一类基于荧光“双响应”机制的一氧化氮的探针及其合成和应用的制作方法
本发明属于有机光化学技术领域,尤其涉及一类基于荧光“双响应”机制的探针分子材料,具体来说是一类由邻位芳香胺和其他共轭单元经不同桥联单元耦合的推-拉电子型共轭半导体有机分子材料,其实现了对一氧化氮分子双重信号响应的检测和荧光成像应用。
背景技术:
传感技术是现代科技的前沿技术,许多国家已经将传感技术列为与通信技术和计算机技术同等重要的位置,成为信息技术的三大支柱之一。所谓传感技术,是人们为了对被测对象所包含的信息进行定性的了解和定量掌握所采取的一系列技术措施,而传感器就是完成相应传感功能的器件活装置。传感器大致分为三种:(一)物理传感器(二)化学传感器(三)生物传感器。荧光探针是化学传感技术领域在上个世纪八十年代的一项重大发现,目前已有愈来愈多的荧光探针应用于分子水平上进行实时监测。荧光检测技术由于灵敏度高、操作简便、可视性强,且对细胞、生物体的损伤小,成为了用于临床分析、环境监测、生物分析及生命科学等领域不可缺少的检测工具。
一氧化氮(NO)在心血管系统、神经系统及免疫功能调节中起着重要的作用,是细胞间和细胞内信息传递的重要调节因子,广泛参与调节生物体内的生理和病理机制。近几年来,许多与一氧化氮(NO)相关的疾病已被报道。由于一氧化氮(NO)的关键生物学作用,所以对于一氧化氮(NO)的研究已经迫在眉睫。
因为信息传递在人的生理运转和疾病机理中有着重要的作用,所以对于活性氮化合物(RNS)的研究,尤其是一氧化氮(NO)的研究成为了一个热门的领域。人们在研究过程中所遇到的问题主要集中在缺乏必要的工具对信息传递过程进行的实时跟踪。荧光检测技术的应用,特别是在结合了微观成像技术后,荧光成像技术的运用对研究对象进行实时的分析与跟踪进入了一个全新的领域。
现有的一氧化氮(NO)探针是基于邻位二胺结构的,但是,通常基于邻位二胺的NO探针往往通过直接利用探针分子本身荧光光谱发射强度变化进行分析检测,这样的方法会受限于探针本身的背景荧光干扰,导致检测灵敏度低。此外,传统基于邻苯二胺取代基的NO探针由于本身背景干扰的缘故,很少能在细胞成像中做半定量、定量检测。
因此,发明和设计一种具有高灵敏度的、可用于定量或半定量检测的一氧化氮(NO)探针是生物监测领域亟待解决的问题。
技术实现要素:
鉴于现有技术中存在上述技术问题,本发明提供一种基于荧光“双响应”机制的一氧化氮的探针分子及其制备方法和应用,所述探针分子拟采用构筑基于共轭电子给体单元和受体单元、具有推-拉电子效应并含有与以往NO检测类似的邻位二胺单元的新型探针分子。所述探针分子具有灵敏度高、选择性好的探针分子材料,同时,还具备了响应时间短、膜透性好、背景噪音低、可直接观察等优点。本发明采用的技术方案如下所述:
本发明提供一种基于荧光“双响应”机制的一氧化氮的探针分子材料,所述探针分子材料具有下述通式(Ⅰ)所示结构:
其中,Spacer为炔键、噻吩或取代芳基;Ar为芳基、取代芳基、杂环芳基或取代杂环芳基,其中,所述取代基是醚链、烷基链、磺酸基或季铵盐阳离子中的一种。
进一步的,在式(Ⅰ)通式中,上述芳基或取代芳基是未取代或取代的苯、联苯、萘、苊、蒽、菲、芘、苝、芴中的一种;上述杂环芳基或取代杂环芳基是未取代或取代的吡啶、噻吩、咔唑、硅芴、吲哚、噻唑、苯并噻唑或芳基胺中的一种;芳基或杂环芳基的取代基可以是醚链、长度不超过6个碳原子的烷基链、磺酸基或季铵盐阳离子中的一种或多种,取代基个数为1-4个。
该探针分子在与NO发生作用时,会导致邻位二胺由强给电子特性转为弱吸电子特性,从而深度影响共轭体系的电子云分布,从而导致不仅荧光强度变化,而且荧光发射峰波长同样改变的双响应特性。
上述探针分子材料,在传统的基于邻位二胺结构的探针分子结构中引入了共轭结构,形成了一类由邻位芳香胺和其他共轭单元经不同桥联单元耦合的推-拉电子型共轭半导体有机分子材料。其中,邻苯二胺基团与一氧化氮相互作用发生关环反应,引起荧光光谱的变化从而实现对一氧化氮检测;同时,这种邻位二胺单元与一氧化氮的选择性反应能对共轭分子推-拉电子效应产生影响,共轭分子的紫外吸收光谱和荧光光谱在短波和长波区域的峰值发生变化,从而可通过比率法定量或半定量检测一氧化氮,实现对一氧化氮分子双重信号响应的检测和荧光成像应用。
本发明还提供上述探针分子材料的制备方法,首先是由4位和7位被卤素取代的苯并噻二唑A通过取代反应或偶联反应制得4位和7位被炔键、噻吩或取代芳基取代的苯并噻二唑B,然后是苯并噻二唑B与卤代芳香化合物反应得到炔键、噻吩或取代芳基上与Ar相连接的苯并噻二唑C,最后是苯并噻二唑C在硼氢化钠和六水合氯化钴的作用下制得具有苯二胺结构的探针分子材料D。或者首先制备具有炔键、噻吩或取代芳基结构与Ar相连接结构的化合物,然后将该化合物与4位和7位被卤素取代的苯并噻二唑A通过取代反应或偶联反应制得苯并噻二唑C,最后是苯并噻二唑C在硼氢化钠和六水合氯化钴的作用下制得具有苯二胺结构的探针分子材料D。
根据探针分子结构的不同,在制备时稍有不同,下面分别举例说明。
对于间隔段为噻吩结构的探针分子材料即Spacer为噻吩的探针分子材料,这类材料的制备方法的合成路线如“制备方法(一)”所示,该方法具体包括以下步骤:
i)在碱水溶液条件下,卤代苯并噻二唑与噻吩硼酸在钯催化剂作用下,于有机溶剂中反应2-8h,得到如式(2)所示的4,7-二噻吩苯并噻二唑;
其中,所述卤素X1为Cl、Br、I原子;所述碱可以是碳酸钾、碳酸钠、碳酸氢钠、碳酸铯或四丁基氢氧化铵等;所述有机溶剂可以是二氧六环、四氢呋喃、甲苯、甲基乙二醇等;所述钯催化剂是四(三苯基膦)钯、醋酸钯或二氯二三苯基膦钯等。
ii)在碱性条件下,上述4,7-二噻吩苯并噻二唑和卤代芳香化合物在钯催化剂和三甲基乙酸的作用下,于有机溶剂中反应6-12h,得到一类如式(4)所示共轭分子;
其中,所述卤素X2为Cl、Br、I原子;所述碱可以是碳酸钾、碳酸钠、碳酸氢钠、碳酸铯或四丁基氢氧化铵等;所述的有机溶剂是N.N-二甲基甲酰胺、N.N-二甲基乙酰胺或甲苯,且使用的有机溶剂须经无水无氧处理;所述钯催化剂是四(三苯基膦)钯、醋酸钯或二氯二三苯基膦钯等;反应温度为100-130℃。
iii)式(4)所示共轭分子溶于有机溶剂,加入还原剂和相当于还原剂2-10%摩尔比的六水合氯化钴,搅拌反应液,得到如通式(I)所示的一类基于荧光“双响应”机制的一氧化氮的探针分子,产率最高可达90%以上。
其中,步骤iii)的所述有机溶剂是甲醇、乙醇、乙酸乙酯中的一种或其混合溶液,所述还原剂为硼氢化钠、氢化铝锂等。
通过上述制备方法(一)能够制得间隔段为噻吩结构的探针分子材料,其中,X1、X2为Cl、Br、I原子,Ar为芳基、取代芳基、杂环芳基或取代杂环芳基;所述的芳基或取代芳基是未取代或取代的苯、联苯、萘、苊、蒽、菲、芘、苝、芴或螺芴中的一种;所述杂环芳基或取代杂环芳基是未取代或取代的吡咯、吡啶、呋喃、噻吩、咔唑、硅芴、吲哚、噻唑、苯并二唑或苯并噻唑中的一种;所述芳基或杂环芳基的取代基可以是醚链、长度不超过6个碳原子的烷基链、磺酸基或季铵盐阳离子中的一种,取代芳基或取代杂环芳基的取代基个数为1-4个。
对于间隔段为炔键结构的探针分子材料即Spacer为炔键的探针分子材料,这类材料的制备方法的合成路线如“制备方法(二)”所示,该方法具体包括以下步骤:
i)将卤代苯并噻二唑溶于有机溶剂,加入钯催化剂、碘化亚铜和三甲基硅乙炔,在50-80℃下搅拌4-24h,得到一类如式(2)所示的三甲基硅乙炔取代的苯并噻二唑化合物;
其中,所述有机溶剂可以为四氢呋喃、甲苯、苯和二异丙胺、三乙胺、二乙基乙胺、乙二胺等有机碱的混合溶剂,体积比约为1:1-2:3;所述催化剂是四(三苯基膦)钯、醋酸钯、二氯二三苯基膦钯等。
ii)将三甲基硅乙炔取代的苯并噻二唑化合物溶于有机溶剂,加入强碱,在20-40℃下搅拌2-12h,得到一类如式(3)所表示的一类乙炔取代的苯并噻二唑化合物;
其中,所述有机溶剂可以为甲苯、四氢呋喃、二氧六环、甲醇或乙醇等的混合溶剂;所述强碱是氢氧化钾、氢氧化钠、碳酸钾等。
iii)将卤代芳香化合物和乙炔取代的苯并噻二唑化合物溶于有机溶剂,加入钯催化剂和碘化亚铜,20-60℃进行Sonogashira偶联反应,反应4-24h,得到如通式(5)所示的共轭分子;
其中,所述有机溶剂可以为四氢呋喃、甲苯和二异丙胺、三乙胺、二乙基乙胺、乙二胺等有机碱的混合溶剂,体积比约为1:1-2:3,所述催化剂可以是四(三苯基膦)钯、醋酸钯、二氯二三苯基膦钯等。
iv)式(5)所示共轭分子溶于有机溶剂,加入还原剂和相当于还原剂2-10%摩尔比的六水合氯化钴,搅拌反应液,得到如通式(I)所示的一类基于荧光“双响应”机制的一氧化氮的探针分子,产率可达80%以上;
其中,所述有机溶剂是甲醇、乙醇、乙酸乙酯中的一种或其混合溶液,所述还原剂为硼氢化钠、氢化铝锂等。
通过上述制备方法(二)能够制得间隔段为炔键结构的探针分子材料,制备过程如“制备方法(二)”所示,其中,X1、X2为Cl、Br、I原子,Ar为芳基、取代芳基、杂环芳基或取代杂环芳基;所述的芳基或取代芳基是未取代或取代的苯、联苯、萘、苊、蒽、菲、芘、苝、芴或螺芴中的一种;所述杂环芳基或取代杂环芳基是未取代或取代的吡咯、吡啶、呋喃、噻吩、咔唑、硅芴、吲哚、噻唑、苯并二唑或苯并噻唑中的一种;所述芳基或杂环芳基的取代基可以是醚链、长度不超过6个碳原子的烷基链、磺酸基或季铵盐阳离子中的一种,取代芳基或取代杂环芳基的取代基个数为1-4个。
在上述所有制备方法中,制得的探针分子两端的芳香取代基团Ar可以在制备过程中或制备完成后通过取代反应或偶联反应进行更改。
此外,本发明还提供上述基于荧光“双响应”机制的一氧化氮的探针分子材料在生物检测领域的应用,所述生物监测的具体步骤如下:
A)将上述探针分子材料配成浓度高于10-6M稀溶液,溶剂可以是PBS混合缓冲液、去离子水或四氢呋喃、二甲亚砜、乙醇等构成混合溶剂;
B)将待测NO储备液加入探针分子材料溶液中,定容,其中探针分子材料测试中的最终浓度为10-6M;
C)测定在不同NO浓度存在下,探针分子材料的紫外可见吸收光谱、荧光发射光谱,根据共轭分子材料荧光光谱在短波和长波区域峰值变化,可对待测物进行检测;
通过对紫外吸收光谱中特定吸收峰强度变化值或荧光光谱中特定的发射峰强度变化的比率进行分析,确定NO的量。
本发明还提供上述基于荧光“双响应”机制的一氧化氮的探针分子材料在细胞成像领域的应用,所述细胞成像的具体步骤如下:
A)将探针分子或利用再沉淀法制备的具有探针分子的荧光纳米颗粒配制0.5-20μM的溶液;
B)将上述溶液与细胞在培养基中共培养30min到4h,通过流式细胞仪检测细胞中染色物质荧光光谱,并与探针分子与NO作用的光谱曲线对比,确认其中NO的比例。
本发明具有如下有益效果:1、设计一类具有灵敏度高、选择性好的探针分子材料,同时,还具备了响应时间短、膜透性好、背景噪音低、可直接观察等优点;2、本发明中所涵盖的共轭分子材料具有良好的发光性能,化学稳定性、热稳定性和光稳定性,是一类全新的传感检测平台,具有良好的应用前景;3、所述共轭分子材料,具有结构简单,分子结构简单,实验步骤少,易于合成与纯化等优点。
附图说明
图1.共轭分子材料B-1与不同量NO储备液作用后的紫外图谱。
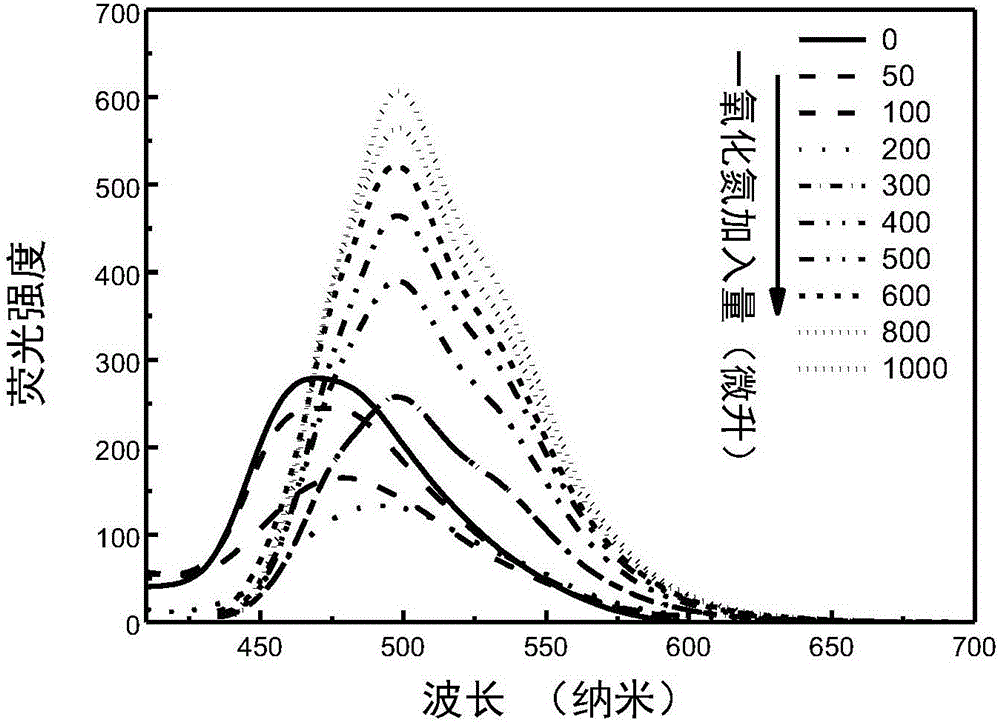
图2.共轭分子材料B-1与不同量NO储备液作用后的荧光图谱。
图3.共轭分子材料B-1与不同量NO储备液作用后,分别在420nm、362nm处吸收光谱强度变化拟合曲线图。
图4.共轭分子探针B-1应用与Hela细胞成像,与未加P1的共培养细胞对照图。
具体实施方式
为了更清楚的说明本发明的技术方案,下面结合具体实施例和附图进行进一步的说明。应当知道的是,以下实施例仅是对本发明的部分实施方式,而不是对本发明的限制。
实施例1:
本实施例制备的探针分子材料A-1的分子结构为:
本实施例1采用制备方法(一)制备得到,具体步骤为:
i)两口瓶(100mL)中,将4,7-二溴-2,1,3-苯并噻二唑(2.01g,6.84mmol),2-噻吩硼酸(1.42g,11mmol)溶解在20mL甲苯中,注入2M碳酸钾溶液15mL.抽换气三次,氮气保护下,加入四(三苯基膦)钯(150mg,0.14mmol),加热至75℃,反应2-8h。自然冷至室温,二氯甲烷萃取,干燥,浓缩,柱层析分离得到红色晶体4,7-二噻吩-2,1,3-苯并噻二唑1.73g,产率84.3%。1H NMR(400MHz,CDCl3)δ8.12(dd,J=3.7,1.1Hz,2H),7.88(s,2H),7.46(dd,J=5.1,1.1Hz,2H),7.24–7.20(m,2H).13C NMR(101MHz,CDCl3)δ152.61,139.36,128.02,127.51,126.81,125.75。
ii)在两口瓶(250ml)中,将3-溴丙酸叔丁基(6.7g,32mmol),二溴芴(2.9g,12mmol),碘化钾(200mg,1.2mmol)溶解在DMSO(100ml)和50%(w/w)NaOH水溶液(30ml)中。反应混合物在60℃下搅拌6h。二氯甲烷萃取,干燥,浓缩,柱层析分离得到淡黄色液体9,9-双(3,3'-二丙酸叔丁酯)-2-溴芴5.2g,直接用于下步。
在100mL的Schlenk管中,三甲基乙酸(33mg,0.32mmol),碳酸钾(400mg,2.9mmol),4,7-二噻吩-2,1,3-苯并噻二唑(340mg,1.13mmol),9,9-双(3,3'-二丙酸叔丁酯)-2-溴芴(1g,2mmol),溶解在35ml无水N,N-二甲基乙酰胺中,抽换气三次,氮气保护下,加入醋酸钯(60mg,0.27mmol),加热至100-130℃,反应6-12h。二氯甲烷萃取,干燥,浓缩,柱层析分离得到红色晶体TBBT-1 1.6g,产率70.2%.1H NMR(400MHz,CDCl3)δ8.13(d,J=3.7Hz,2H),8.08–8.07(m,1H),7.92(s,2H),7.72(s,8H),7.50(d,J=3.7Hz,2H),7.45–7.32(m,6H),2.50–2.38(m,8H),1.63–1.48(m,8H),1.31(s,36H).13C NMR(101MHz,CDCl3)δ172.80,152.58,149.04,148.35,145.83,140.98,140.66,138.61,133.52,128.62,127.87,127.68,125.72,125.43,125.30,124.18,123.13,120.50,120.10,120.07,80.19,34.72,28.03。
iii)在两口瓶(100ml)中,将TBBT-1(200mg,0.4mmol)溶解在乙酸乙酯(10ml)和甲醇(3ml)混合物中,慢慢加入硼氢化钠(10mg,0.26mmol)及适量的六水合氯化钴,反应混合物在0℃下搅拌2h。二氯甲烷萃取,干燥,浓缩,柱层析分离得到黄色晶体A-1 113.5mg,产率60.2%。1H NMR(400MHz,CDCl3)δ7.73–7.68(m,4H),7.64(dd,J=9.7,1.6Hz,4H),7.44(d,J=3.6Hz,2H),7.42–7.31(m,6H),7.24(d,J=3.7Hz,2H),7.02(s,2H),2.39(t,J=8.3Hz,8H),1.52(dd,J=18.4,8.5Hz,8H),1.30(s,36H).
实施例2:
本实施例制备的探针分子材料B-1的分子结构为:
本实施例2采用制备方法(一)制备得到,具体步骤为:
i)4,7-二噻吩-2,1,3-苯并噻二唑的制备过程与实施例1步骤i)相同。
ii)在两口瓶(250ml)中,将1-溴-2-(2-(2-甲氧基乙氧基)乙氧基)乙烷(7.3g,27.4mmol),二溴芴(2.9g,12mmol),碘化钾(200mg,1.2mmol)溶解在DMSO(100ml)和50%(w/w)NaOH水溶液(30ml)中。反应混合物在60。反下搅拌6h。二氯甲烷萃取,干燥,浓缩,柱层析分离得到淡黄色液体9,9-双(3,3'-2-(2-(2-甲氧基乙氧基)乙氧基)乙烷)-2-溴芴4.9g,产率79.1%。1H NMR(400MHz,CDCl3)δ7.65(dd,J=5.6,3.0Hz,1H),7.56–7.52(m,2H),7.46(dd,J=8.0,1.8Hz,1H),7.38(d,J=2.6Hz,1H),7.33(dd,J=6.2,2.8Hz,2H),3.55–3.45(m,8H),3.40(t,J=4.9Hz,4H),3.21(dd,J=8.3,4.7Hz,4H),2.74(d,J=7.0Hz,4H),2.39–2.32(m,4H).
在100mL的Schlenk管中,三甲基乙酸(33mg,0.32mmol),碳酸钾(400mg,2.9mmol),4,7-二噻吩-2,1,3-苯并噻二唑(340mg,1.13mmol),9,9-双(3,3'-2-(2-(2-甲氧基乙氧基)乙氧基)乙烷)-2-溴芴(1g,2mmol),溶解在35ml无水N,N-二甲基乙酰胺中,抽换气三次,氮气保护下,加入醋酸钯(60mg,0.27mmol),加热至105℃,反应6-12h。二氯甲烷萃取,干燥,浓缩,柱层析分离得到红色晶体TBBT-2 1.48g,产率68.9%.1H NMR(400MHz,CDCl3)δ8.17(d,J=3.9Hz,2H),7.96(s,2H),7.77–7.68(m,8H),7.51(d,J=3.9Hz,2H),7.46–7.42(m,2H),7.39–7.30(m,4H),3.54–3.49(m,8H),3.48–3.43(m,9H),3.43–3.39(m,8H),3.33–3.30(m,12H),3.26–3.21(m,8H),2.87–2.76(m,8H),2.51–2.42(m,8H).
iii)在两口瓶(100ml)中,将TBBT-2(200mg,0.4mmol)溶解在乙酸乙酯(10ml)和甲醇(3ml)混合物中,慢慢加入硼氢化钠(10mg,0.26mmol)及适量的六水合氯化钴,反应混合物在0℃下搅拌2h。二氯甲烷萃取,干燥,浓缩,柱层析分离得到墨绿色固体B-1 80.4mg,产率41.1%。
实施例3:
本实施例制备的探针分子材料C-1的分子结构为:
本实施例3采用制备方法(二)制备得到,具体步骤为:
i)两口瓶(100mL)中,将4,7-二溴-2,1,3-苯并噻二唑(2.01g-3.05,6.84-10mmol),三甲基硅乙炔(0.67g-1.0g,15-22.5mmol)溶解在20-40mL四氢呋喃中,注入15-30mL三乙胺。抽换气三次,氮气保护下,加入四(三苯基膦)钯(50-150mg,0.05-0.14mmol),50-80℃,反应4-24h。自然冷至室温,二氯甲烷萃取,干燥,浓缩,柱层析分离得到黄绿色晶体4,7-二三甲基硅乙炔基-2,1,3-苯并噻二唑,产率77-84.3%。1H NMR(400MHz,CDCl3)δ8.22(dd,J=3.7,1.1Hz,2H),0.14(m,18H).
ii)在两口瓶(150mL)中,将4,7-二三甲基硅乙炔基-2,1,3-苯并噻二唑溶解于四氢呋喃与甲醇的混合溶剂中,加入碳酸钾(或者氢氧化钾等碱),20-40℃氮气保护下搅拌2-12h。二氯甲烷萃取,干燥,浓缩,柱层析分离得到橙黄色晶体4,7-二乙炔基-2,1,3-苯并噻二唑,直接用于后续步骤。
在100mL的Schlenk管中,4,7-二乙炔基-2,1,3-苯并噻二唑(270mg,1.13mmol),2-碘-N-(三乙氧基甲基)咔唑(1.2g,2mmol)溶解在20-30mL四氢呋喃中,注入15-20mL三乙胺。抽换气三次,氮气保护下,加入四(三苯基膦)钯(50-150mg,0.05-0.14mmol),20-60℃,反应4-24h。自然冷至室温,二氯甲烷萃取,干燥,浓缩,柱层析分离得到橘红色粘液,产率85-95%。1H NMR(400MHz,CDCl3)δ8.13(d,J=3.7Hz,2H),7.96(s,2H),7.76(s,4H),7.42(d,J=3.7Hz,4H),7.45–7.32(m,4H),2.50–2.38(m,8H),1.63–1.48(m,8H),1.31(s,36H).13C NMR(101MHz,CDCl3)δ173.81,155.68,148.07,148.35,145.83,140.98,140.66,138.61,133.52,128.62,127.87,127.68,125.72,125.43,125.30,124.18,123.13,120.50,120.10,119.2,118.7,81.2,33.7,26.23。
iii)在两口瓶(100ml)中,将上述步骤橘红色粘液(165mg,0.4mmol)溶解在乙酸乙酯(10ml)和甲醇(3ml)混合物中,慢慢加入硼氢化钠(10-18mg)及适量的六水合氯化钴,反应混合物在0℃下搅拌2h。二氯甲烷萃取,干燥,浓缩,柱层析分离得到C-1纯品98.5mg,产率67%。1H NMR(400MHz,CDCl3)δ7.94(s,2H),7.75–7.68(m,4H),7.64(dd,J=9.7,1.6Hz,4H),7.76(s,4H),7.42(d,J=3.7Hz,4H),7.45–7.32(m,4H),2.50–2.38(m,8H),1.52(dd,J=18.4,8.5Hz,8H),1.30(s,36H).
实施例4:
本实施例制备的探针分子材料D-1的分子结构为:
本实施例4采用芳基与邻位二胺直接相连,具体步骤为:
i)在两口瓶(250ml)中,烷氧基取代的噻吩并吲哚的三丁基锡试剂(7.1g,22.4mmol),4,7-二溴-2,1,3-苯并噻二唑(2.01g-3.05,6.84-10mmol)溶解在甲苯(70ml)中,加入四(三苯基膦)钯(50-150mg,0.05-0.14mmol)。反应混合物在110-130℃反下4-12h。浓缩,柱层析分离得到深红色液体,产率75-85.1%。1H NMR(400MHz,CDCl3)δ7.65(dd,J=5.6,3.0Hz,1H),7.56–7.52(m,2H),7.46(dd,J=8.0,1.8Hz,1H),7.38(d,J=2.6Hz,1H),7.33(dd,J=6.2,2.8Hz,2H),3.55–3.45(m,8H),3.40(t,J=4.9Hz,4H),3.21(dd,J=8.3,4.7Hz,4H),2.74(d,J=7.0Hz,4H),2.39–2.32(m,4H).
ii)在两口瓶(100ml)中,将上述深红色样品(400mg,1.3mmol)溶解在乙酸乙酯(10ml)和甲醇(3ml)混合物中,慢慢加入硼氢化钠(30-50mg)及适量的六水合氯化钴,反应混合物在0-40℃下搅拌2-4h。二氯甲烷萃取,干燥,浓缩,柱层析分离得到橘红色蜡状固体D-1纯品325mg,产率83%。1H NMR(400MHz,CDCl3)δ7.94(s,2H),7.75–7.68(m,4H),7.64(dd,J=9.7,1.6Hz,4H),7.76(s,4H),7.42(d,J=3.7Hz,4H),7.45–7.32(m,4H),2.50–2.38(m,8H),1.52(dd,J=18.4,8.5Hz,8H),1.30(s,36H).
实施例5:
以下实施例是根据本发明的选材选择了其中一种共轭分子材料B-1(实施例2所述),应用于生物分子检测。
1)在DMSO和PBS混合缓冲溶液(1:5,v/v,PH=7.04)中配制3μM的B-1,测其紫外吸收光谱谱图。
2)在PBS混合缓冲溶液(pH=7.04)中加入B-1与不同量的NO饱和储备液(v/v=0-1),室温搅拌0.5-4h。
3)测定每组溶液的紫外、荧光图谱,并对吸收曲线进行拟合,如图1、图2和图3所示。
通过图1可知,随着NO储备液加入B-1溶液,原B-1在362纳米出的吸收峰逐渐减弱,同时,在420纳米出的吸收峰逐渐增强,且较B-1在362纳米处的吸收强度更强,表明新的共轭体系的形成以及新形成的共轭结构具有更强的电子离域程度。由图2可知,随着NO储备液的加入,B-1在466纳米处蓝色荧光逐渐减弱,同时,在500纳米处产生对应于420纳米新共轭结构的荧光峰,且其荧光强度更高。这种因为共轭结构变化导致的吸收光谱和荧光光谱位置、强度变化非常有利于本发明所述探针对NO的有效检测和分析。图3为利用362纳米和420纳米激发光分别激发B-1与一氧化氮储备液共混溶液后记录的466纳米和520纳米出荧光强度变化趋势,是对图1和图2的补充。
实施例6:
以下实施例是根据本发明的选材选择了其中一种共轭分子材料B-1,应用于细胞成像。
1)在DMSO和PBS混合缓冲溶液(1:5,v/v,PH=7.04)中配制3μM的B-1。
2)将上述溶液与细胞在培养基中共培养30min到2h,通过流式细胞仪检测细胞中染色物质荧光光谱,并与探针分子与NO作用的光谱曲线对比,确认其中NO的比例。
如图4所示,其中A和B是加入B-1在明场和360nm激发下,未加NO刺激剂,显然,这是,仅能观察到B-1的蓝色荧光(图4-B中亮的区域);C和D是加入B-1在明场和360nm激发下,加NO刺激剂,此时,因为NO在细胞中的释放,与进入细胞质的B-1发生作用,B-1的蓝色荧光减弱,产生黄绿色荧光;E和F是加入B-1在明场和360nm激发下明场和360nm激发下,加NO刺激剂及血红蛋白抑制剂,由于血红蛋白抑制剂的存在,细胞释放的NO优先于血红蛋白结合,因此,图4-F显示依然为B-1本身的蓝色荧光;G和H是未加入P1在明场和360nm激发下明场和360nm激发下空白对照。结合以上图片显示的信息,可知,本发明所述的NO探针可应用与细胞内NO的有效探测。
- 还没有人留言评论。精彩留言会获得点赞!