一种替加环素的晶型及其制备方法与流程
本发明涉及晶型药物领域,尤其涉及一种替加环素的晶型及其制备方法。
背景技术:
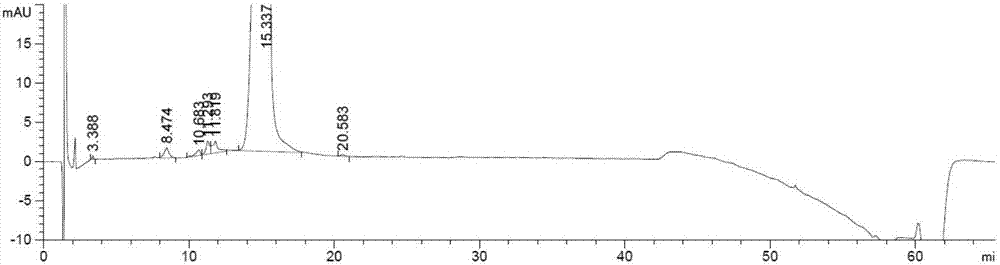
:替加环素,英文名为tigecycline,分子式:c29h39n5o8,化学名称为(4s,4as,5ar,12as)-9-(2-(叔丁基氨基)乙酰氨基)-4,7-双(二甲基氨基)-1,4,4a,5,5a,6,11,12a-四羟基-1,11-二氧-2-萘并萘甲酰胺,cas号:220620-09-7,分子量:585.65,其结构式如式i:替加环素由美国惠氏公司开发,最早公开在美国专利us5494903和5284963中,是第一个新一代的称作甘氨酰基环素的四环素抗生素药物。用于18岁及18岁以上复杂皮肤和皮肤结构感染或者复杂腹内感染患者的治疗。目前来看,替加环素制备工艺中大都存在中间体或粗品质量难以控制的不足。替加环素产品中主要的杂质为替加环素的差向异构体,其结构和性质与替加环素非常相似,生产中难以去除,通常需要多次精制产品纯度才能勉强达到99%,但在此基础上继续提高其纯度非常困难,尤其是需要将单一杂质控制在0.1%以下是无法达到的,难以满足注射剂原料的质量要求。并且,多次纯化步骤也导致了收率的降低,每次纯化过程都造成大量产品的损失。同时,现有文献中公开的晶型存在纯度难以提高、工艺繁琐、成本偏高、对环境不友好等劣势,不适宜工业化生产,迫切需要一种高纯度、操作工艺简单、低成本的替加环素晶型。技术实现要素:有鉴于此,本发明要解决的技术问题在于提供一种替加环素的晶型及其 制备方法,本发明提供的替加环素晶型纯度高,其制备方法所得产品收率高。本发明提供的替加环素的晶型,x-射线粉末衍射图在2θ±0.2°位置有衍射峰,所述2θ为8.5°、10.3°和20.0°。本发明中,2θ为8.5°、10.3°、12.1°、20.0°和24.7°。本发明中,2θ为8.5°、10.3°、12.1°、12.4°、14.2°、15.7°、16.9°、17.3°、17.9°、18.2°、18.8°、20.0°、20.6°、22.0°、23.2°、23.4°、24.7°、25.0°、26.4°和27.9°。根据x-射线粉末衍射图,本发明所述的替加环素晶型的x-射线衍射的详细参数如表1,表现为衍射峰位置:2θ值(°),衍射峰相对强度:峰高值(height%)。表1本发明提供的替加环素晶型的x射线衍射参数峰2θ(±0.2°)18.5210.3312.1412.4514.2615.7716.9817.3917.91018.21118.81220.01320.61422.01523.21623.41724.71825.01926.42027.9x-射线粉末衍射图的2θ值可在机器之间或样品之间稍有变化,其数值可 能相差大约0.2个单位,或者相差大约0.1个单位,因此所引用的数值不能解释为绝对值。同样应该理解,峰高的大小同样可能相差大约5个单位,或者相差大约4个单位,或者相差大约3个单位,或者相差大约2个单位,或者相差大约1个单位,因此包括于本发明中的xrpd迹线(trace)强度为说明性的,并非意欲用于绝对比较。x-射线粉末衍射图显示,本发明提供的替加环素晶型与惠氏开发替加环素的晶型i或晶型ii的2θ值皆不同,峰高值也不同,二者属于不同的晶型。尽管本发明提供的替加环素的晶型与惠氏开发的晶型在化学稳定性、溶解性、生物活性方面并无显著差异,研究表明,本发明提供的晶型同样能够用于18岁及18岁以上复杂皮肤和皮肤结构感染或者复杂腹内感染患者的治疗。但实验表明,本发明制备的晶型中杂质的含量非常低,总杂的含量不超过0.1%,且收率很高,可达89.5%。而在现有技术制备的替加环素产品,不论是惠氏开发的晶型i、晶型ii,还是无定型替加环素,其中都含有替加环素的差向异构体,纯度很难超过99%,且纯化过程往往需要经过多次重结晶的步骤,导致收率仅为50%左右。本发明通过改善重结晶条件,获得了高纯度的替加环素新晶型,降低了杂质的含量,提高了用药的安全性。本发明提供的替加环素晶型的制备方法,为将替加环素以低碳醇和丙酮的混合溶剂重结晶,获得本发明提供的晶型;所述低碳醇为c1~c3的醇类。在本发明中,制备替加环素晶型使用的替加环素原料为替加环素晶型i、替加环素晶型ii或替加环素无定型晶。影响替加环素产生不同晶型的主要因素来自于各种物理条件参数的变化,例如,结晶时的温度、晶体生长的时间,晶体生长的条件(搅拌或静置)、重结晶的溶剂、溶剂与替加环素的比例等。作为优选,低碳醇为甲醇、乙醇、丙醇或异丙醇。优选的,低碳醇为异丙醇。作为优选,低碳醇与丙酮的体积比为(1:3)~(3:1)。作为优选,替加环素与低碳醇和丙酮的混合溶剂的质量-体积比为(1:2)~(1:10)。作为优选,重结晶的温度为-10℃~-25℃。作为优选,重结晶的时间为0.5h~10h。作为优选,重结晶在氮气保护下进行。作为优选,重结晶的条件为搅拌。本发明中,重结晶是指搅拌后,经过滤、真空干燥的过程。在一些实施例中,替加环素晶型的制备方法为,将替加环素与甲醇和丙酮混合,氮气氛围下,0~10℃搅拌6h后,过滤,真空干燥,制得本发明提供的替加环素晶型;甲醇和丙酮的体积比为3:1;甲醇和丙酮的体积和与替加环素的质量比为2:1(体积-质量比)。在一些实施例中,替加环素晶型的制备方法为,将替加环素与乙醇和丙酮混合,氮气氛围下,-10℃~0℃搅拌1h后,过滤,真空干燥,制得本发明提供的替加环素晶型;乙醇和丙酮的体积比为1:3;乙醇和丙酮的体积和与替加环素的质量比为4:1(体积-质量比)。在一些实施例中,替加环素晶型的制备方法为,将替加环素与丙醇和丙酮混合,氮气氛围下,5~15℃搅拌0.5h后,过滤,真空干燥,制得本发明提供的替加环素晶型;丙醇和丙酮的体积比为1:1;丙醇和丙酮的体积和与替加环素的质量比为4:1(体积-质量比)。在一些实施例中,替加环素晶型的制备方法为,将替加环素与异丙醇和丙酮混合,氮气氛围下,-10℃~0℃搅拌4h后,过滤,真空干燥,制得本发明提供的替加环素晶型;异丙醇和丙酮的体积比为1:1;异丙醇和丙酮的体积和与替加环素的质量比为10:1(体积-质量比)。在一些实施例中,替加环素晶型的制备方法为,将替加环素与甲醇、乙醇和丙酮混合,氮气氛围下,10℃~25℃搅拌3h后,过滤,真空干燥,制得本发明提供的替加环素晶型;甲醇、乙醇和丙酮的体积比为1:1:2;甲醇、乙醇和丙酮的体积和与替加环素的质量比为8:1(体积-质量比)。在一些实施例中,替加环素晶型的制备方法为,将替加环素与乙醇、丙醇和丙酮混合,氮气氛围下,0℃~10℃搅拌10h后,过滤,真空干燥,制得本发明提供的替加环素晶型;乙醇、丙醇和丙酮的体积比为1:1:2;乙醇、丙醇和丙酮的体积和与替加环素的质量比为10:1(体积-质量比)。在一些实施例中,替加环素晶型的制备方法为,将替加环素与甲醇和丙酮混合,氮气氛围下,0℃~10℃搅拌2h后,过滤,真空干燥,制得本发明提供的替加环素晶型;甲醇和丙酮的体积比为1:1;甲醇和丙酮的体积和与替加 环素的质量比为3:1(体积-质量比)。在一些实施例中,替加环素晶型的制备方法为,将替加环素与甲醇和丙酮混合,氮气氛围下,0℃~10℃搅拌5h后,过滤,真空干燥,制得本发明提供的替加环素晶型;异丙醇和丙酮的体积比为1:1;异丙醇和丙酮的体积和与替加环素的质量比为5:1(体积-质量比)。在一些实施例中,替加环素晶型的制备方法为,将替加环素与甲醇和丙酮混合,氮气氛围下,0℃~10℃搅拌3h后,过滤,真空干燥,制得本发明提供的替加环素晶型;异丙醇和丙酮的体积比为1:1;异丙醇和丙酮的体积和与替加环素的质量比为6:1(体积-质量比)。在一些实施例中,替加环素晶型的制备方法为,将替加环素与甲醇和丙酮混合,氮气氛围下,0℃~10℃搅拌2h后,过滤,真空干燥,制得本发明提供的替加环素晶型;异丙醇和丙酮的体积比为1:1;异丙醇和丙酮的体积和与替加环素的质量比为5:1(体积-质量比)。本发明提供的替加环素的晶型,x-射线粉末衍射图在2θ±0.2°位置有衍射峰,所述2θ为8.5°、10.3°和20.0°。本发明通过改善重结晶条件,获得了高纯度的替加环素新晶型,降低了杂质的含量,提高了用药的安全性。实验表明,本发明制备的晶型中杂质的含量非常低,总杂的含量不超过0.1%,且收率很高,可达89.5%。附图说明图1示实施例1制得产品的x-粉末颜色图谱;图2示实施例1制得产品的hplc检测图谱;图3示实施例2制得产品的hplc检测图谱;图4示实施例3制得产品的hplc检测图谱;图5示实施例4制得产品的hplc检测图谱;图6示实施例5制得产品的hplc检测图谱;图7示实施例6制得产品的hplc检测图谱;图8示实施例7制得产品的hplc检测图谱;图9示实施例8制得产品的hplc检测图谱。具体实施方式本发明提供了一种替加环素的晶型及其制备方法,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本
发明内容、精神和范围内对本文的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。本发明采用的材料、试剂、仪器皆为普通市售品,皆可于市场购得。下面结合实施例,进一步阐述本发明:实施例1:取无定形替加环素2.0g(hplc纯度为97.82%),加入到0~10℃的混合溶剂(4ml,甲醇:丙酮=3:1,体积比)中,氮气氛围下保温搅拌6h,过滤,室温下真空干燥,获得产品1.764g,x-射线粉末衍射检测,图谱如图1。hplc测得(图谱如图2)其纯度为99.58%,收率为88.2%。根据x-射线粉末衍射图,本发明所述的替加环素晶型的x-射线衍射的详细参数如表1,表现为衍射峰位置:2θ值(°),衍射峰相对强度:峰高值(height%)。表1本发明提供的替加环素晶型的x射线衍射参数实施例2:取无定形替加环素2.0g(hplc纯度为97.82%),加入到-10~0℃的混合溶剂(6ml,乙醇:丙酮=1:3,体积比)中,氮气氛围下保温搅拌1h,过滤,室温下真空干燥。获得产品1.728g,x-射线粉末衍射检测,图谱与图1相同。hplc测得(图谱如图3)其纯度为99.65%,收率为86.4%。实施例3:取无定形替加环素2.0g(hplc纯度为97.82%),加入到5~15℃的混合溶剂(8ml,丙醇:丙酮=1:1,体积比)中,氮气氛围下保温搅拌0.5h,过滤,室温下真空干燥,获得产品1.714g,x-射线粉末衍射检测,图谱与图1相同。hplc测得(图谱如图4)其纯度为99.75%,收率为85.7%。实施例4:取无定形替加环素2.0g(hplc纯度为97.82%),加入到-10~0℃的混合溶剂(20ml,异丙醇:丙酮=1:1,体积比)中,氮气氛围下保温搅拌4h,过滤,室温下真空干燥,获得产品1.79g,x-射线粉末衍射检测,图谱与图1相同。hplc测得(图谱如图5)其纯度为99.54%,收率为89.5%。实施例5:取无定形替加环素2.0g(hplc纯度为97.82%),加入到10~25℃的混合溶剂(16ml,甲醇:乙醇:丙酮=1:1:2,体积比)中,氮气氛围下保温搅拌3h,过滤,室温下真空干燥,获得产品1.652g,x-射线粉末衍射检测,图谱 与图1相同。hplc测得(图谱如图6)其纯度为99.79%,收率为82.6%。实施例6:取无定形替加环素2.0g(hplc纯度为97.82%),加入到0~10℃的混合溶剂(20ml,乙醇:丙醇:丙酮=1:1:2,体积比)中,氮气氛围下保温搅拌10h,过滤,室温下真空干燥,获得产品1.536g,x-射线粉末衍射检测,图谱与图1相同。hplc测得(图谱如图7)其纯度为99.77%,收率为76.8%。实施例7:取无定形替加环素2.0g(hplc纯度为97.82%),加入到0~10℃的混合溶剂(6ml,甲醇:丙酮=1:1,体积比)中,氮气氛围下保温搅拌2h,过滤,室温下真空干燥,获得产品1.766g,x-射线粉末衍射检测,图谱与图1相同。hplc测得(图谱如图8)其纯度为99.56%,收率为88.3%。实施例8:取无定形替加环素2.0g(hplc纯度为97.82%),加入到0~10℃的混合溶剂(10ml,甲醇:丙酮=1:1,体积比)中,氮气氛围下保温搅拌5h,过滤,室温下真空干燥,获得产品1.664g,x-射线粉末衍射检测,图谱与图1相同。hplc测得(图谱如图9)其纯度为99.63%,收率为83.2%。实施例9:取惠氏ⅰ晶型的替加环素2.0g(hplc纯度为96.26%),加入到0~10℃的混合溶剂(12ml,甲醇:丙酮=1:1,体积比)中,氮气氛围下保温搅拌2h,过滤得到本发明所述晶型的替加环素,获得产品1.610g,x-射线粉末衍射检测,图谱与图1相同。hplc测得其纯度为99.71%,收率为80.5%。实施例10:取惠氏ii晶型的替加环素2.0g(hplc纯度为95.05%),加入到0~10℃的混合溶剂(10ml,甲醇:丙酮=1:1,体积比)中,氮气氛围下保温搅拌2h,过滤得到本发明所述晶型的替加环素,获得产品1.705g,x-射线粉末衍射检 测,图谱与图1相同。hplc测得其纯度为99.65%,收率为85.3%。对比例1:搅拌替加环素粗品(110g)与乙酸乙酯的混合物,加热到30-35℃,历经15分钟加入甲醇(550ml)。于30-35℃保持后,在硅藻土(36g)上过滤温热溶液,滤饼用乙酸甲酯(2×106g)洗涤。通过蒸馏(20℃,150mmhg)浓缩滤液至550ml。加入乙酸甲酯(1.1l),通过蒸馏(20℃,150mmhg)将所得混悬液浓缩至550ml。重复该步骤,然后将浓缩物冷却至0-4℃达1小时。过滤收集所得固体,用0-5℃乙酸甲酯(2×150ml)洗涤。将固体在真空中干燥(65~70℃,10mmhg)100小时,获得98.0g(产率89.1%)所需产物。hplc检测纯度为98.8%。可见,对比例1采用梯度温度制备浆液,升温溶解、降温结晶,制浆工艺较为繁琐,同时操作时间相对延长,所用有机溶剂与粗品体积质量比达到20倍,最终产品的c-4差向异构体处于较高水平;而实施例1~10所涉及到纯化工艺,可在短时间内有效纯化产品,收率稳定,且溶剂使用量小,操作过程简单,成本低廉,适合大规模的工业化生产。以上仅是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1