氨基丁三醇前列腺素F2α的合成方法与流程
本发明涉及一种化合物的合成方法,特别是氨基丁三醇前列腺素F2α的合成方法。
背景技术:
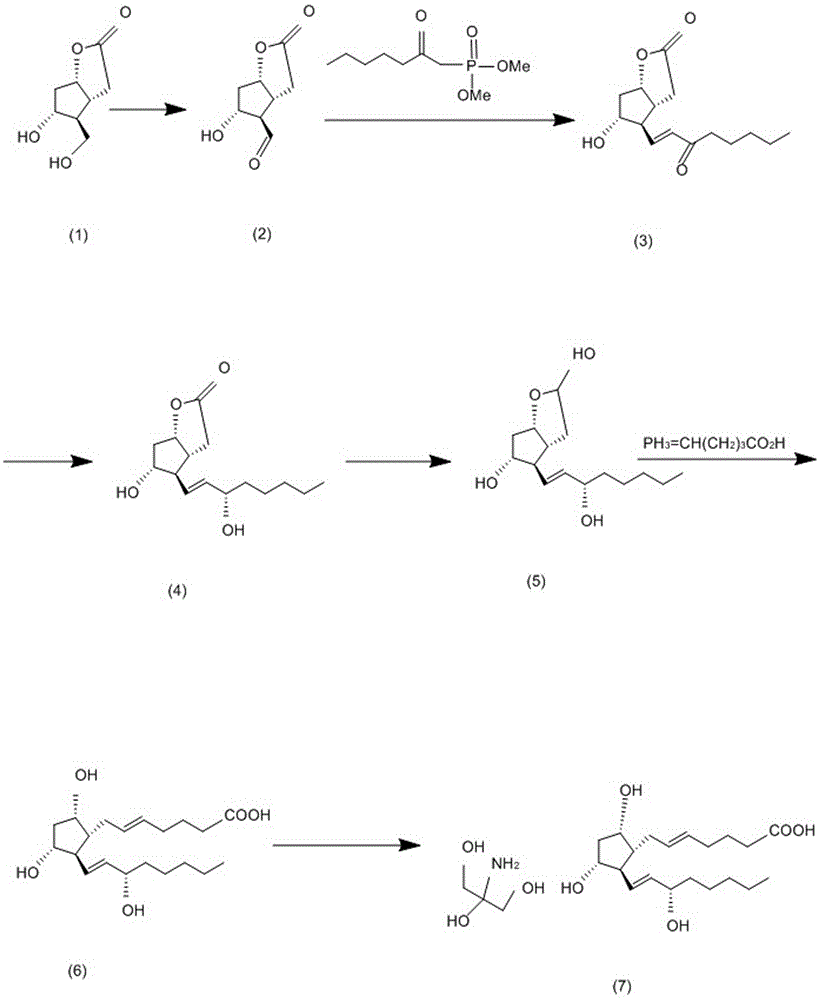
:前列腺素(prostaglandin,PG)是存在于动物和人体中的一类不饱和脂肪酸组成的、具有多种生理作用的活性物质。最早发现它存在于人的精液中,当时以为这一物质是由前列腺释放的,因而定名为前列腺素。现已证明精液中的前列腺素主要来自精囊,全身许多组织细胞都能产生前列腺素。前列腺素(PG)在体内由花生四烯酸所合成,结构为一个五环和两条侧链构成的20碳不饱和脂肪酸。按其结构,前列腺素分为A、B、C、D、E、F、G、H、I等类型。不同类型的前列腺素具有不同的功能,如前列腺素E能舒张支气管平滑肌,降低通气阻力;而前列腺素F的作用则相反。前列腺素的半衰期极短(1~2分钟),除前列腺素I2外,其他的前列腺素经肺和肝迅速降解,故前列腺素不像典型的激素那样,通过循环影响远距离靶向组织的活动,而是在局部产生和释放,对产生前列腺素的细胞本身或对邻近细胞的生理活动发挥调节作用。前列腺素对内分泌、生殖、消化、血液呼吸、心血管、泌尿及神经系统均有作用。由于天然的前列腺素多存在于动物的精囊中,由于含量低,提取困难,稳定性差,于是人们相继全合成了一系列前列腺素物质或类似物,以满足临床需求。其中氨基丁三醇前列腺素F2α是人工合成的前列腺素化合物之一,在畜牧业具有广泛的使用前景。然而目前氨基丁三醇前列腺素F2α的合成通用的是以下两种合成路线,一种是先合成带有任意一条侧链的五元环结构,再通过1,4-加成引入另一侧链,这种合成方法收率较高,但是需要用到贵金属催化剂,贵金属催化剂不但价格昂贵,不易回收,且容易对环境造成污染,限制了在工业领域的使用,另一种是三组分偶联法,即“一锅法”同时引入α和ω位两个侧链,这种合成方法虽然路线简短、原料廉价易得,成本优势明显,但是这个方法关键步骤副反应多,收率低,反应时间难以掌控,中间体化学稳定以及收率都有待进一步研究,亦不适合于工业化生产。技术实现要素:为了解决上述现有技术的不足,本发明的目的是提供一种适于工业化大生产的氨基丁三醇前列腺素F2α的合成方法。为了实现上述目的,本发明所设计的氨基丁三醇前列腺素F2α的合成方法,以化合物(-)-Corey内酯二醇为原料,经氧化反应后制得结构式为的内酯醛,内酯醛与结构式为的下侧链经维蒂希-霍纳尔反应拼接得到结构式为的烯烃,所述烯烃中的双羰基经还原后得到结构式为的醇类,再与结构式为的上侧链经叶立德-维蒂希反应得到前列腺素F2α,再将前列腺素F2α溶解于氨基丁三醇后析晶得到氨基丁三醇前列腺素F2α,其合成路线如下:。优选的,所述的氨基丁三醇前列腺素F2α的合成方法,包括以下合成步骤:步骤一、在有机溶剂中,加入原料(-)-Corey内酯二醇(式1)、氧化剂以及催化剂进行氧化反应得到含有内酯醛(式2)的反应液;步骤二、向上述反应液中加入饱和硫代硫酸钠搅拌分层,分出水层,往水相中分批加入下侧链混合溶液与碳酸钾水溶液,并搅拌,然后用第一萃取液进行萃取得到有机相,对有机相进行干燥、过滤、蒸干、柱层析得到烯烃(式3)的油状物;步骤三、将上述烯烃油状物混合于甲苯溶液内,然后通过还原剂对其双羰基进行还原得到醇类(式5)的油状物;步骤四、将式(5)的油状物滴加到上侧链混合溶液中,滴加后搅拌并用水进行淬灭反应,然后用第二萃取液进行萃取,得到有机相,有机相再用第三萃取液进行萃取,合并水相,并调节水相pH值,之后用第四萃取液萃取取其有机相,干燥,过滤得到滤饼,用乙酸乙酯洗涤得到滤液,对滤液进行浓缩,柱层析得到前列腺素F2α(式6)的油状物;步骤五、将上述前列腺素F2α(式6)的油状物与乙腈混合后回流至溶,并搅拌过滤得到滤液,往滤液中加入氨基丁三醇溶液,冷却过滤得到晶体,晶体用乙腈洗涤后,真空干燥至恒重,得到氨基丁三醇前列腺素F2α(式7)。优选的,所述步骤一中的有机溶剂为二氯甲烷,催化剂为二乙酸碘苯,氧化剂为四甲基砒啶氮氧化物。优选的,所述步骤二中的下侧链混合溶液为下侧链与丙酮混合而成,并且分三批加入下侧链混合溶液与碳酸钾水溶液,并且每批均先加入下侧链混合溶液,再加入碳酸钾水溶液。优选的,所述步骤二中的第一萃取液为乙酸乙酯溶液。优选的,所述步骤三的烯烃油状物混合于甲苯溶液内,然后滴加到混合体系中,滴加之后搅拌并用乙酸乙酯进行萃取得到有机相,然后对有机相分别用饱和碳酸氢钠与水洗涤,再用乙酸乙酯反萃水相,合并有机相,并对有机相进行干燥浓缩柱层析,得到醇类(式4)的油状物,其中混合体系由2,6-二叔丁基-4-甲基苯酚溶于无水甲苯之后,再滴加二异丁基氢化铝制得;然后将醇类(式4)的油状物溶于四氢呋喃与甲苯的混合溶液中,冷却至-70℃,再滴入二异丁基氢化铝,控制温度小于-65℃,并搅拌,然后移除冷槽并用饱和氯化铵溶液淬灭反应,接着用乙酸乙酯进行萃取得到有机相,对有机相进行干燥过滤,浓缩得到醇类(式5)的油状物。优选的,所述步骤四中水相的pH值通过盐酸调节,并控制pH值等于2。优选的,所述步骤四中的第二萃取液为甲基叔丁基醚溶液,第三萃取液为饱和碳酸氢钠溶液,第四萃取液为二氯甲烷。优选的,所述结构式为的下侧链通过以下步骤制得:正己酸混合于二氯甲烷中,加入二甲基甲酰胺,并用干燥管与外界隔离,然后室温滴加草酰氯与二氯甲烷的混合溶液,滴加完成后搅拌至正己酸消失,然后滴加甲醇之后再回流至原料消失,再依次加入饱和碳酸氢钠溶液与二氯甲烷溶液,并用碳酸氢钠固体调节pH值至8~9,分出有机相,水相反萃,合并有机相,然后干燥,减压蒸去二氯甲烷得到乙酸甲酯油状物;将四氢呋喃冷却至-70℃,然后加入丁基锂正己烷溶液,再滴加甲基磷酸二甲酯搅拌,控制温度不超过-60℃,再滴加上述乙酸甲酯油状物与四氢呋喃的混合溶液,亦控制温度不超过-60℃,滴加完毕后搅拌,接着升温至室温搅拌,再蒸干溶剂,并加入乙酸乙酯溶液,用饱和食盐水与水分别洗涤,之后干燥,过滤,浓缩,柱层析得到下侧链油状物。优选的,所述结构式为的上侧链通过以下步骤制得:将5-溴戊酸和三苯基膦混合,然后加入甲苯,搅拌加热至回流,冷却过滤,滤液浓缩出去甲苯,再加入四氢呋喃,搅拌加热至回流,冷却过滤,得到固体,再真空抽干,即得上侧链固体。与现有技术相比,本发明得到的氨基丁三醇前列腺素F2α的合成方法,其有益效果是:(1)、以(-)-Corey内酯二醇为原料,经氧化反应后制得内酯醛,再经维蒂希-霍纳尔反应拼接得到烯烃,烯烃中的双羰基经还原后得到醇类,再经叶立德-维蒂希反应得到前列腺素F2α,将前列腺素F2α溶解于氨基丁三醇后析晶得到氨基丁三醇前列腺素F2α,其中(-)-Corey内酯二醇市场有售,采购方便,无需贵金属催化剂,副反应少,收率高,成本低,污染少,适合于工业化大生产;(2)在烯烃中的双羰基还原过程中,二异丁基氧化铝、2,6二叔丁基-4甲基苯酚作为还原剂,对复杂或多功能基的分子非常有用,具有以下优点:收率高、选择性出众;无副反应;市场有售,采购方便,价格便宜;试剂稳定,贮存方便;工艺简单,操作方便,适合放大生产;(3)、通过维蒂希反应得到化合物前列腺素F2α,维蒂希反应是磷叶立德与醛或酮综合为一个分子,产生烯烃基团,连接原有的两个分子,同时失去相应的氧化磷的过程,而其中此工艺中的磷叶立德以5-溴代戊酸和三苯基膦得到磷盐,再以磷盐得到叶立德,如此一来整个维蒂希反应优势综合起来在于:几种试剂市场有售,便于购买;叶立德与多种醛类在室温下缩合,产物以顺式不饱和羧酸为主,它的副产物三苯基氧膦可通过水洗后处理很容易除去,并且对对烯烃几何构造的有效控制;(4)通过大量的试验,得知该化合物氨基丁三醇前列腺素F2α的收率在90%左右,非常稳定,适合工业化大生产。附图说明图1是氨基丁三醇前列腺素F2α结构示意图。图2是氨基丁三醇前列腺素F2α合成路线图。图3是本发明中下侧链的合成路线图。图4是本发明中上侧链的合成路线图。具体实施方式下面结合附图和实施例对本发明进一步说明。如图1、2所示,本发明提供的氨基丁三醇前列腺素F2α的合成方法,以化合物(-)-Corey内酯二醇为原料,经氧化反应后制得结构式为的内酯醛,内酯醛与结构式为的下侧链经维蒂希-霍纳尔反应拼接得到结构式为的烯烃,所述烯烃中的双羰基经还原后得到结构式为的醇类,再与结构式为的上侧链经叶立德-维蒂希反应得到前列腺素F2α,再将前列腺素F2α溶解于氨基丁三醇后析晶得到氨基丁三醇前列腺素F2α。如图3所示,其中结构式为的下侧链通过以下步骤制得:464g正己酸混合于1520ml二氯甲烷中,加入8ml二甲基甲酰胺,并用干燥管与外界隔离,自然室温滴加含有草酰氯(分子量:127,相对密度:1.488,400ml,1.05eq)的二氯甲烷溶液800ml,滴加过程中有气泡产生,滴加完成后,搅拌半小时,用薄层色谱法进行观测,该薄层色谱法观测的体系取至热的甲醇中后再加热至沸,即观测的体系是将上述正己酸、二氯甲烷等混合溶液溶于热的甲醇后再加热至沸的体系,石油醚与乙酸乙酯体积比为10:1的混合溶液作为展开剂,溴甲酚绿作为显色剂,观测正己酸消失,该薄层色谱法属于现有技术,不再赘述,然后用冰水冷却体系至0~5℃,滴加甲醇328ml,控制温度不超过5℃,滴加完成后回流1h,用薄层色谱法(直接点板,石油醚与乙酸乙酯体积比为10:1的混合溶液作为展开剂),显示原料几乎消失。再加入饱和碳酸氢钠4000ml,二氯甲烷50ml,并用碳酸氢钠固体调节体系pH值至8~9,分出有机层,用水进行反萃2次,每次2400ml,合并有机层,硫酸钠干燥,减压蒸去二氯甲烷,得到己酸甲酯油状物;另一个反应瓶中加入四氢呋喃2560ml,通氩气保护,冷却至-70℃,加入2.4M丁基锂正己烷溶液2080ml,滴加甲基磷酸二甲酯448ml,控制温度不超过-60℃,搅拌30min时后,滴入己酸甲酯的四氢呋喃溶液1600ml(即是将上述得到的己酸甲酯油状物溶于1600ml四氢呋喃溶液中制得),控制温度不超过-60℃,完毕后于-60~-70℃搅拌20min后自然升至室温,搅拌过夜,蒸干溶剂后加入乙酸乙酯4000ml,用2000ml饱和食盐水和2000ml水分别洗涤一次,硫酸钠干燥,过滤,浓缩,柱层析(体积比为5:1的石油醚与乙酸乙酯混合溶液作为展开剂,分去小极性杂点后再分别用体积比1:1、2:1的乙酸乙酯与石油醚的混合溶液进行分离)得到(2-氧代庚基)膦酸二甲酯的油状物400g,收率为77%,即为下侧链。如图4所示,所述结构式为的上侧链通过以下步骤制得:将5-溴戊酸300g和三苯基膦420g加入到3000ml三口瓶中,并加入甲苯3000ml,在搅拌下加热至回流15min,冷却过滤。滤液浓缩除去甲苯。再加入四氢呋喃2000ml,搅拌下加热至回流,冷却过滤,所得白色固体再真空抽干,即为上侧链,该上侧链的熔点为202-206℃。如图2所示,氨基丁三醇前列腺素F2α的合成方法,包括以下步骤:步骤一、306g(-)-Corey内酯二醇(式1)(分子量:172,0.06mol)混合于4680ml二氯甲烷中,自然室温搅拌,分批加入63g二乙酸碘苯(分子量:322,1.1eq)和3.6g四甲基哌啶氮氧化物(分子量:156,0.13eq)的混合物,约1h加完,完毕后室温搅拌过夜,次日TLC监测反应终点,得到含有内酯醛(式2)的反应液,该步骤中二氯甲烷作为有机溶剂,二乙酸碘苯作为催化剂,四甲基砒啶氮氧化物作为氧化剂;步骤二、向上述反应液中加入约1050ml的饱和硫代硫酸钠破坏氧化性,搅拌30min左右分层,分出水层,有机相再用水反萃2次,每次600ml,合并水相,再往水相中分批加入下侧链混合溶液与碳酸钾水溶液,并搅拌1~1.5h,本实施例中,分三批加入,第一批是先加234g下侧链与1800ml丙酮形成的溶液,再加558g碳酸钾与900ml水形成的溶液;第二批是先加54g下侧链与22ml丙酮形成的溶液,再加4.8g碳酸钾与12ml水形成的溶液;第三批是先加1.2g下侧链与660ml丙酮形成的溶液,再加54g碳酸钾与360ml水形成的溶液,每批间隔约5min,然后用乙酸乙酯溶液进行萃取三次得到有机相,每次乙酸乙酯3000ml,合并有机相,干燥、过滤、蒸干、柱层析得到烯烃(式3)的油状物210g,收率为43.8%,其中柱层析中洗脱剂用体积比为30:10:1的石油醚、二氯甲烷、甲醇的混合溶液;步骤三、1080g2,6-二叔丁基-4-甲基苯酚(分子量:220,2.6g,4.4eq)溶于4400ml无水甲苯中,通氩气保护,冷却至0~5℃,滴加3150ml二异丁基氢化铝溶液,约0.5h滴加完成,滴加完成后于0~5℃搅拌1h,冷却至-60~-70℃,滴加由上述烯烃(式3)油状物的210g溶于2100ml甲苯溶液得到混合液,滴加完成后,于此温度搅拌2h,然后升至-40℃搅拌1h,由原先很深的红棕色变浅,用薄层色谱法监测反应终点。然后倒入冰水冷却的4000ml1M盐酸中,5000ml乙酸乙酯萃取,有机相分别用3000ml饱和碳酸氢钠溶液及3000ml水各洗涤一次,再用3000ml乙酸乙酯分别反萃上述水相一次,重复反萃一次,用薄层色谱法检测水相没有产品即可,所有有机相合并,干燥,浓缩,柱层析(先以二氯甲烷为洗脱剂,洗除2,6-二叔丁基-4-甲基苯酚,再以体积比为1:1的乙酸乙酯与二氯甲烷混合液为洗脱剂)得到醇类(式4)油状物195g,收率93%;1.5g醇类(式4)油状物(分子量:268)混合于3260ml四氢呋喃和1130ml甲苯中,通氩气保护,冷却至-70℃,滴入4500ml二异丁基氢化铝,控制温度小于-65℃,搅拌0.5h,薄层色谱法(展开剂用体积为15:1的乙酸乙酯与甲醇混合液)监测反应终点。反应转化完成后,移出冷槽,滴入780ml饱和氯化铵溶液淬灭反应,用780ml的乙酸乙酯萃取3次得到有机相,合并有机相,干燥,有胶体析出,且放热,用硅藻土过滤胶体,所有有机层合并,浓缩,得醇类(式5)油状物240g;步骤四、取上侧链约237g混于1890ml四氢呋喃中,冰水冷却至0~5℃,通氩气保护,分两次加入叔丁醇钾,每次60g,溶液变成橙色,于此温度搅拌40min,滴入195g醇类(式5)与4800ml四氢呋喃溶液的混合物,滴加后,于室温搅拌1h,加水3000ml淬灭反应,用1500ml甲基叔丁基醚萃取2次,有机相再用饱和碳酸氢钠溶液萃取3次,每次1500ml,然后将所有水相合并,用2M盐酸调节体系pH值,使pH值控制在2,然后二氯甲烷萃取2次,每次3000ml,再用硫酸镁干燥,过滤,浓缩,加入乙酸乙酯900ml,室温搅拌1h,固体杂质析出,过滤,滤饼用乙酸乙酯洗涤至无产品,所有滤液浓缩,柱层析(洗脱剂:体积比为60:35:5的乙酸乙酯、石油醚、乙酸的混合溶液),共得前列腺素F2α(式6)油状物150g左右,收率54%;步骤五、将上述150g前列腺素F2α(式6)的油状物与5L乙腈混合后回流加热至溶,温度一般控制在43~47℃,并保温搅拌15min,然后趁热过滤得到滤液,然后用乙腈洗涤,使乙腈总体积达到21L,合并滤液,并搅拌5min左右,在搅拌过程中,往滤液中加入氨基丁三醇溶液,氨基丁三醇溶液由49.2g氨基丁三醇加90ml水,并升温至53~57℃保温搅拌5min制得,加入后立即会有结晶析出,加完后继续搅拌18~24h,然后自然冷却到室温,过滤,用乙腈洗涤结晶3次,每次100ml,放在有五氧化二磷的干燥箱内真空干燥至恒重,一般需要5h以上,得到氨基丁三醇前列腺素F2α(式7)约180g,收率89%。其中,(-)-Corey内酯二醇为(3aR,4S,5R,6aS)-(-)-六氢-5-羟基-4-羟甲基-2H-环戊并[b]呋喃-2-酮,按干燥品计算,含(C8H12O4)应为98.0%~102.0%,本品为白色结晶,其比旋度应为-38°至-45°,熔点为114~118℃。在此基础上,对氨基丁三醇前列腺素F2α的合成进行中试放大试验,得到结果见表1:表1:中试放大试验结果序号投料量g产品产量g摩尔收率%1150185.592.22150186.292.53150182.990.9投料量为(-)-Corey内酯二醇的投料量,产品产量为氨基丁三醇前列腺素F2α的产量,由表1可知,该合成方法的摩尔收率基本在90%,非常稳定,适合工业化大生产。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1