所述差异表达基因的在两种作物中的相对表达量,若二者的相对表达量达到预定目标值,再转入步骤(6)。
进一步地,所述步骤(5)还包括:结合基因组测序数据和共表达分析,排除由于因上游转录因子变化而引起表达变化的差异表达基因。
进一步地,所述特异性包括:盐诱导特性、低温诱导特性、高温诱导特性以及损伤诱导特性。
进一步地,所述特异性包括:叶、茎、种子、花的组织特异性。
另一方面,本发明提供一种作物冷响应启动子分离方法,所述方法包括下述步骤:
(1)选择耐冷水稻品种作为待分离作物并选择常规或冷敏感的水稻品种作为对照作物;
(2)分别对二者的植株进行低温处理,并对二者转录组进行测序和分析;
(3)、将待分离作物植株的转录组测序结果与对照作物植株的转录组测序结果进行比对,从中选出两个品种中受低温诱导差异明显的基因;
(4)结合基因组测序数据和共表达分析,排除由于因上游转录因子变化而引起表达变化的基因;
(5)基于基因组测序数据,从测序结果获得在待分离作物和对照作物中表达差异明显基因的差异表达基因,并分析其碱基多态性是否与表达变化相关;
(6)若所述差异表达基因的碱基多态性与表达变化相关,则将其作为候选启动子;
(7)克隆分离所筛选的候选启动子,转入常规或冷敏感水稻品种中鉴 定表达特性,若鉴定正确,则作为目标冷诱导启动子。
进一步地,在所述步骤(3)中,选择耐冷品种中受诱导而冷敏感水稻品种中不受诱导的基因,或是在耐冷品种中受诱导表达量高而冷敏感水稻品种中受诱导表达量低的基因,或是在耐冷品种中受诱导表达量低而冷敏感水稻品种中受诱导表达量高的基因。
进一步地,在所述步骤(6)中,以转录起始位点上游1~2kb及下游100bp的范围作为候选启动子。
进一步地,在所述步骤(7)中,采用基因克隆的方法,分离候选启动子序列,并构建候选启动子与报告基因的融合表达载体,用于水稻的稳定遗传转化或烟草的瞬时转化,通过对报告基因的表达情况检测,来鉴定所述候选启动子的功能。需要说明的是,本发明步骤(4)中所提到的“诱导所述特异性表达”并不仅限于通过外界环境的改变来使所述特异性表达,其指的是能够实现所研究的特异性表达的任何方式,比如,对于组织特异性而言,则该组织部位的自发表达也包含在本发明的范围内。上面步骤中所提到的“排除”指的是根据共表达分析,如果与其共同表达的转录因子的表达量上升,则排除该基因。
技术效果
本发明的方法能够规模化分离植物基因启动子。本发明的方法特别适用于同一作物不同品种在生物或非生物胁迫条件下,或在生长发育过程中,辨认哪些基因启动子在起主导作用,进而实现有效分离和获取,找出那些可以帮助实现高产、优质、抗逆的调控因子,为作物分子育种和基因工程改良提供服务。与其他的大规模分离启动子方法相比,本发明的创新点如下:
(1)对品种资源创新利用,拓宽了作物启动子研究的范围,现有启动子的研究都是主要集中于对单一品种的基因测序基础上进行的,目前,尚没有人通过跨品种研究的方式来进行启动子的筛选。
(2)所分离的启动子与性状差异直接相关,表达强度更可能是生理上需要的强度,同时启动子可在不同背景下保持特异性,更适于基因工程的使用。
(3)本发明的方法既可批量分离新型启动子,又能直接阐明启动子上核酸多态性的调控作用。此外,其研究成果将帮助我们理解启动子的进化对于作物进化的作用。
附图说明
以下,结合附图来详细说明本发明的实施方案,其中:
图1表示日本晴和padipohonbatu在15℃下萌发图(a图)和相应的根长和茎长(b图)。
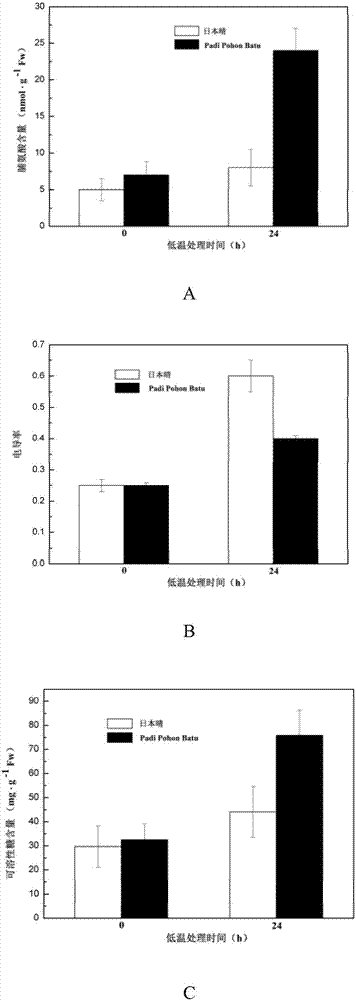
图2表示日本晴和padipohonbatu幼苗在4℃下处理24小时前后的脯氨酸含量、电导率及还原性糖含量的变化(图中:a-脯氨酸含量、b-电导率、c-还原性糖含量)。
图3表示rt-qpcr分析日本晴和padipohonbatu中os06g0165600基因受低温诱导表达情况。
图4表示日本晴和padipohonbatu中os06g0165600基因启动子的序列比对结果。其中,“*”表示碱基一致的序列,“.”表示碱基错配的序列,“—”表示碱基缺失的序列。
图5表示克隆的日本晴和padipohonbatu中os06g0165600基因启动子电泳图,其中:m表示marker,1表示日本晴中os06g0165600基因启动子p1,2表示padipohonbatu中os06g0165600基因启动子p2。
图6表示日本晴和padipohonbatu中的os06g0165600基因启动子的低温诱导活性图,其中:p1表示日本晴中os06g0165600基因启动子,p2表示padipohonbatu中os06g0165600基因启动子。
具体实施方式
实施例1
以下参照具体的实施例来说明本发明。本领域技术人员能够理解,这些实施例仅用于说明本发明,其不以任何方式限制本发明的范围。
下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的原料、试剂材料等,如无特殊说明,均为市售购买产品。
为简单叙述本发明的过程,本发明以水稻冷响应启动子的分离鉴定为例进行说明。
1、耐冷水稻品种的获得
水稻是原产热带或亚热带的一种作物,绝大多数栽培水稻品种的耐冷性不强。从国家水稻数据中心及世界种质资源中心,我们查找得出耐低温水稻品种padipohonbatu,该品种来自于世界微核心种质资源,为安徽省农科院水稻所保存。为研究其低温萌发能力,以日本晴为对照,分别取100粒种子在15℃培养箱中萌发15天,观察这两个品种的发芽率、根长和芽长。结果显示,padipohonbatu在低温下的萌发率达到100%,而日本晴的萌发率只有85%;且padipohonbatu的根长和芽长分别为0.45cm和0.6cm,显著高于日本晴(图1)。
为了进一步验证padipohonbatu的耐冷能力,对其10天幼苗在冷胁迫条件下(4℃)的生理指标如脯氨酸含量、电导率及还原性糖含量等进行检测。结果表明,冷处理24h时,padipohonbatu和日本晴在冷处理条件下脯氨酸含量及还原性糖含量与对照相比都有显著提高,且冷胁迫下padipohonbatu中的脯氨酸含量是日本晴的2倍,电导率比日本晴下降了约30%(说明植物的耐冷性增强),还原性糖含量提高了近1倍(图2)。综合以上冷胁迫生理性状分析数据,进一步验证padipohonbatu和日本晴相比具有极强的耐冷性。因此,我们选用padipohonbatu为后续的耐冷水稻研究品种材料,以常规栽培稻日本晴为对照品种。
2、转录组测序和实时荧光定量pcr验证低温响应基因的表达模式
取10天的padipohonbatu和日本晴幼苗,分别在4℃下处理24小时,以未处理的植株为对照,每种材料至少取三个重复,取样后用锡箔纸包裹,迅速放置于液氮中,-80℃保存。总rna采用商用试剂盒qiangen提取,用于构建cdna文库的材料。提取的rna送至测序公司,进行转录组测序和数据组装及分析,计算差异表达基因。差异表达基因由dchip软件lowerboundfoldchange方法计算,候选基因表达差异倍数为2倍。在本研究中发现,与各自的对照相比,padipohonbatu在冷处理下有5,484个基因差异表达,日本晴在冷处理下有4,856个基因差异表达,而且754个基因在两个品种中都上调表达。在这上调的基因中,go分析结果发现其中相当一部分为转录因子,包括ap2/erebp、myb、hsf和nac蛋白等,表明转录因子在冷胁迫反应中起重要作用。其中,我们发现转录因子os06g0165600基因在日本晴冷处理后上调的倍数(log2-foldchange为3.2)显著低于padipohonbatu(log2-foldchange为7.9),因此,将其选为候选基因。
为了对其进行进一步验证,分别将两种材料在4℃下处理0、4、8、12、24小时,提取rna并反转成cdna,我们对差异表达基因os06g0165600进行实时荧光定量pcr分析(rt-qpcr)。根据候选基因的cds序列,设计相应的rt-qpcr引物,序列如表1所示。同时以actin为内参基因,荧光定量pcr仪是7500(ab公司)。pcr反应每组实验均完成三个生物学重复,每个生物学重复至少做三个技术重复。荧光定量pcr反应体系为20μl:含有sybrmix10μl,正反向引物(10μmol/l)各0.8μl,模板2μl,depc处理过的无菌水补足至20μl。扩增条件为:94℃,5min;94℃,15s;60℃,20s;72℃,30s;40个循环;每个循环于72℃复性末端进行荧光检测。反应结束后先加热到95℃,然后降至72℃,再缓慢升温至95℃,记录荧光信号的变化,得出扩增产物的熔解曲线。用比较ct法(△△ct)计算候 选基因的相对表达量。通过2-δδct估算目的基因的相对表达量和系统误差。实验结果表明,在日本晴中该基因的表达量随冷处理时间的延长而升高,在24小时达到33.5倍,而在padipohonbatu中该基因在低温处理24小时达到120.6倍(图3)。芯片鉴定的基因表达谱和荧光定量pcr结果的符合度非常高,相关系数为0.892,由此验证了上述方法在选择候选基因时的可靠性和稳健性。
表1定量pcr分析引物
3、os06g01656000基因及启动子的序列分析
os06g0165600基因不含内含子,因此从日本晴和padipohonbatu基因组测序数据中直接获取基因序列,同时获取转录起始位点上游1kb及下游100bp的范围os06g0165600基因启动子序列。其中,将日本晴的os06g0165600基因的启动子命名为p1,将padipohonbatu中该启动子命名为p2(核苷酸序列如seqidno.1和no.2中所示)。基因在ncbi网站(http://www.ncbi.nlm.nih.gov/)上进行blast比对分析;p1和p2在启动子分析网站plantpan(http://plantpan2.itps.ncku.edu.tw/)和place(http://www.dna.affrc.go.jp/htdocs/place/)上进行序列分析。经过比对分析发现,日本晴与padipohonbatu的编码序列之间非常保守,但启动子序列之间则存在较多的单核苷酸多态性(snp)和插入缺失(indel)。顺式作用元件分析表明,两个水稻品种os06g0165600基因的启动子序列中都包含大量的低温应答元件,如有ice1蛋白识别的myc元件(canntg)、低温应 答元件cbfhv(ccgac)。但是,由于两品种间snp的出现,一部分myc元件只存在于padipohonbatu中,而在日本晴中则缺失了,包括日本晴中os06g0165600基因的启动子p1上318bp、336bp位置的myc元件,以及747bp位置的cbfhv元件(图4),导致padipohonbatu的启动子序列中包含有更多的myc和cbfhv元件。padipohonbatu中os06g0165600受冷诱导表达幅度明显高于日本晴,我们初步判断os06g0165600的启动子是低温诱导启动子,且p2比p1的低温诱导活性更强,且该基因的冷诱导表达变化可能与其启动子上的核苷酸多态性相关,为了验证这一假设,我们进行后续的p1和p2启动子克隆,并进行相关的功能验证实验。
4、启动子的功能鉴定
步骤1:启动子及融合表达载体构建
从生物信息学网站ncbi或构建的基因组测序数据中获取os06g0165600基因的启动子序列,以转录起始位点上游1kb及下游100bp的范围作为启动子序列。根据启动子序列信息,设计相关的带酶切位点的扩增引物(fp:aagcttcgcctcgcggaggaggtcatca,带有hindiii酶切位点;rp:gaattccgtccttactatgttgctaatg,带有ecori酶切位点)。
以水稻品种日本晴和padipohonbatu的dna为模板,按常规pcr体系,扩增程序如下:
95℃预变性5min;95℃变性30s,58℃退火30s,72℃延伸1min30s,35个循环;最后72℃延伸10min。
pcr产物经1%琼脂糖凝胶电泳后(图5),回收pcr扩增的目的片段,连接到pgem-t-easy载体(购自promega公司,按载体说明书中的比例混合)上,按照热激法转化大肠杆菌xl-blue感受态细胞后,经菌落pcr筛选获得阳性克隆,挑取单克隆摇菌液提质粒,用hindiii和ecori进行双酶 切验证。经过鉴定的阳性克隆送交invitrogen公司测序,最终获得1002bp的日本晴中该基因启动子p1,及1004bp的padipohonbatu中该基因启动子p2。
回收hindiii和ecori双酶切后的启动子片段,同时hindiii和ecori线性化处理、回收pcambia1391,将上述的两个片段用t4连接酶(购于takara公司)进行连接,得到启动子与gus基因融合的植物表达载体p1-1391和p2-1391,利用冻融法分别将表达载体转入根癌农杆菌(agrobacteriumtumefaciens)eha105中,在-80℃保存。
步骤2:烟草瞬时转化
采取烟草瞬时转化实验快速验证启动子的功能,具体步骤如下:
(1)烟草种子经消毒后,接种于1/2ms固体培养基上,于30℃、16h光照/8h黑暗条件下培养一个月后,移栽至温室中,用于农杆菌瞬时表达分析。
(2)配制烟草注射培养液:10.5g/lk2hpo4,4.5g/lkh2po4,1.0g/l(nh4)2so4,0.5g/lnacitrate·2h2o,1mmmgso4·7h2o,0.5%glycerol和10mmmes(ph=5.6)。高压灭菌后,加入0.2%glucose和50μmacetosyringone。
并且,准备烟草注射重悬液:10mmmes(ph=5.6)。
(3)将含有各个载体的农杆菌在50mg/lkan及10mg/lrif的lb固体培养基上划线,28℃培养过夜。
(4)挑取单个克隆,接种于5ml含有50mg/lkan及10mg/lrif的lb液体培养基中,28℃、210rpm下培养过夜。
(5)按1:100将上述液体接种于烟草注射培养液中,并加入10mg/lrif,在28℃、210rpm下培养过夜。
(6)4000rpm,离心10min收集上述培养后的菌体,并用重悬液反复 清洗2次。用重悬液调成od600=0.5~0.6。加入150μmacetosyringone后,室温静置15~30min。
(7)用2ml注射器在烟草叶片背面打上小孔,将农杆菌重悬液经小孔注射到叶片中。注射后的烟草正常培养24h。
(8)将注射后的叶片取下,分别浸泡在ddh2o中,于4℃放置24h,用于低温处理。未处理样品做对照。收集样品用液氮速冻存于-80℃,用于提取蛋白。
步骤3:启动子驱动的报告基因gus的活性测定
通过检测瞬时转化中启动子驱动的报告基因gus的表达情况,来确定启动子的功能。可采用测定组织中gus蛋白的含量,定量的测定启动子活性。gus蛋白测定方法参照文献(juanli,rui-yingqinetal.identificationandanalysisofthemechanismunderlyingheat-inducibleexpressionofriceaconitase1.plantscience[j].2015,233:22–31)。
结果显示,p1和p2启动子在冷诱导前的活性约为550.2和431.84-mupmol/min/mgprotein,经冷诱导后p1和p2启动子的活性分别升高至5061.8和6649.74-mupmol/min/mgprotein。相比于各个启动子诱导前的活性,p1和p2诱导后的活性约是处理前的9.2倍和15.4倍。由此说明,p1和p2启动子均为低温诱导启动子,且p2启动子的低温诱导活性更强。
经过以上的启动子筛选、克隆及功能鉴定,我们成功分离了非常规水稻品种padipohonbatu中受低温诱导的强启动子p2,由此说明这一启动子分离方法的可行性。我们也可借用这一方法,用于分离更多的低温诱导启动子,或其他品种中的诱导性启动子或组织特异启动子。
虽然上面结合本发明的优选实施例对本发明的原理进行了详细的描述,本领域技术人员应该理解,上述实施例仅仅是对本发明的示意性实现 方式的解释,并非对本发明包含范围的限定。实施例中的细节并不构成对本发明范围的限制,在不背离本发明的精神和范围的情况下,任何基于本发明技术方案的等效变换、简单替换等显而易见的改变,均落在本发明保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!