结晶形式的制作方法
本发明涉及一种(s)-2-(8-((5-氯嘧啶-2-基)(甲基)氨基)-2-氟-6,7,8,9-四氢-5h-吡啶并[3,2-b]吲哚-5-基)乙酸(后文亦称为“化合物”)的新颖结晶形式、其制备方法、包括该结晶形式的医药组合物、由此结晶形式制备的医药组合物及它们在多种前列腺素介导的疾病及病症的治疗中作为前列腺素d2受体(“dp受体”)调节剂,最特定言之作为crth2受体(“dp2受体”)调节剂的用途。特定言之,呈结晶形式的该化合物可单独使用或在医药组合物中使用以用于治疗慢性及急性过敏性/免疫疾病/病症,诸如哮喘、过敏性哮喘、嗜酸性粒细胞性哮喘、严重型哮喘、鼻炎、过敏性鼻炎、血管性水肿、昆虫毒液过敏、药物过敏、过敏性窦炎、过敏性肾炎、过敏性结膜炎、异位性皮肤炎、支气管哮喘、食物过敏、全身性细胞肥大病症、过敏性休克、荨麻疹、湿疹、溃疡性结肠炎、慢性阻塞性肺病(copd)、发炎性肠病、风湿性关节炎及鼻息肉病;嗜酸性粒细胞相关的疾病,其包括小血管炎如彻奇-斯全司症候群(churg-strausssyndrome)、韦格纳氏(wegener's)肉芽肿病、显微镜下多血管炎(及后者的器官特异性子集)、嗜酸性粒细胞增多症候群如嗜酸性粒细胞性肺炎、嗜酸性粒细胞性食道炎、逆流性食道炎、嗜酸性粒细胞性心内膜炎(吕佛勒氏(loeffler’s)心内膜炎)、嗜酸性粒细胞增多-肌痛症候群、嗜酸粒细胞性筋膜炎、嗜酸性粒细胞性脓疱性毛囊炎(太藤氏(ofuji's)疾病)、嗜酸性粒细胞性溃疡、血管淋巴样增生伴嗜酸性粒细胞增多症(alhe)、嗜酸性粒细胞性蜂窝性组织炎(韦尔斯(wells)症候群)、慢性嗜酸性粒细胞性白血病、dress症候群(药疹伴嗜酸性粒细胞增多及全身症状)及斯蒂尔病(still’sdisease)(幼年特发性关节炎);嗜碱性粒细胞相关的疾病,其包括嗜碱性粒细胞性白血病及嗜碱性白细胞增多;及囊肿性纤维化。
背景技术:
作为于过敏病状中对过敏原暴露的应答,肥大细胞将被活化且释放如组织胺、血栓素a2(txa2)、半胱胺酰基白三烯素(cyslt)及前列腺素d2(pgd2)的介体。此等介体与它们各自的受体相互作用且造成生理效应诸如血管渗透性增强、水肿、瘙痒、鼻及肺充血、支气管收缩及黏液分泌。以血管渗透性增强为例,其允许嗜酸性粒细胞及嗜碱性粒细胞白血球过量渗入至组织且因此放大过敏性反应。
目前对过敏性疾病的治疗包括可阻断或以其他方式中断此相互作用的药剂,例如抗组织胺(组织胺h1受体拮抗剂)、白三烯素受体拮抗剂、β-肾上腺素能受体激动剂及皮质类固醇。一般而言,运用抗组织胺及白三烯素拮抗剂的治疗是疗效有限的,且长期使用皮质类固醇经常与非所欲的副作用相关联。
pgd2是已知作用于两种g蛋白偶联的受体,pgd2的受体dp1及新近识别的crth2(在th2细胞中表现的化学引诱剂受体同源分子)受体(亦称为“dp2受体”)的激动剂。
如于过敏性疾病诸如过敏性鼻炎、过敏性哮喘、过敏性结膜炎、异位性皮肤炎及类似疾病中所观察到,认为pgd2水平升高将造成发炎。因此,认为阻断pgd2与其受体的相互作用是用于治疗此等疾病的有用治疗策略。
gb2388540揭示雷马曲班(ramatroban)((3r)-3-(4-氟苯-磺酰氨基)-1,2,3,4-四氢咔唑-9-丙酸)(一种具有对crth2的附加拮抗活性的txa2受体(亦称为“tp受体”)拮抗剂)的用途,它们用于预防及治疗过敏性疾病,诸如哮喘、过敏性鼻炎或过敏性结膜炎。t.ishizuka等人在cardiovasculardrugrev.2004,22(2),71-90中描述雷马曲班在发炎晚期阶段的效应。另外,雷马曲班的口服生物可利用性及其抑制前列腺素d2引发的嗜酸性粒细胞在活体外迁移的能力已经报告(journalofpharmacologyandexperimentaltherapeutics,305(1),第347至352页(2003))。
在wo2010/054113、wo2010/054114及b.a.stearns等人,bioorg.med.chem.lett.2009,19,4647-4651中已揭示具有crth2拮抗活性的氮杂吲哚乙酸衍生物。
wo2011/117798及wo2012/140612各自揭示(3-杂芳基氨基-1,2,3,4-四氢-9h-咔唑-9-基)乙酸及(7-杂芳基氨基-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)乙酸衍生物,其衍生物具有crth2拮抗活性。
已出乎意料地发现,化合物在对原代培养的大鼠肝细胞的活体外细胞毒性分析中具有显著改良的性质。因此我们预期化合物具有改良的活体内毒性特性。
现已发现,在特定条件下可发现化合物的特定结晶形式。鉴于化合物作为活性医药成分的潜在用途,化合物的该结晶形式是新颖且可具有有利性质。此等优点可包括更佳流动性质;更低吸湿性;更佳制造再现性(例如更佳过滤参数、更佳形成再现性及/或更佳沉降);及/或限定的形态。化合物的此等结晶形式可特别适用于制造某些医药组合物的方法。
附图说明
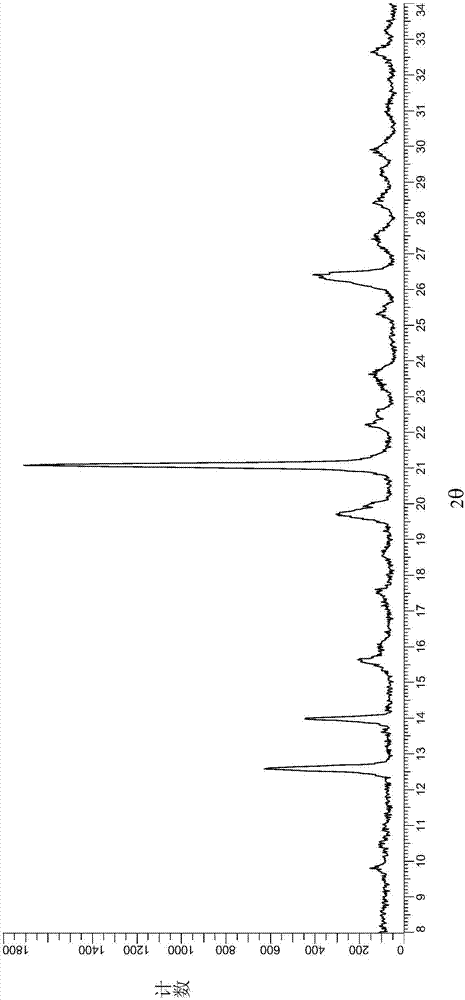
图1显示在5%rh下且在25℃下所测得的呈结晶形式1的化合物的x射线粉末衍射图,其中该x射线粉末衍射图是针对cukα辐射显示。该x射线粉末衍射图显示在指定折射角2θ下具有以下百分比(括号中给出相对峰强度)的与该图的最强峰相比的相对强度的峰(选择记录位于8至30°2θ范围内具有大于10%的相对强度的峰):12.6°(27%)、14.0°(28%)、15.6°(11%)、19.8°(14%)、20.0°(11%)、21.1°(100%)及26.4°(27%)。
图2显示在20%rh下且在25℃下所测得的呈结晶形式1的化合物的x射线粉末衍射图,其中该x射线粉末衍射图是针对cukα辐射显示。该x射线粉末衍射图显示在指定折射角2θ下具有以下百分比(括号中给出相对峰强度)的与该图的最强峰相比的相对强度的峰(选择记录位于8至30°2θ范围内具有大于10%的相对强度的峰):12.6°(34%)、14.0°(23%)、19.7°(14%)、21.1°(100%)及26.4°(19%)。
图3显示在50%rh下且在25℃下所测得的呈结晶形式1的化合物的x射线粉末衍射图,其中该x射线粉末衍射图是针对cukα辐射显示。该x射线粉末衍射图显示在指定折射角2θ下具有以下百分比(括号中给出相对峰强度)的与该图的最强峰相比的相对强度的峰(选择记录位于8至30°2θ范围内具有大于10%的相对强度的峰):12.6°(54%)、14.0°(27%)、19.5°(30%)、21.1°(100%)、21.4°(12%)、23.0°(11%)及26.1°(43%)。
图4显示在95%rh下且在25℃下所测得的呈结晶形式1的化合物的x射线粉末衍射图,其中该x射线粉末衍射图是针对cukα辐射显示。该x射线粉末衍射图显示在指定折射角2θ下具有以下百分比(括号中给出相对峰强度)的与该图的最强峰相比的相对强度的峰(选择记录位于8至30°2θ范围内具有大于10%的相对强度的峰):12.6°(62%)、14.0°(24%)、16.2°(13%)、18.9°(11%)、19.5°(32%)、21.1°(100%)、21.5°(17%)、22.9°(18%)及26.0°(47%)。
在图1至图4的x射线粉末衍射图中,折射角2θ(2θ)是描绘于水平轴上且计数是描绘于纵轴上。
为明确起见,以上所列的峰描述显示于图1至图4的x射线粉末衍射的实验结果。应了解,相对于以上峰列表而言,仅需要选择特征峰来充分且明白地表征本发明的呈各自结晶形式的化合物。
图5显示如自实例1获得的呈结晶形式1的化合物的重量水分吸附图。
在图5的重量水分吸附图中,相对湿度(%rh)是描绘于水平轴上且质量变化(%dm)是描绘于纵轴上。
图6显示呈结晶形式1的化合物的dsc迹线。
在图6的dsc图中,温度(℃)是描绘于水平轴上且功率(mw)是描绘于纵轴上。
发明详述
1)本发明的第一实施方式涉及一种(s)-2-(8-((5-氯嘧啶-2-基)(甲基)氨基)-2-氟-6,7,8,9-四氢-5h-吡啶并[3,2-b]吲哚-5-基)乙酸(化合物)的结晶形式;其特征在于在x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°及21.1°,其中该x射线粉末衍射图是在约5%相对湿度下、在约20%相对湿度下、在约50%相对湿度下或在约95%相对湿度下且在约25℃温度下测量。
应了解,如实施方式1)的该结晶形式包括呈游离酸(即,非呈盐形式)的结晶形式的化合物。另外,该结晶形式可包括非配位及/或配位溶剂(特别是非配位及/或配位水)。配位溶剂(特别是配位水)是作为结晶溶剂化物(特别是结晶水合物)的术语用于本文中。为明确起见,在本申请案中术语“结晶水合物”包括非化学计量水合物。同样地,非配位溶剂是作为经物理吸附或物理截留的溶剂(根据polymorphisminthepharmaceuticalindustry(ed.r.hilfiker,vch,2006),第8章:u.j.griesser:theimportanceofsolvates的定义)的术语用于本文中。应进一步了解,该结晶形式可根据相对湿度含有不同量的配位水且x射线粉末衍射图可因此随相对湿度而变化。为明确起见,本发明包括根据相对湿度可彼此可逆地转化的该结晶形式的所有亚结晶形式且它们的特征在于在具体给定的相对湿度下且在约25℃下的x射线粉末衍射图中存在具体给定的峰。应了解,提及在给定相对湿度下且在给定温度下的测量意指该测量是在该结晶形式已适应具体相对湿度及温度后(即在平衡时间后)进行;通常平衡时间为约0.5h至约24h,尤其1h至12h且特别1h至6h。
2)另一实施方式涉及如实施方式1)的化合物的结晶形式,其特征在于:
a.在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°、19.8°、20.0°、21.1°及26.4°,其中该x射线粉末衍射图是在约5%相对湿度下且在约25℃温度下测量;或
b.在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°、19.7°、21.1°及26.4°,其中该x射线粉末衍射图是在约20%相对湿度下且在约25℃温度下测量;或
c.在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°、19.5°、21.1°、21.4°及26.1°,其中该x射线粉末衍射图是在约50%相对湿度下且在约25℃温度下测量;或
d.在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°、19.5°、21.1°、21.5°及26.0°,其中该x射线粉末衍射图是在约95%相对湿度下且在约25℃温度下测量。
3)另一实施方式涉及如实施方式1)的化合物的结晶形式,其特征在于:
a.在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°、15.6°、19.8°、20.0°、21.1°、23.7°、26.4°、27.5°及28.4°,其中该x射线粉末衍射图是在约5%相对湿度下且在约25℃温度下测量;或
b.在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°、15.6°、19.7°、21.1°、23.3°、23.6°、26.4°、27.4°及28.4°,其中该x射线粉末衍射图是在约20%相对湿度下且在约25℃温度下测量;或
c.在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°、15.2°、16.1°、19.5°、21.1°、21.4°、23.0°、26.1°及27.0°,其中该x射线粉末衍射图是在约50%相对湿度下且在约25℃温度下测量;或
d.在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°、16.2°、18.9°、19.5°、21.1°、21.5°、22.9°、26.0°及27.0°,其中该x射线粉末衍射图是在约95%相对湿度下且在约25℃温度下测量。
4)另一实施方式涉及如实施方式1)的化合物的结晶形式,其特征在于在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°及21.1°(且尤其12.6°、14.0°、19.8°、20.0°、21.1°及26.4°),其中该x射线粉末衍射图是在约5%相对湿度下且在约25℃温度下测量。
5)另一实施方式涉及如实施方式1)的化合物的结晶形式,其特征在于在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°及21.1°(且尤其12.6°、14.0°、19.7°、21.1°及26.4°),其中该x射线粉末衍射图是在约20%相对湿度下且在约25℃温度下测量。
6)另一实施方式涉及如实施方式1)的化合物的结晶形式,其特征在于在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°及21.1°(且尤其12.6°、14.0°、19.5°、21.1°、21.4°及26.1°),其中该x射线粉末衍射图是在约50%相对湿度下且在约25℃温度下测量。
7)另一实施方式涉及如实施方式1)的化合物的结晶形式,其特征在于在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°及21.1°(且尤其12.6°、14.0°、19.5°、21.1°、21.5°及26.0°),其中该x射线粉末衍射图是在约95%相对湿度下且在约25℃温度下测量。
8)另一实施方式涉及如实施方式1)的化合物的结晶形式,
a.其基本上显示如图1中所描绘的x射线粉末衍射图,其中该x射线粉末衍射图是在约5%相对湿度下且在约25℃温度下测量;或
b.其基本上显示如图2中所描绘的x射线粉末衍射图,其中该x射线粉末衍射图是在约20%相对湿度下且在约25℃温度下测量;或
c.其基本上显示如图3中所描绘的x射线粉末衍射图,其中该x射线粉末衍射图是在约50%相对湿度下且在约25℃温度下测量;或
d.其基本上显示如图4中所描绘的x射线粉末衍射图,其中该x射线粉末衍射图是在约95%相对湿度下且在约25℃温度下测量。
9)另一实施方式涉及如实施方式1)的化合物的结晶形式,其基本上显示如图1中所描绘的x射线粉末衍射图,其中该x射线粉末衍射图是在约5%相对湿度下且在约25℃温度下测量。
10)另一实施方式涉及如实施方式1)的化合物的结晶形式,其基本上显示如图2中所描绘的x射线粉末衍射图,其中该x射线粉末衍射图是在约20%相对湿度下且在约25℃温度下测量。
11)另一实施方式涉及如实施方式1)的化合物的结晶形式,其基本上显示如图3中所描绘的x射线粉末衍射图,其中该x射线粉末衍射图是在约50%相对湿度下且在约25℃温度下测量。
12)另一实施方式涉及如实施方式1)的化合物的结晶形式,其基本上显示如图4中所描绘的x射线粉末衍射图,其中该x射线粉末衍射图是在约95%相对湿度下且在约25℃温度下测量。
13)另一实施方式涉及化合物(s)-2-(8-((5-氯嘧啶-2-基)(甲基)氨基)-2-氟-6,7,8,9-四氢-5h-吡啶并[3,2-b]吲哚-5-基)乙酸的结晶形式,诸如基本上纯的结晶形式,其可藉由以下步骤获得:
a)制备25mg/ml的化合物于thf中的溶液;
b)将0.2ml该溶液分配至4ml玻璃小瓶中;
c)藉由使用设定为30℃及100mbar的仪器蒸发thf30分钟,该仪器允许藉由组合使用红外辐射、涡旋及真空蒸发(例如购自hettichag,瑞士的combidancer);
d)添加0.02ml选自乙酸乙酯、乙腈、丙酮或异丙醇(尤其丙酮)的溶剂至固体残余物并允许其在环境温度下于该密闭小瓶中培养3天;及
e)分离所得的固体残余物。
14)另一实施方式涉及如实施方式13)的化合物的结晶形式,其特征在于在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°及21.1°,其中该x射线粉末衍射图是在约5%相对湿度下、在约20%相对湿度下、在约50%相对湿度下或在约95%相对湿度下且在约25℃温度下测量。
15)另一实施方式涉及如实施方式13)的化合物的结晶形式,其特征在于:
a.在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°、19.8°、20.0°、21.1°及26.4°,其中该x射线粉末衍射图是在约5%相对湿度下且在约25℃温度下测量;或
b.在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°、19.7°、21.1°及26.4°,其中该x射线粉末衍射图是在约20%相对湿度下且在约25℃温度下测量;或
c.在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°、19.5°、21.1°、21.4°及26.1°,其中该x射线粉末衍射图是在约50%相对湿度下且在约25℃温度下测量;或
d.在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°、19.5°、21.1°、21.5°及26.0°,其中该x射线粉末衍射图是在约95%相对湿度下且在约25℃温度下测量。
16)另一实施方式涉及如实施方式13)的化合物的结晶形式,其特征在于:
a.在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°、15.6°、19.8°、20.0°、21.1°、23.7°、26.4°、27.5°及28.4°,其中该x射线粉末衍射图是在约5%相对湿度下且在约25℃温度下测量;或
b.在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°、15.6°、19.7°、21.1°、23.3°、23.6°、26.4°、27.4°及28.4°,其中该x射线粉末衍射图是在约20%相对湿度下且在约25℃温度下测量;或
c.在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°、15.2°、16.1°、19.5°、21.1°、21.4°、23.0°、26.1°及27.0°,其中该x射线粉末衍射图是在约50%相对湿度下且在约25℃温度下测量;或
d.在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°、16.2°、18.9°、19.5°、21.1°、21.5°、22.9°、26.0°及27.0°,其中该x射线粉末衍射图是在约95%相对湿度下且在约25℃温度下测量。
17)另一实施方式涉及如实施方式13)的化合物的结晶形式,其特征在于在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°及21.1°(且尤其12.6°、14.0°、19.8°、20.0°、21.1°及26.4°),其中该x射线粉末衍射图是在约5%相对湿度下且在约25℃温度下测量。
18)另一实施方式涉及如实施方式13)的化合物的结晶形式,其特征在于在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°及21.1°(且尤其12.6°、14.0°、19.7°、21.1°及26.4°),其中该x射线粉末衍射图是在约20%相对湿度下且在约25℃温度下测量。
19)另一实施方式涉及如实施方式13)的化合物的结晶形式,其特征在于在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°及21.1°(且尤其12.6°、14.0°、19.5°、21.1°、21.4°及26.1°),其中该x射线粉末衍射图是在约50%相对湿度下且在约25℃温度下测量。
20)另一实施方式涉及如实施方式13)的化合物的结晶形式,其特征在于在该x射线粉末衍射图中于以下折射角2θ处存在峰:12.6°、14.0°及21.1°(且尤其12.6°、14.0°、19.5°、21.1°、21.5°及26.0°),其中该x射线粉末衍射图是在约95%相对湿度下且在约25℃温度下测量。
21)另一实施方式涉及如实施方式13)的化合物的结晶形式,
a.其基本上显示如图1中所描绘的x射线粉末衍射图,其中该x射线粉末衍射图是在约5%相对湿度下且在约25℃温度下测量;或
b.其基本上显示如图2中所描绘的x射线粉末衍射图,其中该x射线粉末衍射图是在约20%相对湿度下且在约25℃温度下测量;或
c.其基本上显示如图3中所描绘的x射线粉末衍射图,其中该x射线粉末衍射图是在约50%相对湿度下且在约25℃温度下测量;或
d.其基本上显示如图4中所描绘的x射线粉末衍射图,其中该x射线粉末衍射图是在约95%相对湿度下且在约25℃温度下测量。
22)另一实施方式涉及如实施方式13)的化合物的结晶形式,其基本上显示如图1中所描绘的x射线粉末衍射图,其中该x射线粉末衍射图是在约5%相对湿度下且在约25℃温度下测量。
23)另一实施方式涉及如实施方式13)的化合物的结晶形式,其基本上显示如图2中所描绘的x射线粉末衍射图,其中该x射线粉末衍射图是在约20%相对湿度下且在约25℃温度下测量。
24)另一实施方式涉及如实施方式13)的化合物的结晶形式,其基本上显示如图3中所描绘的x射线粉末衍射图,其中该x射线粉末衍射图是在约50%相对湿度下且在约25℃温度下测量。
25)另一实施方式涉及如实施方式13)的化合物的结晶形式,其基本上显示如图4中所描绘的x射线粉末衍射图,其中该x射线粉末衍射图是在约95%相对湿度下且在约25℃温度下测量。
26)另一实施方式涉及如实施方式1)至实施方式25)中任一项的化合物的结晶形式,其使用如本文所述的方法藉由差示扫描量热法测定显示在约260℃至276℃的范围内的吸热事件。
27)另一实施方式涉及如实施方式1)至实施方式26)中任一项的化合物的结晶形式,其基本上显示如图5所描绘的重量水分吸附曲线,其中该重量水分吸附曲线是在25℃下测定。
28)另一实施方式涉及如实施方式1)至实施方式12)中任一项的化合物的结晶形式,其可藉由如实施方式13)的方法获得。
基于如上文所揭示的不同实施方式1)至28)的相依性,下列实施方式是因此可行及预期的且在此以个性化形式具体揭示:
1、2+1、3+1、4+1、5+1、6+1、7+1、8+1、9+1、10+1、11+1、12+1、13、14+13、15+13、16+13、17+13、18+13、19+13、20+13、21+13、22+13、23+13、24+13、25+13、26+1、26+2+1、26+3+1、26+4+1、26+5+1、26+6+1、26+7+1、26+8+1、26+9+1、26+10+1、26+11+1、26+12+1、26+13、26+14+13、26+15+13、26+16+13、26+17+13、26+18+13、26+19+13、26+20+13、26+21+13、26+22+13、26+23+13、26+24+13、26+25+13、27+1、27+2+1、27+3+1、27+4+1、27+5+1、27+6+1、27+7+1、27+8+1、27+9+1、27+10+1、27+11+1、27+12+1、27+13、27+14+13、27+15+13、27+16+13、27+17+13、27+18+13、27+19+13、27+20+13、27+21+13、27+22+13、27+23+13、27+24+13、27+25+13、27+26+1、27+26+2、27+26+3、28+1、28+2+1、28+3+1、28+4+1、28+5+1、28+6+1、28+7+1、28+8+1、28+9+1、28+10+1、28+11+1、28+12+1;
在上文列表中,数字是指根据上文提供的编号的实施方式,而“+”指示来自另一实施方式的相依性。用顿号隔开不同的个别实施方式。换言之,例如“26+2+1”是指附属于实施方式2)的实施方式26),该实施方式2)附属于实施方式1),即实施方式“26+2+1”相当于藉由实施方式2)及26)的特征进一步表征的实施方式1)。
为明确起见,每当一个上述实施方式提及“在x射线粉末衍射图中于以下折射角2θ处的峰”时,该x射线粉末衍射图是藉由使用组合的cukα1及kα2射线获得,而无需剥除kα2;且应了解,本文所提供的2θ值的精确度是在+/-0.1至0.2°的范围内。特别地,当在本发明实施方式及权利要求中指定峰的衍射角2θ(2θ)时,给定2θ值应理解为自该值减0.2°至该值加0.2°(2θ+/-0.2°)的区间,且较佳自该值减0.1°至该值加0.1°(2θ+/-0.1°)的区间。
在针对化合物、固体、医药组合物、疾病及类似使用复数形式的情况下,此亦旨在意指单个化合物、固体、医药组合物、疾病或类似。
除非另外明确规定的定义提供更广泛或更狭隘的定义,否则在整个说明书及权利要求中,本文所提供的任何定义旨在一致地应用于在实施方式1)至28)中任一项所定义的标的及加上必要修正。应熟知,术语或表述的定义或较佳定义界定且可替代独立于本文所定义的任何或所有其他术语或表述的任何定义或较佳定义之外(及与其组合)的各自术语或表述。
术语“对映体上富集”在本发明语境中应理解为特别意指至少90重量百分比,较佳至少95重量百分比,且最佳至少99重量百分比的化合物是以一种该化合物的对映体形式存在。应了解,该化合物是以对映体富集的绝对(s)构型存在。
术语“基本上纯”在本发明语境中应理解为尤其意指至少90重量百分比,较佳至少95重量百分比,且最佳至少99重量百分比的该化合物是以本发明结晶形式存在。
当定义例如在x射线粉末衍射图中的峰的存在时,常用方法为根据s/n比(s=信号,n=噪音)进行此定义。根据此定义,当阐述一个峰必须存在于x射线粉末衍射图中时,应理解在x射线粉末衍射图中的该峰是藉由具有大于x(x为大于1的数值),通常大于2,特别大于3的s/n比(s=信号,n=噪音)而定义。
在阐述结晶形式基本上显示如图1、图2、图3或图4各自所描绘的x射线粉末衍射图的语境中,术语“基本上”意指至少在该等图中所描绘的图的主峰(即相比图中的最强峰具有多于20%,特别是多于10%的相对强度的彼等)必须存在。然而,熟习x射线粉末衍射的本领域技术人员应认识到x射线粉末衍射图中的相对强度可因较佳定向效应而经受强烈的强度变化。
除非有关相对湿度及温度使用,否则本申请案中置于数值“x”前的术语“约”是指自x减x的10%扩展至x加x的10%的区间,且较佳是指自x减x的5%扩展至x加x的5%的区间。在相对湿度的特定情况下,本申请案中置于相对湿度“y”前的术语“约”是指自相对湿度y减3扩展至y加3的区间,且较佳是指自y减1扩展至y加1的区间;例如术语“约5%相对湿度”是指相对湿度在2%与8%之间,且较佳是指相对湿度在4%与6%之间。在温度的特定情况下,本申请案中置于温度“y”前的术语“约”是指自温度y减5℃扩展至y加5℃的区间,较佳是指自y减3℃扩展至y加3℃的区间。室温意指约25℃的温度。
每当使用词语“在...之间”或“至”来描述数值范围,应了解所指示范围的端点是明确地包含于该范围内。例如:若所描述的温度范围为在40℃与80℃之间(或40℃至80℃),则此描述意指端点40℃及80℃是包含于该范围内;或若所定义的变量为在1与4之间(或1至4)的整数,此定义意指该变量为整数1、2、3或4。
表述重量%是指与所考虑的组合物的总重量相比的重量百分比。同样地,表述v/v是指所考虑的两种组分的体积的比。
如实施方式1)至28)中任一项的化合物的结晶形式,特别是其基本上纯的结晶形式可用作药物,例如呈用于经肠(此等特别是口服的)或非经肠投与(包括局部施用或吸入)的医药组合物的形式。
29)另一实施方式涉及如实施方式1)至28)中任一项的化合物(s)-2-(8-((5-氯嘧啶-2-基)(甲基)氨基)-2-氟-6,7,8,9-四氢-5h-吡啶并[3,2-b]吲哚-5-基)乙酸的结晶形式,其用作药物。
如实施方式1)至28)中任一项的化合物的固体,特别是其基本上纯的固体可呈单一组分或呈与化合物的其他结晶形式或非晶形式的混合物使用。
该等医药组合物可以任何本领域技术人员已知的方式如下制造(例如,参见remington,thescienceandpracticeofpharmacy,第21版(2005),第5部分,“pharmaceuticalmanufacturing”[lippincottwilliams&wilkins出版]):藉由将本发明结晶形式(视需要与其他具有治疗价值的物质组合)连同合适的非毒性惰性医药上可接受的固体或液体载体材料,及若需要,连同常用药物佐剂变成盖伦投药形式。
30)本发明的另一实施方式涉及包含作为活性成分的如实施方式1)至28)中任一项的化合物的结晶形式及至少一种医药上可接受的载体材料的医药组合物。
31)本发明的另一实施方式涉及如实施方式1)至28)中任一项的化合物的结晶形式,其用于制造医药组合物,其中该医药组合物包含作为活性成分的该化合物及至少一种医药上可接受的载体材料。
32)本发明的另一实施方式涉及如实施方式1)至28)中任一项的化合物的结晶形式,其用于预防或治疗选自由以下组成的群的疾病:慢性及急性过敏性/免疫疾病/病症,其包括哮喘、过敏性哮喘、嗜酸性粒细胞性哮喘、严重型哮喘、鼻炎、过敏性鼻炎、血管性水肿、昆虫毒液过敏、药物过敏、过敏性窦炎、过敏性肾炎、过敏性结膜炎、异位性皮肤炎、支气管哮喘、食物过敏、全身性细胞肥大病症、过敏性休克、荨麻疹、湿疹、溃疡性结肠炎、慢性阻塞性肺病(copd)、发炎性肠病、风湿性关节炎及鼻息肉病;嗜酸性粒细胞相关的疾病,其包括小血管炎如彻奇-斯全司症候群(churg-strausssyndrome)、韦格纳氏(wegener's)肉芽肿病、显微镜下多血管炎(及后者的器官特异性子集)、嗜酸性粒细胞增多症候群如嗜酸性粒细胞性肺炎、嗜酸性粒细胞性食道炎、逆流性食道炎、嗜酸性粒细胞性心内膜炎(吕佛勒氏(loeffler’s)心内膜炎)、嗜酸性粒细胞增多-肌痛症候群、嗜酸粒细胞性筋膜炎、嗜酸性粒细胞性脓疱性毛囊炎(太藤氏(ofuji's)疾病)、嗜酸性粒细胞性溃疡、血管淋巴样增生伴嗜酸性粒细胞增多症(alhe)、嗜酸性粒细胞性蜂窝性组织炎(韦尔斯(wells)症候群)、慢性嗜酸性粒细胞性白血病、dress症候群(药疹伴嗜酸性粒细胞增多及全身症状)及斯蒂尔病(still’sdisease)(幼年特发性关节炎);嗜碱性粒细胞相关的疾病,其包括嗜碱性粒细胞性白血病及嗜碱性白细胞增多;及囊肿性纤维化。
33)本发明的一较佳实施方式涉及如实施方式1)至28)中任一项的化合物的结晶形式,其用于预防或治疗选自由以下组成的群的疾病:哮喘、嗜酸性粒细胞性哮喘、过敏性鼻炎、异位性皮肤炎、鼻息肉病、食物过敏(尤其是ige所介导的食物过敏)、荨麻疹(尤其是慢性荨麻疹)、嗜酸性粒细胞性食道炎、彻奇-斯全司症候群、嗜酸性粒细胞增多症候群、嗜酸性粒细胞性肺炎(尤其是慢性嗜酸性粒细胞性肺炎)、dress症候群、斯蒂尔病、copd及囊肿性纤维化(且尤其是哮喘、嗜酸性粒细胞性哮喘、过敏性鼻炎、异位性皮肤炎、ige介导的食物过敏、慢性荨麻疹、嗜酸性粒细胞性食道炎及彻奇-斯全司症候群)。
34)本发明的另一实施方式涉及如实施方式1)至28)中任一项的化合物的结晶形式,其用于制造用于预防或治疗选自由以下组成的群的疾病的医药组合物:慢性及急性过敏性/免疫疾病/病症,其包括哮喘、过敏性哮喘、嗜酸性粒细胞性哮喘、严重型哮喘、鼻炎、过敏性鼻炎、血管性水肿、昆虫毒液过敏、药物过敏、过敏性窦炎、过敏性肾炎、过敏性结膜炎、异位性皮肤炎、支气管哮喘、食物过敏、全身性细胞肥大病症、过敏性休克、荨麻疹、湿疹、溃疡性结肠炎、慢性阻塞性肺病(copd)、发炎性肠病、风湿性关节炎及鼻息肉病;嗜酸性粒细胞相关的疾病,其包括小血管炎如彻奇-斯全司症候群、韦格纳氏肉芽肿病、显微镜下多血管炎(及后者的器官特异性子集)、嗜酸性粒细胞增多症候群如嗜酸性粒细胞性肺炎、嗜酸性粒细胞性食道炎、逆流性食道炎、嗜酸性粒细胞性心内膜炎(吕佛勒氏心内膜炎)、嗜酸性粒细胞增多-肌痛症候群、嗜酸粒细胞性筋膜炎、嗜酸性粒细胞性脓疱性毛囊炎(太藤氏疾病)、嗜酸性粒细胞性溃疡、血管淋巴样增生伴嗜酸性粒细胞增多症(alhe)、嗜酸性粒细胞性蜂窝性组织炎(韦尔斯症候群)、慢性嗜酸性粒细胞性白血病、dress症候群(药疹伴嗜酸性粒细胞增多及全身症状)及斯蒂尔病(幼年特发性关节炎);嗜碱性粒细胞相关的疾病,其包括嗜碱性粒细胞性白血病及嗜碱性白细胞增多;及囊肿性纤维化。
35)本发明的一较佳实施方式涉及如实施方式1)至28)中任一项的化合物的结晶形式,其用于制造用于预防或治疗选自由以下组成的群的疾病的医药组合物:哮喘、嗜酸性粒细胞性哮喘、过敏性鼻炎、异位性皮肤炎、鼻息肉病、食物过敏(尤其是ige介导的食物过敏)、荨麻疹(尤其是慢性荨麻疹)、嗜酸性粒细胞性食道炎、彻奇-斯全司症候群、嗜酸性粒细胞增多症候群、嗜酸性粒细胞性肺炎(尤其是慢性嗜酸性粒细胞性肺炎)、dress症候群、斯蒂尔病、copd及囊肿性纤维化(且尤其是哮喘、嗜酸性粒细胞性哮喘、过敏性鼻炎、异位性皮肤炎、ige介导的食物过敏、慢性荨麻疹、嗜酸性粒细胞性食道炎及彻奇-斯全司症候群)。
36)本发明的另一实施方式涉及如实施方式30)的医药组合物,其用于预防或治疗选自由以下组成的群的疾病:慢性及急性过敏性/免疫疾病/病症,其包括哮喘、过敏性哮喘、嗜酸性粒细胞性哮喘、严重型哮喘、鼻炎、过敏性鼻炎、血管性水肿、昆虫毒液过敏、药物过敏、过敏性窦炎、过敏性肾炎、过敏性结膜炎、异位性皮肤炎、支气管哮喘、食物过敏、全身性细胞肥大病症、过敏性休克、荨麻疹、湿疹、溃疡性结肠炎、慢性阻塞性肺病(copd)、发炎性肠病、风湿性关节炎及鼻息肉病;嗜酸性粒细胞相关的疾病,其包括小血管炎如彻奇-斯全司症候群、韦格纳氏肉芽肿病、显微镜下多血管炎(及后者的器官特异性子集)、嗜酸性粒细胞增多症候群如嗜酸性粒细胞性肺炎、嗜酸性粒细胞性食道炎、逆流性食道炎、嗜酸性粒细胞性心内膜炎(吕佛勒氏心内膜炎)、嗜酸性粒细胞增多-肌痛症候群、嗜酸粒细胞性筋膜炎、嗜酸性粒细胞性脓疱性毛囊炎(太藤氏疾病)、嗜酸性粒细胞性溃疡、血管淋巴样增生伴嗜酸性粒细胞增多症(alhe)、嗜酸性粒细胞性蜂窝性组织炎(韦尔斯症候群)、慢性嗜酸性粒细胞性白血病、dress症候群(药疹伴嗜酸性粒细胞增多及全身症状)及斯蒂尔病(幼年特发性关节炎);嗜碱性粒细胞相关的疾病,其包括嗜碱性粒细胞性白血病及嗜碱性白细胞增多;及囊肿性纤维化。
37)本发明的一较佳实施方式涉及如实施方式30)的医药组合物,其用于预防或治疗选自由以下组成的群的疾病:哮喘、嗜酸性粒细胞性哮喘、过敏性鼻炎、异位性皮肤炎、鼻息肉病、食物过敏(尤其是ige介导的食物过敏)、荨麻疹(尤其是慢性荨麻疹)、嗜酸性粒细胞性食道炎、彻奇-斯全司症候群、嗜酸性粒细胞增多症候群、嗜酸性粒细胞性肺炎(尤其是慢性嗜酸性粒细胞性肺炎)、dress症候群、斯蒂尔病、copd及囊肿性纤维化(且尤其是哮喘、嗜酸性粒细胞性哮喘、过敏性鼻炎、异位性皮肤炎、ige介导的食物过敏、慢性荨麻疹、嗜酸性粒细胞性食道炎及彻奇-斯全司症候群)。
本发明亦涉及一种用于预防或治疗本文所提及的疾病或病症的方法,其包括投与给个体医药上活性量的如实施方式1)至28)中任一项的化合物的结晶形式或如实施方式30)的医药组合物。
本发明亦涉及一种制备呈对映体上富集形式的化合物的方法,及制备并表征如实施方式1)至28)中任一项的化合物的结晶形式的方法。该等方法是描述于实施方式13)中,以及描述于以下实验部分的程序中。
具体实施方式
实验程序:
缩写字(如上文或下文所使用)
ac乙酰基(诸如oac=乙酸酯、acoh=乙酸)
aq.水溶液
boc叔丁氧基羰基
bsa牛血清白蛋白
bu丁基,诸如n-bu=正丁基
conc.浓
dcm二氯甲烷
dea二乙胺
diea二异丙基乙胺
dmfn,n-二甲基甲酰胺
dmso二甲基亚砜
dpm每分钟衰变
edta乙二胺四乙酸
els(d)蒸发光散射(检测)
eq当量
et乙基
etoac乙酸乙酯
etoh乙醇
fc在硅胶上的快速层析
fig图
h小时
hepes4-(2-羟乙基)-哌嗪-1-乙磺酸
1h-nmr质子核磁共振
hplc高效液相层析
lc-ms液相层析–质谱法
m摩尔浓度
m准确质量(用于lc-ms)
me甲基
mecn乙腈
meoh甲醇
mw微波
mw毫瓦
μl微升
min分钟
ms质谱法
n当量浓度
pbs磷酸盐缓冲盐水
ph苯基
pph3三苯基膦
prep.制备型
rh相对湿度
rt室温
sat.饱和
tfa三氟乙酸
thf四氢呋喃
tr滞留时间
tris三-(羟甲基)氨基甲烷缓冲液
uv紫外线
除非另有指示,否则所有所用的溶剂及试剂皆可自商业来源得到。
用摄氏度(℃)指示温度。除非另有指示,否则反应于室温(rt)下进行。
除非另有指示,否则在混合物中,呈液体形式的溶剂或洗脱剂或试剂混合物的份数的关系是以体积关系(v/v)给定。
藉由在硅胶上的快速管柱层析法(fc)或藉由制备型hplc纯化化合物。藉由lc-ms表征本发明所描述的化合物(滞留时间tr是以分钟给定;获得自质谱的分子量是以g/mol给定,使用以下所列的条件)。
分析lc-ms条件是用于以下实例中:
在配有dionexp580二元泵、dionexpda-100光电二极管数组检测器及finniganaqa质谱仪的agilent1100系统上进行lc-ms分析。
使用以下洗脱条件获得lc滞留时间。
-在
制备型hplc/ms纯化(碱性条件)是在配有gilson215自动进样器及洗脱流份收集器、dionexuvd340udad检测器、高分子实验室pl-els1000els检测器及thermomsqplusms检测器的gilson333/334二元高压梯度泵系统上,使用watersxbridgec18管柱(10μm,30×75mm),以水/0.5%25%nh4oh(b)及mecn(a)自80/20开始至5/95(b)/(a)历时5分钟的线性梯度;流速75ml/min进行。
在手性固定相上的分析型hplc是在daicelchiralpakad-h(4.6x250mm,5μm)管柱或chiralpakay-h(4.6x250mm,5μm)管柱上进行。手性hplc的典型条件为50%庚烷+0.05%dea与50%etoh+0.05%dea的等浓度混合物,在0.8ml/min.的流速下,在210nm下检测(手性hplc-1)或40%庚烷与60%etoh+0.1%tfa的等浓度混合物,在1.0ml/min.的流速下,在210nm下检测(手性hplc-2)。
在手性固定相上的制备型hplc是在daicelchiralpakad-h(20x250mm,5μm)管柱上进行。手性hplc的典型条件为50%etoh+0.05%dea与50%庚烷的等浓度混合物,在34ml/min的流速下,在210nm下检测。
x射线粉末衍射分析(xrpd)
在配有用于设定及控制样品上方的温度及相对湿度的anton-paarchcplus+腔室的brukerd8高级x射线衍射仪上收集x射线粉末衍射图。该衍射仪是配有lynxeye检测器、使用平行光束光学器件以cukα辐射操作及以反射模式操作。通常,在40kv/40ma下运行x射线管。应用在3至35°(2θ)的扫描范围内的0.02°(2θ)的步长及95秒的步进时间。将粉末轻压至标准anton-paarttk样品支撑架中。利用cukα1
重量水分吸附(gvs)分析
在于25℃下以步进模式操作的多样品仪器sps-100n(projektmesstechnik,ulm,德国)上进行测量。在开始预定义的湿度程序之前允许样品在40%相对湿度下平衡(应用40-0-95-0-95-40%rh,步长为5%δrh及以24小时/步的最大平衡时间)。使用约20至30mg的各样品。根据欧洲药典技术指导(europeanpharmacopeiatechnicalguide)(1999年,第86页)完成吸湿性分类,例如,轻微吸湿性:质量增加小于2%且等于或大于0.2%质量/质量;吸湿性:质量增加小于15%且等于或大于2%质量/质量。考虑在第一吸附扫描中介于20%相对湿度与80%相对湿度之间的质量改变。
差示扫描量热法(dsc)
在配有34位自动进样器的mettlertoledostare系统(dsc822e模块、具有陶瓷传感器的量测池及13.00版本star软件)上收集dsc数据。使用经认证的铟校正该仪器的能量及温度。除非另外指出,否则通常以10℃min-1自-20℃至320℃加热在经自动穿孔的40μlmettler铝盘中的2mg的各样品。对样品维持20mlmin-1的氮气冲洗。
i-化学
(s)-2-(8-((5-氯嘧啶-2-基)(甲基)氨基)-2-氟-6,7,8,9-四氢-5h-吡啶并[3,2-b]吲哚-5-基)乙酸的合成
1)4-((5-氯嘧啶-2-基)(甲基)氨基)环己酮的合成
(r表示5-氯-嘧啶-2-基)
在0℃下,将甲基胺(8m含于etoh中,1eq)及nabh(oac)3(1.5eq)连续添加至市售1,4-二氧杂螺[4.5]癸烷-8-酮(1eq)含于dcm(20ml/10mmol)中的溶液中。允许将反应混合物升温至rt并搅拌2h。将反应混合物倒入nahco3的饱和溶液,用盐水清洗有机层,在mgso4上干燥并在真空中蒸发以获得n-甲基-1,4-二氧杂螺[4.5]癸烷-8-胺,其无需进一步纯化即可用于后续步骤。
将diea(2eq)及2,5-二氯嘧啶(1.05eq)添加至n-甲基-1,4-二氧杂螺[4,5]癸烷-8-胺(1eq)含于dmf(10.5ml/6mmol)中的溶液中。将反应混合物在90℃下搅拌过夜。冷却至rt后,添加乙酸异丙酯。用水及10%柠檬酸水溶液清洗混合物。干燥(mgso4)有机层并在真空中浓缩。藉由fc(0至15%etoac/庚烷)纯化粗产物,以提供呈固体的所需中间化合物。
在rt下搅拌此中间体(1eq)含于2nhcl(2.7ml/5mmol)与meoh(2.7ml/5mmol)的混合物中的溶液过夜。用dcm萃取水层。干燥(mgso4)有机层并在真空中浓缩。藉由fc(0至17%etoac/庚烷)纯化粗制残余物,以获得呈固体的标题化合物。
lc-ms:tr=0.78min;[m+h]+=240.2。
2.1)n-(5-氯嘧啶-2-基)-2-氟-n-甲基-6,7,8,9-四氢-5h-吡啶并[3,2-b]吲哚-8-胺的合成(方法a)
(r表示5-氯-嘧啶-2-基)
在小瓶中合并3-氨基-2-溴-6-氟-吡啶(1eq)、4-((5-氯嘧啶-2-基)(甲基)氨基)环己酮(1.2eq)、(ph3p)4pd(0.05eq)与吡啶(8.17eq)的溶液。在160℃下用mw照射该小瓶1h。再次添加(ph3p)4pd(0.025eq)并在160℃下再次用mw照射该小瓶30分钟。冷却至rt后,将反应混合物与水合并并用dcm萃取两次。干燥(mgso4)、过滤并在真空中浓缩合并的有机萃取物。藉由制备型hplc(碱性条件)纯化残余物以获得所需产物。
lc-ms:tr=0.87min;[m+h]+=332.09。
2.2)n-(5-氯嘧啶-2-基)-2-氟-n-甲基-6,7,8,9-四氢-5h-吡啶并[3,2-b]吲哚-8-胺的合成(方法b)
a)1-(6-氟吡啶-3-基)肼-1,2-二甲酸二-叔丁酯的合成
在n2氛围下,将在-40℃下的正丁基锂(1.6m含于己烷中,1.1eq)的溶液逐滴添加至5-溴-2-氟-吡啶(1eq)含于乙醚(14.5eq)中的溶液中。在-40℃下搅拌反应混合物20分钟并随后逐滴添加偶氮二甲酸二-叔丁酯(1.1eq)含于thf(18.5eq)中的溶液。在-40℃下搅拌反应混合物30分钟并允许其历时30分钟升温至rt。添加水,接着添加dcm。分离有机相并在mgso4上干燥、过滤并在真空中浓缩。藉由fc(etoac/正庚烷:2/8)纯化残余物以获得所需产物。
lc-ms:tr=0.88min;[m+h]+=328.12。
b)n-(5-氯嘧啶-2-基)-2-氟-n-甲基-6,7,8,9-四氢-5h-吡啶并[3,2-b]吲哚-8-胺的合成
(r表示5-氯-嘧啶-2-基)
在100℃下搅拌1-(6-氟吡啶-3-基)肼-1,2-二甲酸二-叔丁酯(1eq)、4-((5-氯嘧啶-2-基)(甲基)氨基)环己酮(1eq)含于4%h2so4水溶液(10ml/0.04mol)中的溶液150分钟。冷却至rt后,将反应混合物与饱和nahco3合并并用etoac萃取。干燥(mgso4)、过滤并在真空中浓缩合并的有机萃取物。藉由制备型hplc(碱性条件)纯化残余物以获得所需产物。
lc-ms:tr=0.87min;[m+h]+=332.03。
3)(s)-2-(8-((5-氯嘧啶-2-基)(甲基)氨基)-2-氟-6,7,8,9-四氢-5h-吡啶并[3,2-b]吲哚-5-基)乙酸乙酯及(r)-2-(8-((5-氯嘧啶-2-基)(甲基)氨基)-2-氟-6,7,8,9-四氢-5h-吡啶并[3,2-b]吲哚-5-基)乙酸乙酯的合成
将nah(95%,56.1mg,2.22mmol,1.2eq)小心地添加至n-(5-氯嘧啶-2-基)-2-氟-n-甲基-6,7,8,9-四氢-5h-吡啶并[3,2-b]吲哚-8-胺(614mg,1.85mmol,1eq)含于dmf(6.36ml)中的冷溶液(0℃)中。搅拌反应混合物20分钟。缓慢添加溴乙酸乙酯(0.23ml,2.04mmol,1.1eq)并允许反应混合物升温至rt并搅拌2h。将反应混合物溶于etoac中,并用饱和nahco3溶液清洗。在mgso4上干燥、过滤并在真空中浓缩有机萃取物。藉由fc(正庚烷对正庚烷/etoac:7/3)纯化残余物以获得呈外消旋物的所需产物。
lc-ms:tr:0.96min./[m+h]+:418.01。
藉由在手性固定相上的制备型hplc分离所得产物的两种对映体:
(s)-2-(8-((5-氯嘧啶-2-基)(甲基)氨基)-2-氟-6,7,8,9-四氢-5h-吡啶并[3,2-b]吲哚-5-基)乙酸乙酯
(271mg,35%):hplc(手性hplc-1):tr:6.22min;
(r)-2-(8-((5-氯嘧啶-2-基)(甲基)氨基)-2-氟-6,7,8,9-四氢-5h-吡啶并[3,2-b]吲哚-5-基)乙酸乙酯
(273mg,35%):hplc(手性hplc-1):tr:7.66min。
4)(s)-2-(8-((5-氯嘧啶-2-基)(甲基)氨基)-2-氟-6,7,8,9-四氢-5h-吡啶并[3,2-b]吲哚-5-基)乙酸的合成
在rt下将naoh(1n,10ml,10mmol,15.4eq)添加至(s)-2-(8-((5-氯嘧啶-2-基)(甲基)氨基)-2-氟-6,7,8,9-四氢-5h-吡啶并[3,2-b]吲哚-5-基)乙酸乙酯(271mg,0.649mmol,1eq)含于thf(10ml)中的溶液中。在rt下搅拌反应混合物1h。在真空中浓缩反应混合物以仅移除thf。随后用浓hcl将其酸化至ph约5至6并在rt下搅拌。用etoac(4x)萃取悬浮液。在mgso4上干燥、过滤并在真空中浓缩合并的有机层以获得呈米色固体的标题化合物(255mg,100%)。
lc-ms:tr:0.82min./[m+h]+:390.12
hplc(手性hplc-2):tr:4.96min。
ii.化合物的结晶形式的制备
实例1:呈结晶形式1的化合物的制备及表征
将0.1g化合物溶于4mlthf中并将0.2ml溶液移至4ml棕色玻璃小瓶中。藉由使用设定在30℃及100mbar的combidancer(hettichag,瑞士)自小瓶中蒸发thf30分钟。留在玻璃小瓶中的固体残余物是非晶体。将0.02ml丙酮添加至固体残余物中,将该小瓶气密式密封并允许混合物在环境温度下于该密闭小瓶中培养3天。固体残余物为呈结晶形式1的化合物。除丙酮外,该程序可使用乙酸乙酯、乙腈或异丙醇完成。
表1:呈结晶形式1的化合物的表征数据
iii.生物分析
hcrth2受体膜的制备及放射性配位体置换分析:
首先,使用橡胶淀帚将重组hek293-hcrth2细胞自培养板卸离至于5ml缓冲液a/板(缓冲液a:5mmtris,1mmmgcl2-6h2o,ph=7.4)中。随后将细胞转移至离心管中并以400g离心5分钟。将细胞小球再悬浮于相同缓冲液中并在–80℃下冷冻管子。融解细胞并藉由使用polytron均质机均质化(30秒)来产生膜片段。随后将膜片段以3000g离心20分钟并再悬浮于缓冲液c(缓冲液c:75mmtris,25mmmgcl2,250mm蔗糖,ph7.4)中。在–20℃下储存膜片段的等分试样。
以250μl的最终分析体积进行结合分析。首先,将预先在结合缓冲液(结合缓冲液:50mmtris-碱,100mmnacl,1mmedta,0.1%bsa(无蛋白酶),0.01%nan3,10mmmncl2,ph7.0)中稀释的25μl测试化合物置于各孔中。添加75μl结合缓冲液后,将50μl放射性配位体3h-pgd2(以2.5nm(220.000dpm/孔)的量,来自anawaart0662)添加至各孔中。藉由添加100μlcrth2膜片段以达成20μg/孔的最终浓度,开始结合分析。为了非特异性结合,将pgd2添加至反应混合物中至10mm最终浓度。在室温下培养此分析混合物90分钟并随后滤过在0.5%聚乙亚胺(pei)中预浸泡3小时的gf/c过滤器96-孔板。用冰冷的结合缓冲液清洗过滤器-孔3次。接着,将40μlmicroscint-40(packard)添加至各孔中并在topcount(packard)中量化留存放射性。
化合物的拮抗活性:ic50=5.6nm。
放射性配位体置换分析-人类血清白蛋白(hsa):
如上所述,经以下修改进行在人类血清白蛋白(hsa)存在下的放射性配位体置换分析。结合缓冲液-hsa:结合缓冲液+0.5%来自人类血清a1887的σ-白蛋白(取代0.1%bsa)。将预先在结合缓冲液–hsa中稀释的25μl体积的测试化合物置于各孔中。添加75μl结合缓冲液-hsa后,将50μl3h-pgd2(以2.5nm(220.000dpm/孔)的量,来自anawaart0662)添加至各孔中。剩余方案是与如上所述相同。
化合物的拮抗活性:ic50=5.0nm。
利用人类血浆的嗜酸性粒细胞形状改变分析
获得知情同意书后,根据由瑞士巴塞尔(basel)的人体试验委员会(ethicscommittee)批准的方案,藉由静脉穿刺汲取血液样本。使用polymorphpreptm方法(axis-shield)分离多形核白血球(含有嗜酸性粒细胞、嗜碱性粒细胞及嗜中性粒细胞)。简而言之,将经抗凝的全血在polymorphprep梯度(密度1.113g/ml)上分层并以500g离心30分钟。收取多形核细胞部分并藉由低渗盐水溶胞耗尽红血球。
将多形核细胞以5x106个细胞/ml再悬浮于分析缓冲液(具有ca2+/mg2+的1xpbs,补充有0.1%bsa、10mmhepes及10mm葡萄糖,ph7.4)中并用抗-cd49d-apc(apc=别藻蓝蛋白)在rt下染色1小时。在人类血浆(经凝血酶抑制剂抗凝)中预培育不同浓度的测试化合物10分钟。随后,将人类血浆添加至多形核细胞至50%的最终分析体积,其中多形核细胞为4x106个细胞/ml。在37℃下培育10分钟后,在37℃下藉由添加100nm最终浓度的pgd2来活化多形核细胞5分钟。藉由添加0.5ml多聚甲醛(1%)停止活化。
在经多聚甲醛固定不久后,藉由facscanto流式细胞仪(bdbiosciences)分析样品并藉由目标细胞的前向散射(fsc)及侧向散射(ssc)特征识别靶细胞。藉由抗-cd49d-apc信号及其特征性侧向散射(ssc)分析图识别嗜酸性粒细胞。将指示嗜酸性粒细胞活化的形状改变反应量化为具有增强前向散射的细胞的百分比。
化合物的拮抗活性:ic50=3.1nm。
在原代培养的大鼠肝细胞中的体外细胞毒性
1.方法
1.1大鼠肝细胞的分离及培养
用戊巴比妥钠麻醉成年雄性维斯塔(wistar)大鼠并根据标准程序(即藉由使用胶原酶溶液的原位肝脏灌注)分离肝细胞。藉由锥蓝染色排除法检查的纯化肝细胞的存活率为大于85%。将所分离的肝细胞再悬浮于标准威廉姆斯(williams)培养基e中,不含酚红,补充(wme补充)有运铁蛋白(100μg/ml)、三碘甲腺原胺酸(10μg/ml)、庆大霉素(50μg/ml)、氢化可的松半琥珀酸酯(13.36μg/ml)、胰高血糖素(5μg/ml)、hepes(10mm)、纤维核苷(10μg/ml)、胰岛素(10μg/ml)、链霉素(100μg/ml)及青霉素(100u/ml)及10%胎牛血清(fbs)。将细胞以2x105个细胞/孔的起始密度置于经胶原蛋白涂覆的24孔板中。在附着至培养板4h后,将培养基吸出并由不含fbs含测试化合物的新制wme补充更新并在37℃下在95%o2及5%co2的氛围下培养24h。对于各实验(即,各批肝细胞)而言,以测试化合物的处理是以一式四份进行。一式四份对照(仅以溶媒的处理)是亦存在于各培养板上。
1.2活体外曝露于测试化合物
在处理开始前数小时,于dmso中制备测试化合物的储备溶液。在刚处理前,将此等储备液的适当稀释液添加至培养基中,以获得0、3、10、30、100及300μm的最终浓度。溶媒dmso的最终浓度为1%(v/v)。
1.3细胞培养物的存活率
1.3.1单层形态的监测
在曝露于测试化合物24小时后,藉由光学显微镜监测单层肝细胞的形态。如下列分级描述与处理相关的效应:
0当与对照培养物比较时,处理后未观测到形态改变
1至3处理造成任何形态的改变,例如细胞内肉芽发生、空泡形成或细胞死亡。此等改变根据严重程度被视为轻微(1)、中等(2)或强烈(3)。
k处理造成细胞100%死亡及/或单层的完全脱离,从而获得清晰的无细胞盘。
1.3.2乳酸脱氢酶的漏出
在处理肝细胞培养物24h后,小心收集培养基的等分试样并将其用于藉由分光光度计使用购自clontech的ldh细胞毒性检测试剂盒(目录号630117,mountainview,ca,usa)的乳酸脱氢酶(ldh)活性的分析。对于各实验而言,附加培养物是用于在处理开始时的总细胞内ldh活性的测定。出于此目的,使用冷盐水在处理之前清洗4孔细胞培养物/实验、在新制培养基中超声处理并分析均质物的总ldh活性。评估培养基中的酶活性并表示成在开始处理时存在于所培养的肝细胞中的总活性的百分比。
2.数据分析
基于在24h处理后的细胞形态及ldh漏出,给定各化合物的最低细胞毒性浓度(lcc)及无效浓度(noec)。将lcc定义为测试化合物导致在所培养的大鼠肝细胞上明显效应的最低浓度(形态分级≥2或ldh漏出≥2倍增加)。>300μm的lcc值指示在以300μm的最高测试浓度的两个端点上无效应。将noec定义为化合物在所培养的大鼠肝细胞上无效应的最高测试浓度。
3.结果:lcc值及noec值
活体内肝脏毒性
可藉由使用三种不同剂量的该化合物长达4周的口服处理大鼠及非啮齿类物种来分析式(i)化合物的肝脏毒性。可在后续无处理期间(恢复期间)研究可能毒性的可逆性。基于各自物种的剂量范围发现研究选择剂量水平。预期高剂量识别接近最大耐受剂量的器官毒性。基于估计的治疗人类曝露量选择中剂量及低剂量。在每个剂量水平下测量该化合物的曝露量。
在处理结束及恢复结束时,测量在血液中的肝脏生物标志(诸如例如肝脏酶、蛋白质、三酸甘油酯或胆固醇)。此外,在显微镜下检查经苏木色素-曙红(hematoxilin-eosin)染色的肝脏切片以直接评估可能的器官损害。可需要肝脏切片的专用染色以进一步表征可能的肝脏发现结果。
- 还没有人留言评论。精彩留言会获得点赞!