小干扰RNA及其应用和抑制plk1基因表达的方法与流程
技术领域:
本发明涉及一种siRNA及其应用和抑制plk1基因表达的方法。具体而言,本发明涉及一种抑制plk1基因表达的siRNA和应用,以及利用siRNA抑制plk1基因表达的方法。
背景技术:
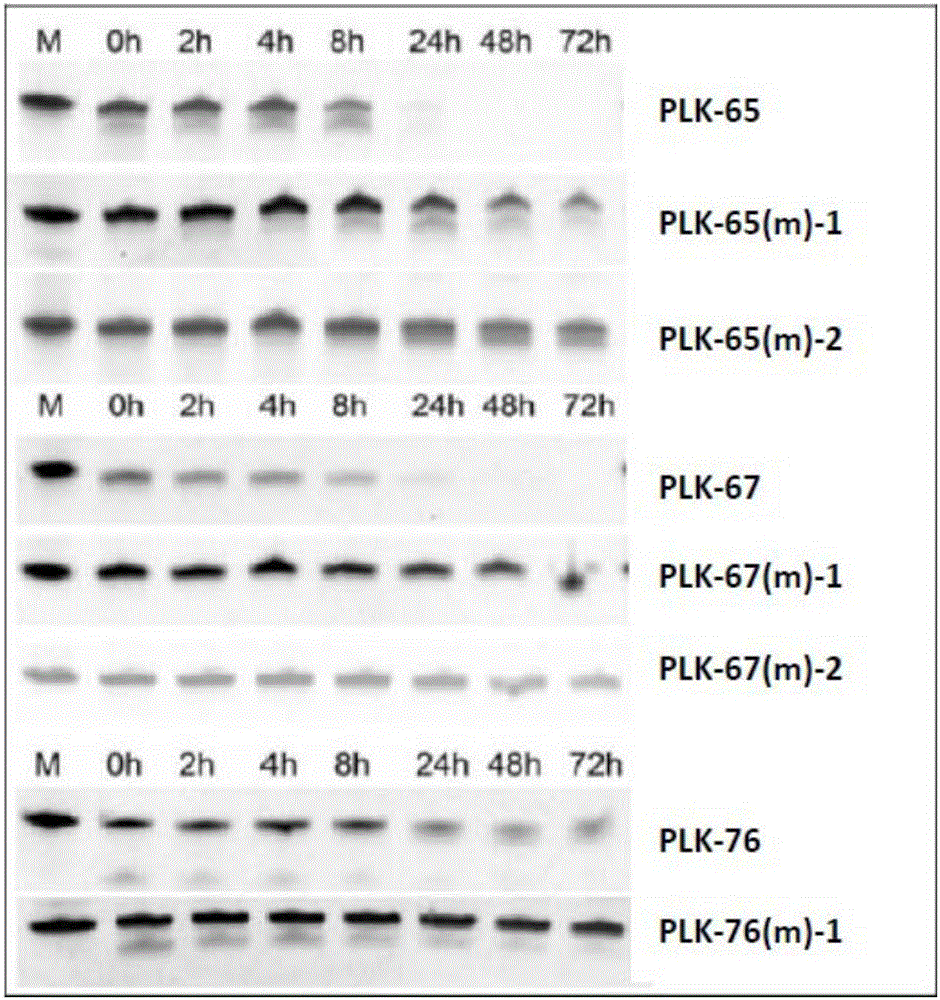
:Polo样激酶1(polo-likekinase-1,plk1)是一种高度保守的丝/苏氨酸激酶。人plk1基因定位于染色体的16p12位点,编码的mRNA长约2.3kb、蛋白质分子量约为67kd。plk1蛋白在其N端有一个高度保守的催化区域,在其C端通常具有3个被称作为Polo盒(polobox)的保守区域。研究发现,plk1有诱导DNA合成、DNA完整性的检修以及防止细胞凋亡方面的作用。plk1还能通过磷酸化p53而抑制其转活性,进而抑制p53发挥检验点(check-point)蛋白和诱导细胞凋亡的功能。p53是G1期主要的调控蛋白,plk1对抑癌基因p53的抑制作用诱导持续的、甚至是永久性的G1期阻滞。另外,plk1与肿瘤的发生与发展密切相关,plk1基因的表达被抑制之后,会抑制细胞的增殖、促进细胞的凋亡,进而抑制肿瘤生长。plk1还可以通过抑制MAVS调节干扰素IFN的诱导生成,破坏先天性免疫。plk1在包括乳腺癌、肝癌、肺癌和结肠癌的大多数人类肿瘤组织中均有高表达。plk1的高表达和肿瘤患者的生存率之间具有统计相关性,肿瘤组织中plk1的表达水平还与肿瘤的转移及预后密切相关。这说明plk1在肿瘤的生成和发展过程中可能发挥着重要的作用,并且是一个潜在的抗肿瘤药物作用靶点。研究进展还表明,阻断plk1的表达或抑制其激酶活性可以有效抑制肿瘤细胞增殖并介导其凋亡,但对正常细胞没有明显影响。目前处于临床前或临床试验阶段的多种plk1抑制剂都表现出了高药性、低毒性的特点。乳腺癌是女性最常见的恶性肿瘤之一。手术、放疗、化疗、内分泌治疗并列成为乳腺癌的四大临床处置手段。对于大多数乳腺癌患者来说,在初诊确定时癌细胞就有可能已迁移至其他组织,而化疗作为重要的全身性干预手段在乳腺癌治疗中占有极其重要的地位。目前主要的化疗手段是小分子药物和大分子靶向药物,但药效和随之产生的耐药性是乳腺癌治疗中常用的小分子药物面临的主要问题,而适用人群范围窄则是大分子靶向药物的面临的主要问题。因此,可抑制癌基因表达的小干扰核酸作为替代型药物有望解决小分子药物和靶向抗体药物不能解决的问题。从抗肿瘤药物提高治疗有效性、耐药性、降低毒副作用等几个方面讲,作为与目前临床使用的主流药物的作用机制完全不同的新兴药物分子,开发有效的siRNA药物已成为目前临床实际应用的迫切要求。技术实现要素:本发明的目的在于提供用于抑制plk1基因表达的siRNA、含有所述siRNA作为药物活性成分的药物组合物、利用所述siRNA或药物组合物抑制plk1基因表达的方法,以及所述siRNA或药物组合物在治疗和/或预防癌症疾病中的应用。即,本发明通过提供以下技术方案得以实现本发明的上述目的。一方面,本发明提供一种具有双链结构的siRNA,所述双链结构由完全互补的第一单链和第二单链组成,其中,所述第一单链具有与由SEQIDNO:1表示的plk1mRNA序列中的靶位点序列相同且由SEQIDNOs:2-133表示的核苷酸序列,与所述第一单链互补的所述第二单链具有与由SEQIDNO:1表示的plk1mRNA序列中的靶位点序列互补且由SEQIDNOs:134-265表示的核苷酸序列。根据本发明的一种实施方式,所述第一单链具有与由SEQIDNO:1表示的plk1mRNA序列中的靶作用位点序列相同且由SEQIDNOs:4、17、38、42、55、65、66、68、77、93、103、104、109或129表示的核苷酸序列,与所述第一单链互补的所述第二单链具有与由SEQIDNO:1表示的plk1mRNA序列中的靶作用位点序列互补且由SEQIDNOs:136、149、170、174、187、197、198、200、209、225、235、236、241或261表示的核苷酸序列。根据本发明一种优选的实施方式,所述第一单链具有与由SEQIDNO:1表示的plk1mRNA序列中的靶作用位点序列相同且由SEQIDNOs:66、68或77表示的核苷酸序列,与所述第一单链互补的所述第二单链具有与由SEQIDNO:1表示的plk1mRNA序列中的靶作用位点序列互补且由SEQIDNOs:198、200或209表示的核苷酸序列。根据本发明的另一种实施方式,所述第一单链和所述第二单链中至少一条单链的3′末端可以连接1至3个核苷酸,从而在所述第一单链和所述第二单链互补形成所述双链结构后,在所述双链结构的至少一个末端形成由所述1至3个核苷酸构成的3′突出端。其中,优选所述3′突出端为由连续的两个脱氧胸腺嘧啶核苷酸dTdT或连续的两个尿嘧啶核苷酸UU构成。根据本发明的另一种实施方式,所述第一单链和所述第二单链中分别包含至少一个被修饰的核苷酸基团。其中,所述被修饰的核苷酸基团为磷酸基团、核糖基团或碱基中的至少一种被修饰的核苷酸基团。优选所述被修饰的核苷酸基团为核糖基团的2′-羟基被甲氧基或氟取代的核苷酸基团。另一方面,本发明提供一种药物组合物,其中含有所述抑制plk1基因表达的siRNA作为药物活性成分,以及还含有阳离子组分、非阳离子组分和药学上可接受的载体。根据本发明中的一种实施方式,所述阳离子组分选自N,N-二羟乙基-N-甲基-N-2-(胆固醇氧羰基氨基)乙基溴化铵、(2,3-二油氧基丙基)三甲基氯化铵、N-(1-(2,3-二油酰氧基)丙基)-N,N,N-三甲基氯化铵、聚乙烯亚胺、聚β-氨基酯和壳聚糖季铵盐中的至少一种,所述非阳离子组分选自聚乙二醇-聚乳酸两嵌段共聚物、聚乙二醇-聚乳酸三嵌段共聚物、聚乙二醇-聚(乳酸-乙醇酸)两嵌段共聚物和聚乙二醇-聚(乳酸-乙醇酸)三嵌段共聚物中的至少一种,所述药学上可接受的载体选自pH值为4.0-9.0的磷酸盐缓冲液、pH值为7.5-8.5的三羟甲基胺基甲烷盐酸盐缓冲液、生理盐水或质量百分比浓度为7-15%的蔗糖溶液。另一方面,本发明提供一种抑制哺乳动物细胞内plk1基因表达的方法,该方法包括向哺乳动物细胞内导入如上所述的siRNA的处置,从而使所述siRNA能够序列特异性地诱导所述plk1基因表达的抑制。根据本发明的一种实施方式,所述处置是将所述siRNA直接地进行导入,或以如上所述的含有siRNA的药物组合物的形式进行导入。另一方面,本发明提供如上所述的siRNA、及药物组合物在制备治疗和/或预防肿瘤的药物中的应用。其中,所述肿瘤为plk1基因异常高表达的乳腺癌、肝癌、肺癌、宫颈癌或结肠癌。本发明提供的siRNA能够序列特异性地介导plk1基因表达的抑制,并具有很好的血清稳定性。通过将本发明的siRNA导入至肿瘤细胞内,可以有效地抑制内源性plk1基因的表达,从而抑制肿瘤细胞生长、促进肿瘤细胞凋亡。附图说明图1示出实施例2的siRNA抑制plk1mRNA表达水平的检测结果。图2示出实施例3的化学修饰前后的siRNA的血清稳定性的检测结果。图3示出实施例4的化学修饰前后的siRNA抑制plk1mRNA表达水平的检测结果。图4为实施例6的通过尾静脉注射方式系统给药含plk1siRNA的药物组合物时对乳腺癌细胞生长的抑制效果图。图5为实施例7的通过尾静脉注射方式系统给药含plk1siRNA的药物组合物时对宫颈癌细胞生长的抑制效果图。具体实施方式对于本说明书中使用的术语,其定义如下。术语“polo样激酶1”、“plk1”或“plk1激酶”意指一种含激酶区域(kinasedomain)及polo盒区域(polo-boxdomain)的丝氨酸/苏氨酸激酶。关于plk1激酶的性质与功能,详细描述可参见Nat.Rev.Mol.CellBiol.2004,5:429-。现已知plk1激酶的活性及细胞内表达水平对于细胞有丝分裂具有关键性调节作用。本发明中所使用的plk1mRNA序列为Genebank注册号为NM_005030.3的序列(SEQIDNO:1)。术语“mRNA(messengerRNA,信使RNA)”意指作为体内蛋白质翻译的模板将基因的编码信息由DNA传导至蛋白质产物的RNA分子。术语“RNA干扰(RNAinterference)”或“RNAi”指一种存在生物体内的转录后基因表达调控现象,该现象由单链或双链RNA介导的靶mRNA特异性降解而引起。关于RNAi调控机制的细节可参见Biotech.Adv.2008,26(3):202-等文献的描述。本发明中,如无特殊说明,术语“小干扰RNA(smallinterference/small-interferingRNA)”、或“siRNA”意指能够序列特异性地诱导RNAi现象、且由两条长度为15-27个核苷酸的单链RNA组成、及具有部分或完全互补的双链结构的RNA分子。本发明所涉及的siRNA中,互补双链结构的长度可以为17-25个、18-22个、或19-21个碱基对。本发明所涉及的siRNA可以是由两条长度为15-27个核苷酸的单链RNA组成的平末端的双链RNA结构,或也可以是在双链结构的至少一个末端具有由1-3个连续的核苷酸组成的3`突出端的结构。本发明中,作为所述3`突出端,优选由两个连续的脱氧胸腺嘧啶核苷酸dTdT、或由两个连续的尿嘧啶核苷酸UU组成。本发明中,如无特殊说明,术语“第一单链”、或“正义链”是指siRNA两条单链中的一条单链,该单链具有与靶mRNA中的该siRNA作用位点核苷酸序列部分或完全相同的核苷酸序列;术语“第二单链”、或“反义链”则指在siRNA两条单链中的另一条单链,该单链具有与靶mRNA中的该siRNA作用位点核苷酸序列部分或完全互补的核苷酸序列。本发明中提到siRNA的第一单链(或正义链)可与相应的第二单链(或反义链)形成部分或完全互补的双链结构。术语“互补”意指两条核酸链的碱基根据鸟嘌呤G与胞嘧啶C、腺嘌呤A与尿嘧啶U/胸腺嘧啶T碱基配对原则形成反平行互补配对的情况。本发明中,如无特殊说明,术语“抑制(suppress/suppressing,inhibit/inhibiting)”意指由于siRNA、或其他小干扰核酸(small-interferingnucleicacid,siNA)抑制剂介导的mRNA降解而使靶基因表达得以显著下调(down-regulation)的情况。所述“显著下调”指靶基因表达相对于正常或处理前水平下降5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%或99%以上或100%的情况。术语“系统给药”、或“系统输送”意指将siRNA等药物活性成分输送至体内广泛组织区域的一种给药方式。为了达到系统给药的效果,通常需要药物活性成分、或药物组合物具有较长的血液滞留时间,且不易被肝、肾等主要代谢器官吸收清除。作为siRNA的系统给药方式,可采用静脉注射、皮下注射、腹腔内注射、或口服等方式。本发明中,优选采用静脉注射方式进行siRNA的系统给药。术语“局部给药”、“局部输送”意指将siRNA等药物活性成分输送至局部组织区域的一种给药方式。例如,可以通过将siRNA等药物活性成分直接注射或涂覆在病变组织区域中来实现siRNA的局部给药。作为适于siRNA等药物活性成分局部给药的组织区域,例如为皮肤、眼部玻璃体腔、肝、肾、肺等器官组织。本发明中,siRNA的设计是以plk1的mRNA序列为模板,针对plk1基因的保守区选取15-27个核苷酸长度的靶序列,从而得到相应的siRNA。本发明中使用的plk1mRNA序列为Genebank注册号为NM_005030.3的序列(SEQIDNO:1),其长度为2204个核苷酸,编码区自54位的起始密码子ATG起始至1865位的终止密码子TAA为止。具体来说,本发明的siRNA的设计按以下原则进行。首先,在plk1mRNA的全长序列范围内选取15-27个核苷酸长度的序列。15-27个核苷酸长度的序列的选取主要参考以下几项原则:1)GC含量在35-60%之间,2)避免处于重复序列或低复杂性序列区域内,3)避免出现4个以上的连续核苷酸序列,4)避免处于包含读码框起始密码和终止密码的50-100个核苷酸序列区域之内。除此之外,还要分析核苷酸序列的组成和热力学性质,确保进入体内后双链容易解链,并避免免疫反应的发生。随后,通过BLAST分析,将候选siRNA的靶位点序列同人类基因组序列进行同一性比对,排除同其它基因具有16个核苷酸长度以上同一性的序列,以确保候选siRNA的靶位点序列不与其他非相关基因的序列之间具有高相似性,从而保证设计的siRNA仅对靶基因plk1具有特异的抑制作用。本发明中,siRNA的设计包括与plk1mRNA中的靶作用位点相同的第一单链的设计、以及与plk1mRNA中的靶作用位点互补的第二单链的设计。本发明中,siRNA的第二单链(或反义链)与plk1mRNA序列中的靶作用位点序列互补,并通过RNAi机制序列特异性地诱导plk1mRNA的降解,从而对plk1基因表达形成抑制。本发明所设计的第一单链和第二单链彼此完全互补,经退火可形成平末端的,即,不具有3`突出端的双链结构。本发明的一个实施方式中,在根据上述原则设计的15-27个核苷酸长度的一条RNA单链的3′末端加上两个脱氧胸腺嘧啶核苷酸dTdT,互补的另一条RNA单链同样也做3′末端加上两个脱氧胸腺嘧啶核苷酸dTdT的处理。这样,两条互补的RNA单链退火形成双链结构后,在所述双链结构的两端可分别形成由两个脱氧胸腺嘧啶核苷酸dTdT构成的3′突出端。本发明的另一个实施方式中,只在siRNA的一条RNA单链的末端加上两个脱氧胸腺嘧啶核苷酸dTdT,由此,两条互补的RNA单链退火形成双链结构后,在所述双链结构的一个末端形成由两个脱氧胸腺嘧啶核苷酸dTdT构成的3′突出端。本发明的siRNA的一条RNA单链可以通过固相或液相核酸合成方法进行合成。这些方法包括四个工艺步骤。即,1)寡聚的合成、2)脱保护、3)纯化分离、及4)脱盐的四个工艺步骤。这四个步骤中的技术细节为本领域技术人员所公知,在此不再赘述。除化学合成外,本发明的siRNA也可通过质粒和/或病毒载体的表达而得到。例如,设计一个长度为50-90个核苷酸的DNA序列,并在其两端加上两个不同的限制酶切位点,例如加上BamHI和EcoRI的酶切位点。由所设计的DNA编码的RNA转录产物具有中间一段序列可形成环(loop)结构,经U字折返(U-turn)后的环两端的序列可形成互补配对的双链结构。通过克隆技术将所设计的DNA插入到用相应限制酶酶切过的表达载体中。将表达载体导入至细胞中,由所设计的DNA序列生成的RNA转录产物可被细胞固有的siRNA加工机制加工成成熟的siRNA。由此,即可在细胞中一时或稳定地表达siRNA。本发明对siRNA进行的化学修饰可以是以下一种或一种以上化学修饰的组合:1)对RNA链骨架结构中连接核苷酸残基的磷酸二酯键的修饰,2)对RNA链骨架结构中核糖的修饰,3)对RNA的核苷酸残基中碱基的修饰。例如,本发明提到的磷酸基团的修饰是指对磷酸基团中的氧进行修饰,包括硫代磷酸修饰(Phosphorthioate)和硼烷化磷酸盐修饰(Boranophosphate)。如下式所示分别用硫和硼烷基置换磷酸基团中的氧,两种修饰都能稳定核酸的结构,保持碱基配对的高特异性和高亲和力。本发明中,核糖基团的修饰是指对核糖基团中2′-羟基(2′-OH)的修饰。在核糖基团的2′-羟基位置引入某些取代基如甲氧基或氟后,使血清中的核糖核酸酶不易切割核酸,由此增加了核酸的稳定性,使核酸具有更强的抵抗核酸酶水解的性能。对核苷酸戊糖中2′-羟基的修饰包括2′-氟修饰(2′-fluromodification)、2′-甲氧基修饰(2′-methoxymodification)、2′-甲氧乙氧基修饰(2'-methoxyethoxymodification)、2′-2,4-二硝基苯酚修饰(2′-DNPmodification)、锁核酸修饰(Lockednucleicacidmodification)、2′-氨基修饰(2′-Aminomodification)、2′-脱氧修饰(2′-deoxymodification)等。本发明中,碱基的修饰是指对核苷酸基团中的碱基进行修饰,如在尿嘧啶的5’位点引入溴或碘的5′-溴尿嘧啶(5′-bromo-uracil)和5′-碘尿嘧啶(5′-iodo-uracil)修饰是常使用的碱基修饰方法,其他还有N3-甲基脲嘧啶(N3-methyl-uracil)修饰、2,6-二氨基嘌呤(2,6-diaminopurine)修饰等。本发明中,核糖基团被修饰的核苷酸基团优选为核糖基团的2′-羟基被甲氧基或氟取代的核苷酸基团。修饰后的siRNA抵抗核酸酶酶解的能力增强,但其抑制plk1基因表达的活性不会因修饰产生明显变化。在本发明的一个实施方式中,为了促进siRNA的脂溶性,可以在siRNA的正义链的5′或3′末端引入如胆固醇、脂蛋白、维生素E、脂肪链等亲脂性基团,这些亲脂性基团可以通过共价键与siRNA结合。所述亲脂性基团也可通过非共价键与siRNA结合,例如,siRNA通过疏水键或离子键与中性磷脂分子、多肽、多糖等结合。已知在siRNA上通过共价结合或非共价结合引入亲脂性基团可以增加siRNA的体内稳定性、血液代谢特性和生物学活性。在本发明的一个实施方式中,siRNA的第一单链(或正义链)的5′末端和/或3′末端连接有5′-和/或3′-帽子(5`-and/or3′-cap)。所述5′-和/或3′-帽子结构可以使siRNA抵御核酸外切酶的攻击,进而提高siRNA的体内稳定性。作为所述5′-和/或3′-帽子,可以列举但不限于甘油、无碱基反向脱氧异核苷(inverteddeoxyabasicmoiety)、4′,5′-亚乙基核苷(4′,5′-methylenenucleotide)等。在本发明的一个实施方式中,siRNA的第二单链(或反义链)的5′末端连接有磷酸基团。已知siRNA的反义链的5′端磷酸基团可以提高siRNA的活性。本发明中,所述抑制plk1基因表达的siRNA可与协助药物体内输送的载体体系形成药物组合物,并以药物组合物形式在哺乳动物体内施用。所述载体体系包括但不限于阳离子组分、非阳离子组分、以及药学上可接受的载体。本发明中,所述阳离子组分可以是但不限于带正电的多肽或蛋白质、阳离子脂质、带正电的聚合物等。作为所述带正电的多肽或蛋白质,可列举寡聚精氨酸、寡聚赖氨酸、鱼精蛋白等。作为所述阳离子脂质,可以是选自二甲基二(十八烷基)溴化铵盐(DDAB)、1,2-二肉豆蔻酰基-3-三甲基铵丙烷、1,2-二油酰基-3-三甲基铵丙烷(DOTAP)、1,2-二油酰基-3-三甲基铵丙烷甲基硫酸盐、1,2-二棕榈酰基-3-三甲基铵丙烷、1,2-二硬脂酰基-3-三甲基铵丙烷、N-(1-(2,3-二油酰氧基)丙基)-N,N,N-三甲基氯化铵(DOTMA)、二肉豆蔻酰基氧基丙基二甲基羟基乙基溴化铵盐(DMRIE)、二油酰氧基丙基二甲基羟基乙基溴化铵(DORIE)、二甲基二(十二烷基)溴化铵、N-(a-三甲基铵基乙酰基)-二(十二烷基)-D-谷氨酰胺盐酸盐、N-(a-三甲基铵基乙酰基)-O,O’-双-(1H,1H,2H,2H-全氟癸烷基)-L-谷氨酰胺盐酸盐、O,O’-二(十二烷酰基)-N-(a-三甲基铵基乙酰基)二乙醇胺盐酸盐、甲基烯丙基二(十二烷基)溴化铵、N-{p-(w-三甲基铵基丁基氧基)-苯甲酰基}-二(十二烷基)-L-谷氨酰胺盐酸盐、9-(w-三甲基铵基丁基)-3,6-双(十二烷酰基)咔唑溴化物、二甲基二(十八烷基)铵盐酸盐、N-w-三甲基铵基癸酰基-二(十六烷基)-D-谷氨酰胺溴化物、N-{p-(w-三甲基铵基己基氧基)-苯甲酰基}-二(十四烷基)-L-谷氨酰胺溴化物、p-(w-三甲基铵基癸基氧基)-p’-辛氧基偶氮苯溴化物盐(MC-1-0810)、p-{w-(b-羟基乙基)二甲基-铵基-癸基氧基}-p’-辛氧基偶氮苯溴化物盐(MC-3-0810)、O,O’,O”-三(十二烷酰基)-N-(w-三甲基-铵基癸酰基)-三(羟基甲基)氨基甲烷澳化物盐(TC-1-12)、1,2-二月桂基-甘油-3-乙基磷酸胆碱、1,2-二肉豆蔻酰基-甘油-3-乙基磷酸胆碱、1,2-二棕榈酰基-甘油-3-乙基磷酸胆碱、1,2-二硬脂酰基-甘油-3-乙基磷酸胆碱、1,2-二油酰基-甘油-3-乙基磷酸胆碱、1-棕榈酰基-2-油酰基-甘油-3-乙基磷酸胆碱、N,N-二羟乙基-N-甲基-N-2-(胆固醇氧羰基氨基)乙基溴化铵(BHEM-Chol)、(2,3-二油氧基丙基)三甲基氯化铵(DOTAP)和N-(1-(2,3-二油酰氧基)丙基)-N,N,N-三甲基氯化铵中的至少一种阳离子脂质。作为所述带正电的聚合物,可以是选自聚乙烯亚胺、聚β-氨基酯、壳聚糖季铵盐中的至少一种正电性聚合物。本发明中,优选N,N-二羟乙基-N-甲基-N-2-(胆固醇氧羰基氨基)乙基溴化铵、(2,3-二油氧基丙基)三甲基氯化铵、N-(1-(2,3-二油酰氧基)丙基)-N,N,N-三甲基氯化铵、或聚己内酯-聚(N,N-二甲氨乙基甲基丙烯酸酯)嵌段共聚物类聚β-氨基酯作为阳离子组分。本发明所述的药物组合物中,所述非阳离子组分可以是但不限于中性的膜融合脂质(fusogeniclipid)、阴离子脂质、两亲性聚合物等。作为所述膜融合脂质,可以列举二油酰基磷脂酰乙醇胺、二油酰基磷脂酰胆碱、反式磷脂酰基乙醇胺、1,2-双(10,12-二十三烷二酰基)-磷酸乙醇胺、1,2-二反油酰基磷酸乙醇胺、1,2-二(十六烷基)磷酸乙醇胺、1,2-二己酰基磷酸乙醇胺、1,2-二月桂酰基磷酸乙醇胺、1,2-二亚油酰基磷酸乙醇胺、1,2-二肉豆蔻酰基磷酸乙醇胺、1,2-二油酰基磷酸乙醇胺、1,2-二棕榈油酰基磷酸乙醇胺、1,2-二棕榈酰基磷酸乙醇胺、1,2-二植烷酰磷酸乙醇胺、1,2-二硬脂酰基磷酸乙醇胺、1-棕榈酰基-2-油酰基磷酸乙醇胺、1-棕榈酰基-2-(10,12-二十三烷二酰基)磷酸乙醇胺、1,2-二油酰基磷酸乙醇胺-N-己酰胺、1,2-二棕榈酰基磷酸乙醇胺-N-己酰胺、N,N-二甲基-1,2-二油酰基磷酸乙醇胺、N,N-二甲基-1,2-二棕榈酰基磷酸乙醇胺、N-十二烷酰基-1,2-二棕榈酰基磷酸乙醇胺、N-十二烷酰基-1,2-二油酰基磷酸乙醇胺、1,2-二油酰基磷酸乙醇胺-N-十二烷基胺、1,2-二棕榈酰基磷酸乙醇胺-N-十二烷基胺、1,2-二油酰基磷酸乙醇胺-N-戊二酰、1,2-二棕榈酰基磷酸乙醇胺-N-戊二酰、1,2-二油酰基磷酸乙醇胺-N-乳糖、1,2-二油酰基磷酸乙醇胺-N-[4(p-马来酰亚胺甲基)环己烷-羧酸盐]、二棕榈酰磷酸乙醇胺-N-[4-(p-马来酸亚胺甲基)环己烷-羧酸盐]、1,2-二棕榈酰基磷酸乙醇胺-N-[4-(p-马来酰亚胺苯基)丁酰胺]、1,2-二油酰基磷酸乙醇胺-N-[4-(p-马来酰亚胺苯基)丁酸盐]、N-甲基-1,2-二油酰基磷酸乙醇胺、N-甲基-二棕榈酰基磷酸乙醇胺、1,2-二油酰基磷酸乙醇胺-N-[3-(2-吡啶二硫)丙酸盐、1,2-二棕榈酰基磷酸乙醇胺-N-[3-(2-吡啶二硫)丙酸盐]、N-(琥珀酰)-1,2-二油酰基磷酸乙醇胺、N-(琥珀酰)-1,2-二棕榈酰基磷酸乙醇胺等。本发明的药物组合物中,通过含有上述膜融合脂质,能够进一步提高所述药物组合物在哺乳动物体内的转运及输送效率。作为所述两亲性聚合物,可以列举聚乙二醇-聚乳酸两嵌段共聚物、聚乙二醇-聚乳酸三嵌段共聚物、聚乙二醇-聚(乳酸-乙醇酸)两嵌段共聚物、或聚乙二醇-聚(乳酸-乙醇酸)三嵌段共聚物、聚己内酯-聚磷酸酯两嵌段共聚物、聚己内酯-聚磷酸酯三嵌段共聚物、聚乙二醇-聚己内酯两嵌段共聚物、聚乙二醇-聚己内酯三嵌段共聚物等。其中,在本发明的药物组合物中优选使用二油酰基磷脂酰乙醇胺、或聚乙二醇-聚乳酸嵌段共聚物作为非阳离子组分。本发明的药物组合物中,所述药学上可接受的载体可以是pH值为4.0-9.0的磷酸盐缓冲液、pH值为7.5-8.5的三羟甲基胺基甲烷盐酸盐缓冲液、生理盐水、或7-15%的蔗糖溶液,其中,优选使用pH值为4.0-9.0的磷酸缓冲液作为本发明的药学上可接受的载体。本发明的药物组合物中还可以含有保护剂和/或渗透压调节剂。保护剂选自肌醇、山梨醇和蔗糖中的一种或几种,渗透压调节剂可以是氯化钠和/或氯化钾。以注射用液体制剂形式的药物组合物为例,所述保护剂的含量可以是0.01-30重量%。对于渗透压调节剂的含量没有特殊限定,只要可保持液体制剂的渗透压为200-700毫渗摩尔/千克即可。向动物或人类个体施用本发明的液体制剂形式的药物组合物时,其剂量可以为本领域常用的剂量。例如,单次注射的剂量可以是在1-10g/kg体重的范围内。具体使用时,剂量选择可以由各种参数、尤其根据待动物或人类个体的年龄、体重和症状等来确定。本发明中,以所述siRNA作为活性成分的药物组合物中还可以包含可增强药物组合物的稳定性、维持和增强siRNA的抑制效果、可促进药物组合物的代谢特性及组织靶向性的辅助组分。作为辅助组分,可列举但不限于胆固醇、多肽、蛋白质、多糖、脂肪链、中性磷脂、及聚乙二醇-脂质(PEG-lipid)中的一种或几种。对于这些辅助组分在本发明的药物组合物中的用量没有特别的限制,只要能够增强药物组合物的稳定性、血液代谢性能、靶向输送效果即可。根据本发明提供的抑制哺乳动物细胞内plk1基因表达的方法,该方法包括向哺乳动物细胞内导入上述提到的抑制plk1基因表达的siRNA,从而使导入的siRNA能够序列特异性地诱导所述plk1基因表达的抑制。本发明中,在体外将siRNA导入到细胞内时可以采用已知方法进行。常用的体外siRNA导入方法如电穿孔法、显微注射法、磷酸钙法、DEAE-葡聚糖法、病毒包裹法及脂质体包裹法等。其中,脂质体包裹法已成为siRNA的常规体外导入方法。本发明中,作为脂质体可利用市售的阳离子脂质体,例如利用Lipofectamin2000(Invitrogen公司制)、Oligofectamine(Invitrogen公司制)及Tfx50(Promega公司制)等。对于siRNA和阳离子脂质体的配合比率没有特殊限定,只要可有效地进行导入且不对细胞显示剂量毒性即可,例如,相对于100重量份的小干扰RNA,阳离子脂质体的含量可以为100-10000000重量份。本发明中,将siRNA导入到哺乳动物体内细胞时的方法有两种。一是将裸的siRNA(nakedsiRNA)直接导入到易于到达的皮肤组织细胞、眼部组织细胞、肺部组织细胞中等;二是以本发明的药物组合物形式进行siRNA系统输送。以下,结合实施例进一步说明本发明。除非特别说明,本发明所用到的试剂、培养基等实验材料均为市售商品。实施例1:siRNA的设计与合成针对人plk1的mRNA序列(Genebank注册号:NM_005030.3,SEQIDNO:1)并根据上述提到的设计原则得到了132个siRNA,得到的siRNA的序列示于表1。其中,127条siRNA(PLK-1~PLK-127)分布于plk1基因的编码区,最后5条siRNA(PLK-128~PLK-132)分布于plk1基因的3’端非翻译区。表1中,对于一个siRNA,分别示出其正义链、及互补的反义链序列,具体如,siRNAPLK-1的正义链具有由SEQIDNO:2表示的序列,且与plk1mRNA序列中相应的靶位点序列相同,反义链具有由SEQIDNO:134表示的序列,与plk1mRNA序列中相应的靶位点序列互补;其他siRNA的两条单链序列也如同siRNAPLK-1依次对各自的两条单链进行序列编号。表1:针对人mRNA的siRNA序列对于上述132个siRNA,进一步依据物种同源性分析得到12个人-小鼠同源的siRNA序列,结果示于表2;同样,得到11个人-大鼠同源的siRNA序列、105个人-猕猴同源的siRNA序列、119个人-黑猩猩同源的siRNA序列,这些结果分别示于表3-表5中。表2:人-小鼠同源的siRNA序列表3:人-大鼠同源的siRNA序列表4:人-猕猴同源的siRNA序列表5:人-黑猩猩同源的siRNA序列对于上述经由设计得到的132个siRNA进一步进行优选。此时,考虑点包括以下六个方面。即,1)需使所选siRNA的作用靶点均匀分布在mRNA全长序列中,2)避免作用靶点位于mRNA复杂的二级或三级结构域中,3)siRNA的作用靶点序列位于plk1mRNA的编码区。plk1mRNA的5′端非编码区只有53个核苷酸长度,这个区域的序列可能会被结合的翻译调控蛋白和核糖体亚基等封闭,因此并不适合于作为siRNA的作用靶点。对于编码区和3′端非编码区的靶点,则应优先考虑位于编码区的靶点,尤其需避免靶点位于3′端非编码区中接近poly(A)尾的低复杂性且长度易变区域,4)优先选择第一单链中第1个碱基为G/C,第13个碱基不是G,第19个碱基是A而不是G的序列,5)优先选择第一单链中第3个碱基是A,第10个碱基是U的序列,6)避免含有单核苷酸多态性位点。基于以上考虑,从132个设计的siRNA序列中最终优选出14个siRNA序列,结果示于表6。siRNA的寡聚核苷酸单链按照本领域公知的方法进行化学合成。合成的寡聚核苷酸的序列示于表6。合成时,在寡聚核苷酸单链的3′末端加上两个脱氧胸腺嘧啶核苷酸dTdT(表6中,用下划线标记)。互补的寡聚核苷酸单链退火形成双链RNA,双链结构的两端分别具有dTdT的3′突出端。表6实施例2:siRNA对plk1基因表达的抑制效果验证(siRNA的转染)将人类肝癌细胞株HepG2用含10%胎牛血清、2mML-谷胺酰胺、100U/ml青霉素、100g/ml链霉素的DMEM完全培养基接种于24孔板,细胞密度为4×105细胞/孔,每孔0.5ml,37℃培养过夜。转染的具体操作步骤如下。将100ng实施例1中合成的14个siRNAPLK-3、16、37、41、54、64、65、67、76、92、102、103、108和128分别稀释于50μlDEME无血清培养基中,同时将1μlLipofectamineTM2000(Invitrogen公司制)稀释于50μlDEME无血清培养基中,将上述两种溶液分别在室温下孵育5分钟后,均匀混合。混合溶液于室温静置20分钟后,把100μl的上述混合溶液加入到接种有HepG2细胞的24孔板中。siRNA的最终浓度约为10nM。细胞于37℃培养4小时,再加入1ml含10%胎牛血清、2mML-谷胺酰胺、100U/ml青霉素、100g/ml链霉素的DMEM完全培养基,然后在37℃再培养24小时。作为阴性对照,同时转染正义链为5′-UUCUCCGAACGUGUCACGUdTdT-3′、互补的反义链为5′-ACGUGACACGUUCGGAGAAdTdT-3′的阴性对照siRNA(N.C.siRNA)。(siRNA对plk1mRNA表达水平的抑制效果)通过荧光定量实时PCR(QuantitativeReal-TimePCR,qRT-PCR)检测分别转染了siRNAPLK-3、16、37、41、54、64、65、67、76、92、102、103、108和128的HepG2细胞中plk1mRNA的表达量,具体步骤如下。即,培养转染的细胞24小时后,用RNeasyminiKit(Qiagen公司制)提取细胞中的总RNA。用紫外分光光度计测定提取的RNA样品的OD280和OD260的吸光度,并利用公式:RNA浓度(μg/μL)=0.04×OD260×稀释倍数,计算RNA样品的浓度。然后用PrimeScriptTM1stStrandcDNASynthesisKit(Takara公司制)合成cDNA,每个样品使用2μg通过上述步骤提取的总RNA。在合成cDNA后,利用PremixExTaqTM(Takara公司制)试剂盒进行荧光定量实时PCR反应。其中,用于扩增plk1和作为定量PCR反应的内参的β-actin的PCR扩增引物如表7所示。表7上游引物下游引物plk15′-GCCCCTCACAGTCCTCAATA-3′5′-TACCCAAGGCCGTACTTGTC-3′β-actin5′-AGCGAGCATCCCCCAAAGTT-3′5′-GGGCACGAAGGCTCATCATT-3′siRNA对plk1mRNA表达水平的抑制率按如下等式计算。抑制率=[1-(实验孔plk1mRNA的表达量/实验孔β-ActinmRNA的表达量)/(阴性对照孔plkmRNA的表达量/阴性对照孔β-ActinmRNA的表达量)]×100%。结果如图1所示。从图1可以看出,本发明的siRNA均具有抑制plk1mRNA表达水平的效果。其中,siRNAPLK-65、siPLK-67和siPLK-76对plk1mRNA表达水平的抑制率均高于60%,分别为64%、68%和78%,这说明本发明的siRNA对plk1基因表达具有抑制活性,可用于抑制plk1基因的表达。制备例1委托苏州瑞博生物技术有限公司合成表8中所列的寡聚核苷酸。这些寡聚核苷酸中包含修饰的核苷酸基团,互补的寡聚核苷酸链退火形成修饰的siRNA,分别标记为PLK(m)-65-1、PLK(m)-65-2、PLK(m)-67-1、PLK(m)-67-2和PLK(m)-76-1。其中,(OMe)代表它左边的核苷酸残基中戊糖基团的2′羟基被甲氧基取代,(F)代表它左边的核苷酸残基中戊糖基团的2′羟基被氟取代。这些siRNA未加修饰前的核苷酸序列分别对应于实施例1中的PLK-65、PLK-67和PLK-76。表8实施例3:化学修饰对小干RNA血清稳定性的影响的评价对制备例1中得到的PLK(m)-65-1、PLK(m)-65-2、PLK(m)-67-1、PLK(m)-67-2和PLK(m)-76-1,以及实施例1中得到的PLK-65、PLK-67和PLK-76测定其在血清环境中的稳定性。具体步骤如下。将10μl上述修饰和未修饰的siRNA(20μmol)分别与50μl胎牛血清(FBS,购自HyClone,货号GTB0060)和40μlPBS混合后,在37℃下孵育0、2、4、8、24、48和72小时后得到处理样品。处理样品取样10μl进行20%的PAGE凝胶电泳。由上述处理样品的电泳条带光强度与0小时的电泳条带光强度的比值计算降解率,结果如图2及表9所示。表9中列出的降解率为由72小时的电泳条带光强度与0小时的电泳条带光强度的比值算出的降解率。由图2及表9可以看出,在血清环境中,修饰的siRNA的稳定性相比未修饰的siRNA的稳定性明显提高。表9实施例4:化学修饰前后siRNA对plk1mRNA表达水平的抑制效果验证按照与实施例2相同的方法,对制备例1中得到的siPLK(m)-65-1、siPLK(m)-67-1和siPLK(m)-76-1,以及实施例1中得到的siPLK-65、PLK-67和PLK-76分别测定其对plk1mRNA表达水平的抑制效果。转染时,对于上述各siRNA采用梯度剂量进行转染,使其终浓度分别为0.1nM、1nM和10nM。作为阴性对照,采用与实施例2中相同的正义链为5′-UUCUCCGAACGUGUCACGUdTdT-3′、互补的反义链为5′-ACGUGACACGUUCGGAGAAdTdT-3′的阴性对照siRNA(N.C.siRNA)。荧光定量实时PCR测定结果如图3所示。由图3可知,修饰的siRNA对于plk1mRNA表达量具有与未修饰的siRNA类似的抑制效果。采用10nM剂量时,PLK(m)-65-1、PLK(m)-67-1相比对应的未修饰的siRNAPLK-65、PLK-67具有更为优异的抑制效果。显然,这是由于所述修饰增强了siRNA的稳定性,并由此加长了siRNA在细胞内存留时间的缘故。实施例5:局部给药siRNA对于乳腺癌细胞生长的抑制在BALB/c裸鼠第二个乳腺的脂肪垫下原位接种人乳腺癌细胞株MDA-MB-435s(5×106个细胞/每只裸鼠),14天左右后形成可见肿瘤。肿瘤的体积按以下公式计算:V=0.5×a×b2,其中,a指肿瘤长径,b指肿瘤短径。每5只接种的裸鼠作为一组,每组裸鼠的肿瘤体积平均约为50mm3。作为处理组,将10μgsiRNAPLK-65、PLK(m)-65-1、PLK(m)-65-2、PLK-67、PLK(m)-67-1、PLK(m)-67-2、PLK-76和PLK(m)-76-1分别溶于100μl的磷酸盐缓冲液(pH值为7.4)中,并直接进行瘤内注射。作为阴性对照组,采用实施例2中提到的阴性对照siRNA(N.C.siRNA,正义链为5′-UUCUCCGAACGUGUCACGUdTdT-3′、互补的反义链为5′-ACGUGACACGUUCGGAGAAdTdT-3′)。每隔一天,再将10μg上述siRNA进行瘤内注射一次。第一次注射起20天后,按照如上所述的方法测量肿瘤体积,结果如表10所示。表10由表10可知,与阴性对照组相比,PLK-65、PLK(m)-65-1、PLK(m)-65-2、PLK-67、PLK(m)-67-1、PLK(m)-67-2、PLK-76和PLK(m)-76-1处理组的肿瘤细胞生长均得到了显著抑制。实施例6:通过尾静脉注射系统给药siRNA药物组合物时对于乳腺癌细胞生长的抑制(siRNA药物组合物的制备)本实施例中,采用聚乙二醇-聚乳酸两嵌段共聚物(PEG-PLA)、及阳离子脂质N,N-二羟乙基-N-甲基-N-2-(胆固醇氧羰基氨基)乙基溴化铵(BHEM-Chol)制备siRNA药物组合物,具体制备步骤参考了YangXZ.,etal.J.Cont.Release2011,156(2):203中的方法。即,将25mgPEG5000-PLA25000和1mgBHEM-Chol溶于0.5mL氯仿中,加入siRNA(0.025mL,0.2mg)水溶液后,冰浴下超声形成初始乳液;随后,将初始乳液加入到1.5mL的1%聚乙烯醇(Polyvinylalcohol,PVA)水溶液中,冰浴下超声乳化得到乳液,将乳液加入到25mL的0.3%PVA水溶液中;减压下蒸除有机溶剂,离心收集沉积物,用水重悬并离心收集2次以除去PVA,从而得到含小干扰核酸的给药组合物。通过上述步骤得到的药物组合物中,各组分的质量比为siRNA/阳离子脂质/聚合物=0.2/1.0/25.0。(siRNA药物组合物尾静脉注射给药对于乳腺癌细胞生长的抑制)在BALB/c裸鼠右侧第二个乳腺的脂肪垫下原位接种人乳腺癌细胞株MDA-MB-435s(5×106个细胞),14天左右后形成可见肿瘤,肿瘤体积平均约为50mm3。将裸鼠随机分成4组,每组为8只裸鼠,分别通过尾静脉注射进行给药。给药剂量以有效siRNA的量计算为1mg/kg(约20μgsiRNA/每只小鼠),隔天给药一次,共给药10次。作为阴性对照组,通过尾静脉注射150μlPBS溶液(阴性对照组1)、及通过上述步骤制备的含20μg实施例2中提到的阴性对照siRNA(正义链为5′-UUCUCCGAACGUGUCACGUdTdT-3′、互补的反义链为5′-ACGUGACACGUUCGGAGAAdTdT-3′)的PBS溶液150μl(N.C.siRNA药物组合物,阴性对照组2)。作为处理组,分别通过尾静脉注射通过上述步骤制备的、含20μgPLK(m)-65-1的PBS溶液150μl(PLK(m)-65-1药物组合物,处理组1)、或含20μgPLK(m)-67-1的PBS溶液150μl(PLK(m)-67-1药物组合物,处理组2)。每次给药后均对肿瘤大小进行测量,得到肿瘤生长数据。肿瘤的大小按以下公式计算:V=0.5×a×b2,其中,a指肿瘤长径,b指肿瘤短径。分析结果时,将每组小鼠第一次给药时的“肿瘤大小平均值”(即给药第0天)定为100%,其标准方差除以肿瘤大小平均值得到相对标准方差。之后的给药过程中,每次测得的各组肿瘤大小平均值及标准方差同样对第0天肿瘤大小平均值进行校正,即得到如图4所示的肿瘤细胞生长抑制曲线。由图4可以看出,与阴性对照组相比,通过尾静脉注射系统给药含PLK(m)-65-1及PLK(m)-67-1的药物组合物可以有效促进乳腺癌细胞凋亡,并抑制肿瘤组织生长。实施例7:通过尾静脉注射给药siRNA药物组合物时对于宫颈癌细胞生长的抑制(siRNA药物组合物的制备)本实施例中,采用聚己内酯-聚(N,N-二甲氨基乙基甲基丙烯酸酯)嵌段共聚物(PCL-PDMAEMA)、聚乙二醇嵌段修饰有叶酸的聚乙二醇-聚谷氨酸嵌段共聚物(folate-PEG-PGA)制备siRNA药物组合物。其中,PCL-PDMAEMA为聚β-氨基酯类两亲性阳离子聚合物,PEG-PGA为辅助性的聚合物。siRNA药物组合物的制备步骤参考了HuangYY.,etal.Biomaterials.2012,(18):4653-中的方法。即,20μlsiRNA脱离子水溶液(含约1μgsiRNA)与50μlPCL5000-PDMAEMA2000的PBS溶液混合,室温静置20分钟后,再加入50μlfolate-PEG5000-PGA46000的水溶液充分混合,室温孵育20分钟,之后用PBS调整体系即得到所需的药物组合物。其中,PCL5000-PDMAEMA2000的氮(N)、siRNA的磷(P)、PEG5000-PGA46000的碳(C)的摩尔比为5:1:8。(siRNA药物组合物尾静脉注射给药对于宫颈癌细胞生长的抑制)在BALB/c裸鼠右腋皮下接种人宫颈癌细胞Hela(5×106个细胞),10天左右后形成可见肿瘤,肿瘤体积平均约为50mm3。将裸鼠随机分成3组,每组为7只裸鼠,分别通过尾静脉注射进行给药。给药剂量以有效siRNA的量计算为2mg/kg(约40μgsiRNA/每只小鼠),每三天给药一次,共给药7次。作为阴性对照组,通过尾静脉注射200μl空白PBS溶液(阴性对照组1)、及用200μlPBS溶解的PLK(m)-67-1(裸PLK(m)-67-1,阴性对照组2)。作为处理组,通过尾静脉注射通过上述步骤制备的、含40μgPLK(m)-67-1的PBS溶液200μl(PLK(m)-67-1药物组合物,处理组)。每次给药后均对肿瘤大小进行测量,得到肿瘤生长数据。肿瘤的大小按以下公式计算:V=0.5×a×b2,其中,a指肿瘤长径,b指肿瘤短径。分析结果时,将每组小鼠第一次给药时的“肿瘤大小平均值”(即给药第0天)定为100%,其标准方差除以肿瘤大小平均值得到相对标准方差。之后的给药过程中,每次测得的各组肿瘤大小平均值及标准方差同样对第0天肿瘤大小平均值进行校正,即得到如图5所示的肿瘤细胞生长抑制曲线。由图5可以看出,与阴性对照组相比,通过尾静脉注射系统给药含PLK(m)-67-1的药物组合物可以有效促进宫颈癌细胞凋亡,并抑制肿瘤组织生长。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种利用短单链DNA抑制昆虫基因表达的方法与流程
- 特异性抑制Eya2基因表达的siRNA及其重组载体和应用的制作方法与工艺
- 抑制罗汉果CAS基因表达的方法与流程
- 特异性抑制CLDN8基因表达的siRNA及其重组载体和应用的制作方法与工艺
- 抑制小鼠促性腺激素抑制激素基因表达的siRNA及其应用的制作方法与工艺
- 一种抑制梨种子赤霉素合成基因表达的方法与流程
- KPNA2基因的应用和抑制KPNA2基因表达的siRNA的应用的制造方法与工艺
- 特异性抑制PADI2基因表达的siRNA及其重组载体和应用的制造方法与工艺
- 一种高效表达小鼠促性腺激素抑制激素基因的慢病毒载体及其应用的制造方法与工艺
- 一种抑制人黑色素瘤RBM3基因表达的siRNA及其质粒载体的制造方法与工艺