一种网络结构成炭剂及其制备方法和应用与流程
本发明属于高分子材料技术领域,涉及一种网络结构成炭剂,具体涉及一种含哌嗪和三聚氰胺的网络结构成炭剂及其制备方法和应用,尤其是在制备阻燃高分子材料中的应用。
背景技术:
随着高分子材料工业的发展,合成材料已广泛应用于国民经济和人民生活的各个领域。但是,由于高分子材料的易燃性和可燃性而引起的火灾事故也给人类的生命财产安全带来了巨大的威胁。在树脂中添加阻燃剂,是提高高分子材料阻燃性能最简单、经济和富有成效的方法,其中阻燃剂是决定材料阻燃性能优劣的关键。传统的卤系阻燃剂燃烧时释放出卤化氢、二噁英等有毒和腐蚀性气体,产生较大的烟雾,已经逐步被限制使用。
随着人们环保意识的提高,阻燃剂及阻燃材料的无卤化成为科学研究和工业应用共同追求的目标。在无卤阻燃剂中膨胀型阻燃剂(IFR)因其低毒、低烟、低腐蚀、抗融滴等特点被认为是具有良好发展前景的绿色阻燃剂之一。IFR一般由酸源、碳源、气源三部分组成,在燃烧过程中IFR可以促使材料表面形成膨胀炭层,起到隔热、隔氧、抑烟的作用,从而使火焰熄灭。目前可用于IFR碳源的成炭剂主要存在以下问题:季戊四醇(PER)的热稳定性较差,吸水性较大,而且在加工和使用过程中易迁出、易反应,最终导致阻燃聚合物易吸潮,进而在日常使用过程中影响产品的电性能、耐候性以及耐久性,最终影响材料使用性能。淀粉、多糖类生物基成炭剂阻燃效率低,且不耐温。这些问题阻碍了IFR在很多领域的工业化应用。因此开发新型成炭剂是IFR发展的一个重要方向。
近年来针对传统成炭剂的缺点,各种新型成炭剂相继被研究报道出来。如:公开号为:CN104693442A和CN103756015等公开了利用含磷二胺或者二羟基化合物与三聚氯氰进行反应制备了一种超支化成炭剂,虽然制备成炭剂的成炭效率高,产物的热稳定性好,但是原料中使用了三聚氯氰,需要进行阶段控温,并且需要加入缚酸剂来保证反应的顺利进行,合成工艺相对繁杂,而且氯容易残留。另有公开号为:CN103992481A公开了“一种超支化聚磷腈阻燃成炭剂及其制备方法”,虽然制得的成炭剂阻燃效果好,加工性能良好,但是,该成炭剂的制备需要20h左右,并且原料六氯环三磷腈易升华,其蒸汽对眼睛和呼吸道有刺激作用,且对水资源有害。
本专利以哌嗪和醛类化合物作为单体,以三聚氰胺为交联剂,在溶剂中进行反应,最后以单官能硅烷偶联剂来封端,制备了具有网络结构的成炭剂。该成炭剂不但具有良好的热稳定性和耐水性,而且具有功能性表面,与聚合物相容性好,此外制备方法简单、环保,产率高,易于工业化生产,所用原材料价格便宜。可作为IFR的成炭剂用于高分子材料的阻燃,与现有的成炭剂比有明显的优势。
技术实现要素:
本发明的一个目的是提供一种新型网络结构成炭剂。该成炭剂不仅成炭性能优异,而且制备方法简单,产率高,工艺过程环保,易于工业化生产。
本发明所述的成炭剂分子结构如式(1):
式中1<n<10000;R的结构为下列之一:R1为含单个羟基或氨基的硅烷偶联剂基团。
本发明的另一目的是提供一种制备该网络结构成炭剂的方法。该方法具体步骤如下:
步骤(1)、将醛基化合物加入到有机溶剂中,调节溶液PH至8~9;其中醛基化合物的浓度为0.6L/mol;
所述的用于调节溶液PH的溶液为有机碱或无机碱中的一种;
所述的有机溶剂为N-N二甲基甲酰胺、N-N二甲基乙酰胺、四氢呋喃、二氧六环、乙腈、二甲亚砜中的一种;
所述的含醛基化合物为下列之一:甲醛溶液、固体甲醛、乙醛、1-丁醛、苯甲醛、糠醛;
步骤(2)、将哌嗪溶解于蒸馏水中得到哌嗪水溶液,滴加于步骤(1)的溶液中,滴加时间为0.5~1h,反应温度控制在0~60℃,反应时间2~10h,得到含有A结构的透明溶液;
所述的无水哌嗪的分子式为C4H10N2;
所述的哌嗪与醛类化合物的投料物质的量比为1:2~3;
所述的哌嗪溶液的浓度为40~50%;
所述A的结构为:其中R的结构为下列之一:
步骤(3)、向步骤(2)所得的溶液中,加入三聚氰胺,温度控制在0~60℃,反应时间1~5h,得到含有B结构的溶液;
所述的三聚氰胺的用量为哌嗪物质的量的1~10%;
所述的B结构如下:
其中R的结构为下列之一:
步骤(4)、向步骤(3)中的溶液中,再次滴加哌嗪的水溶液,温度控制在60~90℃,反应时间2~10h,得到白色固液混合物;
所述的哌嗪投料量与步骤(2)中的哌嗪等量;
步骤(5)、向步骤(4)中的固液混合物中,加入单官能硅烷偶联剂反应1~2h,反应结束后,经过抽滤、水洗、干燥,得到目标产物网络结构成炭剂。
所述的偶联剂的用量为0.5~1%(以总溶液质量计);
所述的硅烷偶联剂为含单个羟基或氨基的硅烷偶联剂,具体是下列偶联剂之一:氨丙基三乙氧基硅烷、氨丙基三甲氧基硅烷、2-氨乙基-氨丙基三甲氧基硅烷、氨乙基氨丙基甲基二甲氧基硅烷、氨乙基氨丙基甲基二乙氧基硅烷;
本发明的又一个目的是将上述网络结构成炭剂应用于聚丙烯、聚乙烯以及乙烯-醋酸乙烯共聚物材料中,得到一种阻燃效果更好的高分子组合物;
本发明为了实现以上目的,采用以下技术方案:
成炭剂与聚磷酸盐按比例充分混合均匀,得到复配后的阻燃剂;复配后的阻燃剂与聚合物在挤出机中通过熔融共混,得到具有优异阻燃性能和高热稳定性能的高分子组合物。
本发明所述的聚合物为聚丙烯、聚乙烯、乙烯-醋酸乙烯酯共聚物。
本发明所述的聚磷酸盐为聚磷酸铵、聚磷酸三聚氰胺中的一种。
本发明所述的成炭剂与聚磷酸盐的重量比为1:5~3:1。
本发明所述的复配后的阻燃剂与聚合物的质量比为:10~50:100。
本发明所述的高分子组合物中也包括一般常见的助剂譬如稳定剂、分散剂、成核剂以及其它无机填料譬如云母石、碳酸钙、硅石或他们的混合物也可包括在内,所有成分加起来为100%重量百分比。
与现有技术相比,本发明的有益效果体现在以下方面:
本发明所制备的成炭剂,具有网络结构,不溶不融,具有优异的耐水性,并且经过硅烷偶联剂修饰后,成炭剂在高分子材料中具有更好的相容性和分散性,克服了小分子成炭剂易迁移、易吸水与基体相容性差的不足。另外,该成炭剂成炭效率高、合成工艺简单,反应产率高,阻燃效果好。
附图说明
图1是本发明网络结构成炭剂的红外谱图;
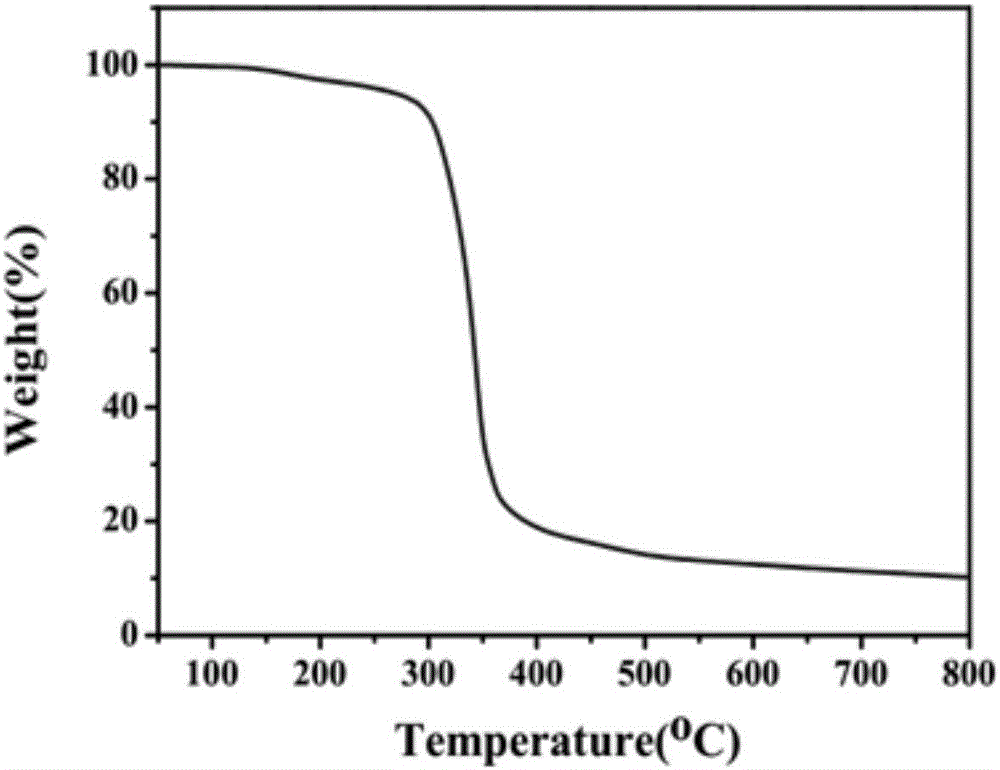
图2是本发明网络结构成炭剂的热重图。
具体实施方式
以下以具体的实施实例来说明本发明的技术方案,但本发明的保护范围不限于此:
燃烧测试标准:GB/T 2408—2008;
氧指数测试标准:GB/T 2406.2—2006。
实施例1:网络结构成炭剂的制备
称取甲醛溶液0.20mol(16.23g),加入到装有冷凝回流装置的500ml三口瓶中,并加入有机溶剂N’N-二甲基甲酰胺120ml,用氢氧化钠调节溶液PH在8-9,控制溶液温度在50℃,于此温度下开始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),约0.5h滴加完毕,在50℃保温反应2h得到澄清透明的溶液,之后向溶液中加入8%的三聚氰胺(1.01g),于50℃反应1h,继续升温至90℃,向反应液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),滴毕后保温反应2h,得到白色固液混合物,反应结束后向溶液中加入0.5%(0.17g)的氨丙基三甲氧基硅烷偶联剂进行功能化处理1h,停止反应,降至室温,抽滤,烘干,得到目标产物成炭剂,产率90%。在上述实验过程中,哌嗪和所制备的含哌嗪结构的成炭剂的红外谱图分别如图1a,b所示,图1(a)中3209cm-1和1551cm-1为N-H的振动吸收峰,2855cm-1、1430cm-1为-C-H的振动吸收峰,1272为C-N基团振动峰吸收峰;而在图1(b)中3209cm-1处N-H振动峰消失,并且出现了1514cm-1处的三个小峰为三聚氰胺碳氮骨架的震动吸收,由此可判断成功合成了含哌嗪结构的成炭剂。所制备的成炭剂的热重曲线如图2所示,从图中可以看出成炭剂具有良好的热稳定性,能够满足高分子材料的加工温度。
实施例2:网络结构成炭剂的制备
称取甲醛溶液0.20mol(16.23g),加入到装有冷凝回流装置的500ml三口瓶中,并加入有机溶剂N’N-二甲基乙酰胺120ml,用氢氧化钠调节溶液PH在8-9,控制溶液温度在20℃,于此温度下开始滴加哌嗪水溶液(哌嗪0.10mol,8.63g,蒸馏水20ml),约0.5h滴加完毕,在20℃保温反应10h得到澄清透明的溶液,之后向溶液中加入1%的三聚氰胺(0.13g),于20℃反应2h,继续升温至90℃,向反应液中滴加哌嗪溶液(哌嗪0.10mol,8.63g,蒸馏水20ml),滴毕后保温反应4h,得到白色固液混合物,反应结束后向溶液中加入0.5%(0.17g)的氨丙基三甲氧基硅烷偶联剂进行功能化处理1h,停止反应,降至室温,抽滤,烘干,得到目标产物成炭剂,产率85%。
实施例3:网络结构成炭剂的制备
称取乙醛0.30mol(13.22g),加入到装有冷凝回流装置的500ml三口瓶中,并加入有机溶剂二氧六环180ml,用三乙胺调节溶液PH在8-9,控制溶液温度在30℃,于此温度下开始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),约0.5h滴加完毕,在30℃保温反应8h得到澄清透明的溶液,之后向溶液中加入8%的三聚氰胺(1.00g),于30℃反应1.5h,继续升温至60℃,向反应液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),滴毕后保温反应5h,得到白色固液混合物,反应结束后向溶液中加入1%(0.31g)的氨丙基三甲氧基硅烷偶联剂进行功能化处理2h,停止反应,降至室温,抽滤,烘干,得到目标产物成炭剂,产率90%。
实施例4:网络结构成炭剂的制备
称取固体甲醛0.40mol(12.01g),加入到装有冷凝回流装置的500ml三口瓶中,并加入有机溶剂乙腈240ml,用氢氧化钠调节溶液PH在8-9,控制溶液温度在45℃,于此温度下开始滴加哌嗪水溶液(哌嗪0.16mol,13.78g,蒸馏水30ml),约1h滴加完毕,在45℃保温反应8h得到澄清透明的溶液,之后向溶液中加入10%的三聚氰胺(2.02g),于45℃反应2h,继续升温至90℃,向反应液中滴加哌嗪溶液(哌嗪0.16mol,13.78g,蒸馏水30ml),滴毕后保温反应2h,得到白色固液混合物,反应结束后向溶液中加入0.8%(0.32g)的氨丙基三乙氧基硅烷偶联剂进行功能化处理1h,停止反应,降至室温,抽滤,烘干,得到目标产物成炭剂,产率86%。
实施例5:网络结构成炭剂的制备
称取苯甲醛0.40mol(42.45g),加入到装有冷凝回流装置的500ml三口瓶中,并加入有机溶剂四氢呋喃240ml,用氢氧化钠调节溶液PH在8-9,控制溶液温度在0℃,于此温度下开始滴加哌嗪水溶液(哌嗪0.16mol,13.78g,蒸馏水30ml),约1h滴加完毕,在0℃保温反应10h得到澄清透明的溶液,之后向溶液中加入10%的三聚氰胺(2.02g),于0℃反应5h,继续升温至80℃,向反应液中滴加哌嗪溶液(哌嗪0.16mol,13.78g,蒸馏水30ml),滴毕后保温反应2h,得到白色固液混合物,反应结束后向溶液中加入0.8%(0.92g)的2-氨乙基-氨丙基三甲氧基硅烷偶联剂进行功能化处理2h,停止反应,降至室温,抽滤,烘干,得到目标产物成炭剂,产率80%。
实施例6:网络结构成炭剂的制备
称取甲醛溶液0.20mol(16.23g),加入到装有冷凝回流装置的500ml三口瓶中,并加入有机溶剂N’N-二甲基甲酰胺120ml,用氢氧化钠调节溶液PH在8-9,控制溶液温度在60℃,于此温度下开始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),约0.5h滴加完毕,在60℃保温反应2h得到澄清透明的溶液,之后向溶液中加入4%的三聚氰胺(0.50g),于60℃反应1h,继续升温至70℃,向反应液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),滴毕后保温反应3h,得到白色固液混合物,反应结束后向溶液中加入0.8%(0.26g)的2-氨乙基-氨丙基三甲氧基硅烷偶联剂进行功能化处理1h,停止反应,降至室温,抽滤,烘干,得到目标产物成炭剂,产率83%。
实施例7:网络结构成炭剂的制备
称取糠醛0.20mol(19.22g),加入到装有冷凝回流装置的500ml三口瓶中,并加入有机溶剂二氧六环120ml,用氢氧化钠调节溶液PH在8-9,控制溶液温度在50℃,于此温度下开始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),约0.5h滴加完毕,在50℃保温反应2h得到澄清透明的溶液,之后向溶液中加入1%的三聚氰胺(0.13g),于50℃反应1h,继续升温至80℃,向反应液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),滴毕后保温反应2.5h,得到白色固液混合物,反应结束后向溶液中加入0.5%(0.19g)的氨丙基三甲氧基硅烷偶联剂进行功能化处理1.5h,停止反应,降至室温,抽滤,烘干,得到目标产物成炭剂,产率78%。
实施例8:网络结构成炭剂的制备
称取固体甲醛0.30mol(9.01g),加入到装有冷凝回流装置的500ml三口瓶中,并加入有机溶剂乙腈180ml,用氢氧化钠调节溶液PH在8-9,控制溶液温度在50℃,于此温度下开始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),约1h滴加完毕,在50℃保温反应2h得到澄清透明的溶液,之后向溶液中加入6%的三聚氰胺(0.76g),于50℃反应1h,继续升温至60℃,向反应液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),滴毕后保温反应5h,得到白色固液混合物,反应结束后向溶液中加入0.5%(0.14g)的氨乙基氨丙基甲基二甲氧基硅烷偶联剂进行功能化处理1h,停止反应,降至室温,抽滤,烘干,得到目标产物成炭剂,产率81%。
实施例9:网络结构成炭剂的制备
称取1-丁醛0.30mol(21.63g),加入到装有冷凝回流装置的500ml三口瓶中,并加入有机溶剂N’N-二甲基乙酰胺180ml,用氢氧化钠调节溶液PH在8-9,控制溶液温度在40℃,于此温度下开始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),约0.5h滴加完毕,在40℃保温反应6h得到澄清透明的溶液,之后向溶液中加入3%的三聚氰胺(0.38g),于40℃反应2h,继续升温至60℃,向反应液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),滴毕后保温反应5h,得到白色固液混合物,反应结束后向溶液中加入0.5%(0.20g)的氨乙基氨丙基甲基二甲氧基硅烷偶联剂进行功能化处理1h,停止反应,降至室温,抽滤,烘干,得到目标产物成炭剂,产率79%。
实施例10:网络结构成炭剂的制备
称取甲醛溶液0.20mol(16.23g),加入到装有冷凝回流装置的500ml三口瓶中,并加入有机溶剂N’N-二甲基甲酰胺120ml,用氢氧化钠调节溶液PH在8-9,控制溶液温度在50℃,于此温度下开始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),约1h滴加完毕,在50℃保温反应8h得到澄清透明的溶液,之后向溶液中加入8%的三聚氰胺(1.00g),于50℃反应2h,继续升温至85℃,向反应液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),滴毕后保温反应3h,得到白色固液混合物,反应结束后向溶液中加入0.8%(0.28g)的氨丙基三甲氧基硅烷偶联剂进行功能化处理2h,停止反应,降至室温,抽滤,烘干,得到目标产物成炭剂,产率83%。
实施例11:网络结构成炭剂的制备
称取苯甲醛0.20mol(21.22g),加入到装有冷凝回流装置的500ml三口瓶中,并加入有机溶剂乙腈120ml,用碳酸钾调节溶液PH在8-9,控制溶液温度在45℃,于此温度下开始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),约0.5h滴加完毕,在45℃保温反应4h得到澄清透明的溶液,之后向溶液中加入8%的三聚氰胺(1.01g),于45℃反应3h,继续升温至60℃,向反应液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),滴毕后保温反应5h,得到白色固液混合物,反应结束后向溶液中加入0.8%(0.32g)的氨乙基氨丙基甲基二乙氧基硅烷偶联剂进行功能化处理1h,停止反应,降至室温,抽滤,烘干,得到目标产物成炭剂,产率80%。
实施例12:网络结构成炭剂的制备
称取固体甲醛0.20mol(6.01g),加入到装有冷凝回流装置的500ml三口瓶中,并加入有机溶剂四氢呋喃120ml,用氢氧化钠调节溶液PH在8-9,控制溶液温度在50℃,于此温度下开始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),约0.5h滴加完毕,在50℃保温反应4h得到澄清透明的溶液,之后向溶液中加入8%的三聚氰胺(1.01g),于50℃反应2h,继续升温至90℃,向反应液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),滴毕后保温反应2h,得到白色固液混合物,反应结束后向溶液中加入1%(0.24g)的氨丙基三甲氧基硅烷偶联剂进行功能化处理2h,停止反应,降至室温,抽滤,烘干,得到目标产物成炭剂,产率88%。
实施例13:网络结构成炭剂的制备
称取乙醛0.30mol(13.21g),加入到装有冷凝回流装置的500ml三口瓶中,并加入有机溶剂N’N-二甲基甲酰胺180ml,用氢氧化钠调节溶液PH在8-9,控制溶液温度在25℃,于此温度下开始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),约0.5h滴加完毕,在25℃保温反应8h得到澄清透明的溶液,之后向溶液中加入10%的三聚氰胺(1.26g),于25℃反应2h,继续升温至75℃,向反应液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),滴毕后保温反应4h,得到白色固液混合物,反应结束后向溶液中加入0.8%(0.25g)的氨乙基氨丙基甲基二乙氧基硅烷偶联剂进行功能化处理1.5h,停止反应,降至室温,抽滤,烘干,得到目标产物成炭剂,产率87%。
实施例14:网络结构成炭剂的制备
称取固体甲醛0.30mol(9.01g),加入到装有冷凝回流装置的500ml三口瓶中,并加入有机溶剂二氧六环180ml,用碳酸钠调节溶液PH在8-9,控制溶液温度在50℃,于此温度下开始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),约0.5h滴加完毕,在50℃保温反应5h得到澄清透明的溶液,之后向溶液中加入8%的三聚氰胺(1.01g),于50℃反应1h,继续升温至60℃,向反应液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),滴毕后保温反应3.5h,得到白色固液混合物,反应结束后向溶液中加入0.8%(0.22g)的氨丙基三甲氧基硅烷偶联剂进行功能化处理2h,停止反应,降至室温,抽滤,烘干,得到目标产物成炭剂,产率76%。
实施例15:网络结构成炭剂的制备
称取甲醛溶液0.20mol(16.23g),加入到装有冷凝回流装置的500ml三口瓶中,并加入有机溶剂乙腈120ml,用氢氧化钾调节溶液PH在8-9,控制溶液温度在50℃,于此温度下开始滴加哌嗪水溶液(哌嗪0.10mol,8.16g,蒸馏水20ml),约0.5h滴加完毕,在50℃保温反应4h得到澄清透明的溶液,之后向溶液中加入5%的三聚氰胺(0.63g),于50℃反应1h,继续升温至90℃,向反应液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸馏水20ml),滴毕后保温反应2h,得到白色固液混合物,反应结束后向溶液中加入0.8%(0.22g)的氨丙基三甲氧基硅烷偶联剂进行功能化处理1h,停止反应,降至室温,抽滤,烘干,得到目标产物成炭剂,产率87%。
实施例1-15得到的目标产物成炭剂分子结构如式(1):
式中1<n<10000;R的结构为下列之一:R1为含单个羟基或氨基的硅烷偶联剂基团。
实施例16网络结构成炭剂的应用
R为网络结构成炭剂、多聚磷酸铵,按重量比1:3混合均匀,得到复配阻燃剂与聚丙烯按重量比10:100进行预混合,然后通过熔融挤出获得阻燃复合物,按标准进行阻燃测试,UL94垂直燃烧为V-0级,极限氧指数LOI为25.2%。
实施例17网络结构成炭剂的应用
R为的网络结构成炭剂、多聚磷酸铵,按重量比2:1混合均匀,得到复配阻燃剂与聚丙烯按重量比30:100进行预混合,然后通过熔融挤出获得阻燃复合物,按标准进行阻燃测试,UL94垂直燃烧为V-0级,极限氧指数LOI为26.2%。
实施例18网络结构成炭剂的应用
R为网络结构成炭剂、多聚磷酸三聚氰胺,按重量比1:5混合均匀,得到复配阻燃剂与聚乙烯按重量比45:100进行预混合,然后通过熔融挤出获得阻燃复合物,按标准进行阻燃测试,UL94垂直燃烧为V-0级,极限氧指数LOI为28.4%。
实施例19网络结构成炭剂的应用
R为的网络结构成炭剂、多聚磷酸铵,按重量比3:1混合均匀,得到复配阻燃剂与聚乙烯按重量比30:100进行预混合,然后通过熔融挤出获得阻燃复合物,按标准进行阻燃测试,UL94垂直燃烧为V-0级,极限氧指数LOI为26.8%。
实施例20网络结构成炭剂的应用
R为的网络结构成炭剂、多聚磷酸铵,按重量比1:2混合均匀,得到复配阻燃剂与乙烯-醋酸乙烯酯共聚物按重量比25:100进行预混合,然后通过熔融挤出获得阻燃复合物,按标准进行阻燃测试,UL94垂直燃烧为V-0级,极限氧指数LOI为25.9%。
实施例21网络结构成炭剂的应用
R为的网络结构成炭剂、多聚磷酸三聚氰胺,按重量比1:3混合均匀,得到复配阻燃剂与乙烯-醋酸乙烯酯共聚物按重量比20:100进行预混合,然后通过熔融挤出获得阻燃复合物,按标准进行阻燃测试,UL94垂直燃烧为V-0级,极限氧指数LOI为24.8%。
实施例22网络结构成炭剂的应用
R为的网络结构成炭剂、多聚磷酸铵,按重量比3:1混合均匀,得到复配阻燃剂与乙烯-醋酸乙烯酯共聚物按重量比50:100进行预混合,然后通过熔融挤出获得阻燃复合物,按标准进行阻燃测试,UL94垂直燃烧为V-0级,极限氧指数LOI为31.2%。
实施例23网络结构成炭剂的应用
R为的网络结构成炭剂、多聚磷酸三聚氰胺,按重量比3:1混合均匀,得到复配阻燃剂与聚丙烯按重量比35:100进行预混合,然后通过熔融挤出获得阻燃复合物,按标准进行阻燃测试,UL94垂直燃烧为V-0级,极限氧指数LOI为26.4%。
实施例24网络结构成炭剂的应用
R为的网络结构成炭剂、多聚磷酸铵,按重量比2:1混合均匀,得到复配阻燃剂与聚乙烯按重量比50:100进行预混合,然后通过熔融挤出获得阻燃复合物,按标准进行阻燃测试,UL94垂直燃烧为V-0级,极限氧指数LOI为30.2%。
实施例25网络结构成炭剂的应用
R为的网络结构成炭剂、多聚磷酸三聚氰胺,按重量比1:2.5混合均匀,得到复配阻燃剂与乙烯-醋酸乙烯酯共聚物按重量比35:100进行预混合,然后通过熔融挤出获得阻燃复合物,按标准进行阻燃测试,UL94垂直燃烧为V-0级,极限氧指数LOI为26.7%。
上述实施例并非是对于本发明的限制,本发明并非仅限于上述实施例,只要符合本发明要求,均属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!