取代的萘二酰亚胺类及其用途的制作方法
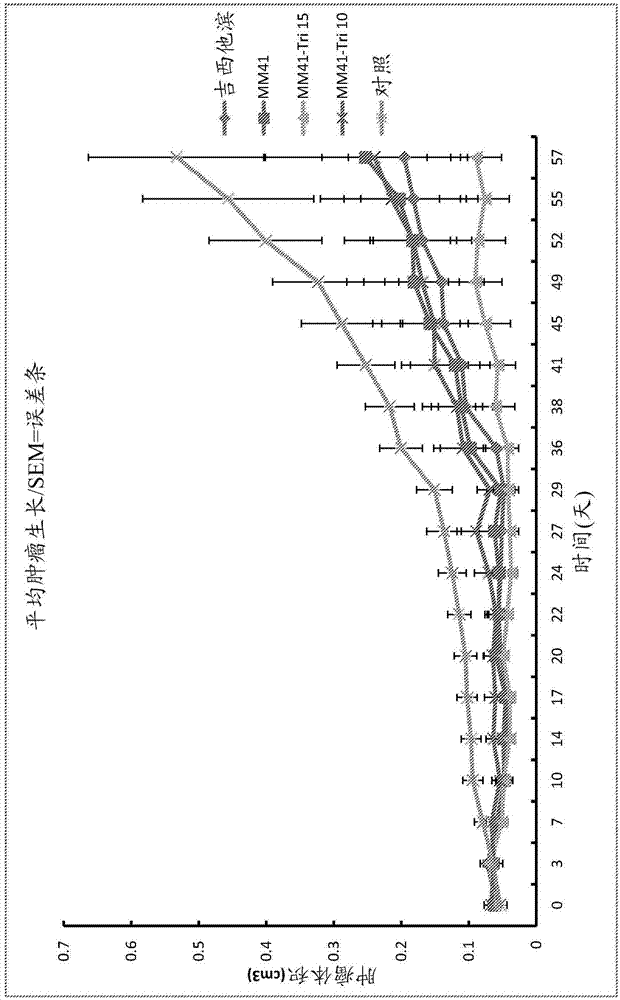
在wo2009/068916中,我们描述了三取代和四取代的萘二酰亚胺及制备它们的方法。没有一种三取代的示例性产物在核心配体的等位和极性位置上具有不同的氨基功能配体。据说适用于制备三取代的化合物的方法是基于以下示意图:其中r1是被任选取代的烷基或芳基并且n是0或1。在实践中,产生了四取代(n=1)的化合物和三取代(n=0)的化合物的混合物。所有取代基(即r1基团)相同。说明书描述了用于从上述使用的二溴化合物的二氯取代的类似物开始制备四取代的化合物的方法。该工艺在一个步骤中进行,在这种情况下,相同的h2nr1试剂在两个酸酐基团和两个氯取代的碳上反应以在产物上得到4个相同的r1取代基,或者在两个步骤中进行,其中在第一步骤中使第一试剂h2nr2在两个酸酐基团上反应和在第二步骤中使第二试剂h2nr3在两个氯取代的碳原子上反应。在酰亚胺取代基上和/或芳族环上具有碱性取代基的化合物具有强大的dna四联体结合性质。已在wo2009/068916、us20140275065a和hampels.m.等人,bioorg.med.chem.lett.(2010)20,6459-6463、micco.m.,等人,j.med.chem.(2013)56,2959-2974、collie,g.w.,等人,j.a.c.s.(2012)134,2723-2731、gunaratnam,m.等人,j.med.chem.(2009)52,3774-3783、gunaratnam,m.等人,bioorg.med.chem.(2011)19,7151-7157及mitchell,t.等人,biochemistry(2013)52,1429-1436中测试了四取代的产物(包括具有不同于基团r3的基团r2的产物)与端粒四联体以及在一些基因的启动子区中存在的那些的结合性质。这些数据显示了数种蛋白的有效下调,所述基因的启动子被二酰亚胺靶向并因此导致来自一组癌细胞系的数种细胞系的生长抑制。我们在这些公开中提出,进一步研究改变取代基基团的性质和阳离子取代基中叔胺基团的碱性对结合特性和强度的影响,并通过包括胰腺癌在内的癌症测试模型来研究该化合物在癌症治疗中的潜力。在scientificreports(2015)5:11385,ohnmacht,s.a.等人中公开了4,9-双((3-(4-甲基哌嗪-l-基)-丙基)氨基)-2,7-双(3-吗啉代丙基)苯并[lmn][3,8]菲罗啉-1,3,6,8(2h,7h)-四酮,也被称为mm41,在人胰腺癌的小鼠模型中的体内活性。在nadai,m.等人,int.j.oncol.(2015)46,369-380中公开了一种三取代的萘二酰亚胺化合物,其具有在每个亚氨基氮原子处的2-二甲基氨基乙基基团取代以及具有作为第三取代基的在ndi核上的4-位置处的2-(4-羟基-3-二甲基氨基甲基苯基)乙基氨基基团取代。其具有稳定端粒g-四联体的活性,从而导致端粒功能障碍和端粒酶下调。在一组细胞系上的全基因表达显示出调控参与端粒功能和癌症机制的基因。然而,作者总结得出仍然缺乏g-4s在细胞背景中的生物相关性的直接证据。由nadai等人报道的三取代的化合物的合成公开于doria等人,orgbiomol.chem,(2012)10,2798-2806中。在本发明中提供了新型的式i的化合物及其盐、水合物和溶剂化物:基团r2是相同的并且选自直链和支链c1-6烷二基;r3选自h和c1-6烷基;r4选自直链和支链c1-6烷二基和c7-12芳烷二基;基团x选自卤素、r1、nr52、conr62、coor7、h和cor8,r1选自h、任选取代的c1-6烷基、任选取代的c5-7环烷基、c5-7杂环烷基和芳基。每个r5选自h、c1-6烷基、芳基和c7-12芳烷基,或基团r5连同它们所附接的n-原子一起形成含n的5-7元杂环基团;基团r6各自选自h和c1-6烷基基团,或基团r6连同它们所附接的n原子一起形成5-7元杂环;r7选自任选取代的c1-6烷基、c7-12芳烷基和芳基;r8选自任选取代的c1-6-烷基、c7-12芳烷基和芳基;条件是当x是r4不同于r2时。从权利要求1的从属权利要求示出可变基团的优选定义。当x是胺基团时,化合物中,即基团nr52特别优选。在此类化合物中,其中两个基团r5连接以形成杂环的那些是优选的,因为它们似乎在癌细胞系测试中具有有用的细胞毒性活性。我们创造了一系列优选的化合物的酸加成盐,其中x是2-(吡咯烷-y-基)乙基,以提供对化合物的水溶解度的控制。优选地,盐是羧酸盐,如二羧酸盐、三羧酸盐,诸如甲酸盐或乙酸盐。它可以是如hcl和羧酸盐的混合盐。本发明还提供了用于治疗动物以抑制实体瘤生长或减小实体瘤例如胰腺肿瘤的大小的方法中的新型化合物。本发明还提供了含有新型化合物以及稀释剂或载体的组合物。该组合物优选药物组合物并且载体是药学上可接受的。本发明还提供了上述组合物,其还包含式ii的化合物:其中r是nh2或n(r13)r14x1,其中r13选自与r3相同的基团,r14选自与r4相同的基团,x1选自与x相同的基团,条件是式ii的化合物以不高于式i的化合物重量的50%的量存在。在本发明的第二方面,提供了用于合成取代的萘二酰亚胺化合物的方法,其包括使式iii的化合物在亲核取代反应中与式iv的胺反应的步骤:其中br是溴;y是h或br;基团r12是相同的且选自直链和支链c1-6烷二基;r13hnr14x2iv其中r13选自h和c1-6烷基;r14选自直链和支链的c1-6烷二基和c7-12芳烷二基;x2选自卤素、or、nr152、conr162、coor17、sh和cor18;r11选自h、任选取代的c1-6烷基、任选取代的c5-7环烷基、c5-7杂环烷基和芳基;每个r15选自h、c1-6烷基、芳基和c7-12芳烷基,或基团r15连同它们所附接的n-原子一起形成5-7个原子的饱和杂环;每个r16选自h和c1-6烷基基团,或基团r16连同它们所附接的n原子一起形成5-7元杂环;r17选自任选取代的c1-6烷基、c7-12芳烷基和芳基;r18选自任选取代的c1-6烷基、c7-12芳烷基和芳基;从而br原子、br原子中的一个或每个br原子被胺试剂的亲核胺氮取代以形成取代的ndi化合物。权利要求12的从属权利要求示出本方法的优选方面。因此,本发明的方法包括使溴化萘二酰亚胺与式iv的胺试剂在芳香族亲核取代反应中反应的步骤,从而溴原子被氨基基团n(r13)r14x2替代。起始二酰亚胺可为单溴化合物(y是h)或二溴化合物(y是br)或两者的混合物。当起始二酰亚胺包括二溴化合物时,芳香族亲核取代反应可导致两个溴原子被胺基团或它们中的仅一个替代。第二溴原子可被h替代,在此情况下产物是根据本发明的第一方面的三取代的化合物。这在高温下更容易发生。优选的是,例如通过使用柱色谱来分离两者的混合物。当二酰亚胺原料具有一个溴取代基时,该产物是本发明的第一方面的新化合物。该产物可以包含取代的ndi的混合物并且还可以含有未反应的ndi。当该产物包含含有或不含有未反应的ndi的取代的ndi的混合物时,与三取代的ndi相比,可以存在不高于50%重量的四取代的ndi。使式iii的化合物(其中y是h)反应的工艺步骤尤其是优选的。如果期望从起始式iii的化合物(其中y是br)形成新型三取代的化合物,则可能需要使用相对高温条件,因为这可以优化h对第二个br原子的替代。通常,然而,此类高温条件可能导致涉及酰亚胺环开环的不希望的副反应。因此,优选地,该工艺在回流下进行。该工艺通常涉及用于从产物混合物中分离所需的终产物的纯化步骤。柱色谱可以用作简单的纯化步骤是本发明的工艺的优点。这可通过选择用于该步骤以及用于合成亚氨基和其前体溴化四羧酸酐及用于其的前体酸酐的准备步骤的反应条件和试剂来实现。本发明的工艺优选地涉及如随附的权利要求20和23及其从属权利要求中所定义的且如工作实施例所示例的准备步骤。本发明的新型化合物(三取代的化合物)具有比我们早期公布的us20140275062中所公开的四取代的化合物更低的分子量。当与四取代的化合物比较时,在相同的摩尔浓度下,在dna四联体稳定测试中,使用fret分析,发现该化合物产生降低的具有与肿瘤生长潜在相关性的一系列dna四联体的稳定化。此外,当针对一组人类癌细胞系测试细胞毒性时,ic50值与我们早期的化合物mm41(即四取代的化合物)的那些ic50值相当。我们令人惊讶地发现当使用mia-paca2胰腺肿瘤细胞系在体内测试(胰腺癌的异种移植小鼠模型)中测试时,结果比使用相同浓度的四取代的化合物的方案更好,这实际上好于当前胰腺癌治疗护理标准品吉西他滨(图1)。动物在治疗后没有任何显著的重量减轻。当在更低浓度下测试时,肿瘤生长的结果等同于我们先前试验中使用的mm41化合物的测试浓度。这些数据导致预期该化合物在例如胰腺肿瘤的实体瘤治疗中是治疗有效的。令人惊讶的是,已发现mia-paca2胰腺癌细胞当用本发明的新型化合物处理时,显示出在相对少量的基因中基因表达的变化,所述基因中的大部分在其启动子中具有推定的四联体形成序列,这与新型化合物选择性靶向核基因的概念一致。还令人惊讶的是,许多这些靶向基因与患者存活率增加相关,其中在人类中,它们的表达被下调,这支持本发明的新型化合物可用于治疗人类癌症的概念。本发明的化合物可以以药学上可接受的组合物的形式提供。该组合物可以通过任何途径施用。对于肿瘤的化疗,该组合物最方便的是静脉内施用。本发明的化合物,尤其是当以酸加成盐的形式呈现时,例如当一些或全部碱性胺基转化为盐形式时,是水溶性的,并且具有大约中性的ph。因此,这些盐适用于以水溶液形式施用,其适用于静脉内施用。药物水溶液优选地包含1至500mg/l的化合物。本发明的化合物可以以适合于例如用载体或稀释剂制成药物组合物的形式提供,例如以干燥的、可再水合的形式。此类干燥形式可以通过结晶和/或蒸发来制备。或者,该化合物可以以浓缩物的形式呈现,例如在施用前在水或有机的、药学上可接受的溶剂中稀释。当用作现有肿瘤的治疗时,本发明的化合物可以使用针对化学治疗剂所开发的方案来进行施用。在随附的实施例中进一步说明了本发明。实施例已经合成了一系列三取代的萘二酰亚胺并对其作为g-四联体配体和作为潜在的抗癌剂进行评估。如下所详述,已经开发了新的合成程序。目前已经将示意图2中的化合物4c鉴定为先导化合物:化学所有化学品、试剂和溶剂均购自sigma-aldrich、alfaaesar、lancastersynthesis和fluorochem(uk),并且未经进一步纯化即可使用。溶剂由vwr和fisherscientific提供。使用bdh硅胶(bdh153325p)进行柱色谱。在玻璃柱中在中压下或在预填充柱(快速二氧化硅柱(40-63μm,))中使用biotagesp4仪器进行色谱。用组合322泵和agilent1100系列检测器的gilson装置,使用c185μm(100mm×4.6mm)柱(41622271(w),ymc,japan)以1ml/min的流速进行hplc分析。用组合322泵和uv/vis-155检测器(在280nm下检测)的gilson装置,使用c185μm(100mm×20mm)柱(201022272)(w),ymc,japan以20ml/min的流速进行制备型hplc。含0.1%甲酸的水和甲醇用作hplc的溶剂。为了纯化化合物4a-4i,使用以下方法:注射后100%水溶液持续5分钟,然后在25分钟内逐渐降至60%水溶液。对于化合物3a和3b,使用以下方法:注射后100%水溶液持续2min,经17min逐渐减少至20%溶液。对于化合物4a-4i的hplc纯度分析,所使用的方法是:注射后100%水溶液持续5min,,至经18min的60%w/v水溶液以及注射后100%水溶液持续5min。最终化合物的纯度大于95%(hplc,280nm)。nmr谱在400mhz或500mhz下在bruker光谱仪上于cdcl3(含0.05%tms,cambridgeisotopelaboratories,usa)中记录。用mestrec4.5.6.0以化学位移使用tms作为标准品(δ=0ppm)分析nmr谱。nmr多重性缩写是s(单重峰)、bs(宽单重峰)、d(双重峰)、t(三重峰)、q(四重峰)、5q(五重峰)和m(多重峰)。如以赫兹(hz)所观察,报道耦合常数j。高分辨率质谱(hrms)在于电喷射电离(esi)下运行的micromassq-ttofultima全局串联质谱仪上测量,并使用masslab3.2软件处理。对于化合物2a和2b,由于溶解度问题,没有获得1h和13cnmr。参考方法1方案1–i)二溴异氰尿酸,h2so4,40℃,5h;ii)3-吗啉代丙胺,乙酸,微波,130℃,25min;iii)胺,nmp,微波,125℃,30min用于4a-4i/胺,net3,nmp,微波,125℃,30min用于4j使用方案1中的程序初始合成化合物4a-4j。2-溴-1,4,5,8-萘四羧酸二酸酐(2a)和2,6-二溴-1,4,5,8-萘四羧酸二酸酐(2b):将萘二酸酐(1)(1g,3.72mmol)溶解于浓硫酸(96%)(38ml)中。将溶解于浓硫酸的二溴异氰尿酸(1.07g,3.72mmol)的溶液(18.5ml)经4h时间段逐滴加入其中。将混合物再搅拌1h,然后倾倒至冰(500ml)上。将形成的黄色固体过滤,用溶于水中的0.5m的hcl(2×10ml)洗涤并真空干燥。将所获得的含有1、2a和2b的混合物的黄色固体不经进一步纯化就使用。由于那些化合物的低溶解度,我们没分离2a和2b。此外,由于溶解度问题没有获得nmr数据。n,n’-双(3-(吗啉代)丙基氨基)-2-溴-1,4,5,8-萘四羧酸二酰亚胺(3a)和n,n’-双(3-(吗啉代)丙基氨基)-2,6-二溴-1,4,5,8-萘四羧酸二酰亚胺(3b):将化合物2(2a和2b的混合物)(250mg,0.720mmol)用超声处理混悬于微波反应容器内的冰乙酸(3ml)中。将3-吗啉代丙胺(311mg,316μl,2.16mmol)逐滴加入至搅拌的混合物中。将反应管密封并在130℃于微波中处理。在去除溶剂后,将残余物通过制备型hplc纯化以得到呈褐色固体的衍生物3a和3b。产量3a(48mg,0.08mmol,11.1%):1hnmr(400mhz,cdcl3)δ2.08-2.13(m,4h),2.77-2.83(m,12h),3.72-3.76(m,8h),4.25-4.31(q,4h),8.75(d,1h,j=8hz),8.80(d,1h,j=7.6hz),8.92(s,1h).13cnmr(100mhz,cdcl3,tms)δ23.31,23.44,38.81,39.11,52.50,55.51,55.45,65.37,65.41,123.97,125.75,125.95,126.01,126.84,128.68,128.76,130.73,131.71,138.44,161.17,161.87,161.98,162.56。hrms(es+)c28h31brn4o6[m+h]+计算值:600.1543。实测值:600.1536。产率3b(49mg,0.0722mmol,10.0%):1hnmr(400mhz,cdcl3)δ:2.00-1.92(m,4h),2.45-2.37(m,8h),2.53-2.50(m,4h),3.54-3.52(m,8h),4.33-4.28(m,4h),8.99-8.76(s,2h).13cnmr(100mhz,cdcl3,tms)δ21.7,38.5,51.8,54.7,63.7,123.4,124.5,126.7,128.2,138.5,161.4,161.5。hrms(es+)c28h30br2n4o6[m+h]+计算值:679.0603。实测值:679.0593。4-((3-(4-甲基哌嗪-1-基)丙基)氨基)-2,7-双(3-吗啉代丙基)苯并[lmn][3,8]菲罗啉-1,3,6,8(2h,7h)-四酮(4a):将化合物3a和3b(150mg,0.25mmol)、1-(3-氨基丙基)-4-甲基哌嗪(78mg,85μl,0.50mmol)和nmp(2ml)的混合物混悬于微波容器中。将管用橡皮杯密封,并在微波辐射下加热至125℃持续30分钟。在去除溶剂后,将粗混合物通过制备型hplc纯化以得到呈红色固体的甲酸盐4a。产率4a(52mg,0.072mmol,28.8%)。1hnmr(400mhz,cdcl3)δ1.99-2.09(m,6h),2.62-2.80(m,17h),3.01(m,8h),3.68-3.76(m,10h),4.20-4.26(m,4h),8.23(s,1h),8.31(d,1h,j=7.6hz),8.61(d,1h,j=7.6hz),10.16(t,1h,exchd20,j=5.8hz).13cnmr(100mhz,cdcl3)δ23.7,23.9,38.2,38.8,41.1,43.3,50.2,52.5,52.7,53.1,54.6,55.7,65.5,65.7,99.9,119.4,119.8,123.4,124.6,126.1,127.9,129.5,131.4,152.3,163.0,163.3,166.0,166.4。hrms(es+)(m+h)+c36h49n7o6计算值676.3823,实测值676.3825。4-((2-(4-甲基哌嗪-1-基)乙基)氨基)-2,7-双(3-吗啉代丙基)苯并[lmn][3,8]菲罗啉-1,3,6,8(2h,7h)-四酮(4b):将化合物3a和3b(150mg,0.25mmol)、1-(2-氨基乙基)-4-甲基哌嗪(72mg,75μl,0.50mmol)和nmp(2ml)的混合物混悬于微波容器中。将管用橡皮杯密封并在微波辐照下加热至125℃持续30分钟。在去除溶剂后,将粗混合物通过制备型hplc纯化以得到呈红色固体的甲酸盐4b。产量4b(9.4mg,0.013mmol,5.3%)。1hnmr(400mhz,cdcl3)δ1.96-2.02(m,4h),2.53-2.68(m,21h),3.03-3.05(m,4h),3.62(t,4h,j=4.6hz),3.67-3.72(m,6h),4.23-4.28(m,4h),8.21(s,1h),8.34(d,1h,j=7.6hz),8.64(d,1h,j=8hz),10.28(t,1h,exchd20,j=4.8hz);13cnmr(100mhz,cdcl3)δ24.0,38.3,39.1,40.2,43.7,50.4,52.9,53.1,53.6,55.8,56.0,56.0,66.0,66.3,100.3,119.5,120.0,123.6,124.6,126.3,128.0,129.5,131.4,152.1,163.0,163.1,163.3,165.9。hrms(es+)(m+h)+c35h47n7o6计算值662.3680,实测值662.3666。2,7-双(3-吗啉代丙基)-4-((2-(吡咯烷-1-基)乙基)氨基)苯并[lmn][3,8]菲罗啉-1,3,6,8(2h,7h)-四酮(4c):将化合物3a和3b(150mg,0.25mmol)、1-(2-氨基乙基)-吡咯烷(57mg,63μl,0.50mmol)和nmp(2ml)的混合物混悬于微波容器中。将管用橡皮杯密封并在微波辐照下加热至125℃持续30分钟。在去除溶剂后,将粗混合物通过制备型hplc纯化以得到呈红色固体的甲酸盐4c。产量4c(25mg,0.037mmol,14.8%)。1hnmr(400mhz,cdcl3)δ1hnmr(400mhz,cdcl3)δ2.02-2.04(m,8h),2.68-2.78(m,12h),3.19(t,4h,j=6.4hz),3.32(t,2h,j=6.6hz),3.71(t,4h,j=4.8hz),3.76(t,4h,j=4.6hz),4.01-4.02(m,2h),4.22(t,4h,j=7hz),8.24(s,1h),8.28(d,1h,j=7.6hz),8.56(d,1h,j=8hz),10.17(t,1h,exchd20,j=4.2hz).13cnmr(100mhz,cdcl3)δ23.3,23.6,23.7,38.2,38.9,40.1,52.6,52.7,53.9,53.9,55.8,65.6,65.8,100.7,119.1,119.5,123.5,124.9,126.1,128.0,129.2,131.4,151.8,162.8,163.2,165.9,166.5。hrms(es+)(m+h)+c34h44n6o6计算值633.3389,实测值633.3401。2,7-双(3-吗啉代丙基)-4-((2-(吡啶-2-基)乙基)氨基)苯并[lmn][3,8]菲罗啉-1,3,6,8(2h,7h)-四酮(4d):将化合物3a和3b(150mg,0.25mmol)、2-(2-氨基乙基)吡啶(61mg,59μl,0.50mmol)和nmp(2ml)的混合物混悬于微波容器中。将管用橡皮杯密封并在微波辐照下加热至125℃持续30分钟。在去除溶剂后,将粗混合物通过制备型hplc纯化以得到呈红色固体的甲酸盐4d。产量4d(12mg,0.017mmol,7%)。1hnmr(400mhz,cdcl3)δ1.90-1.96(m,4h),2.49-2.58(m,12h),3.22(t,2h,j=7hz),3.57(t,4h,j=4.6hz),3.62(t,4h,j=4.6hz),3.97-4.02(m,2h),4.16-4.20(m,4h),7.11-7.15(m,1h),7.29-7.27(m,1h),7.57-7.61(d,1h,j=7.8hz),8.20(s,1h),8.26(d,1h,j=8hz),8.56(d,2h,j=8hz),10.21(t,1h,exchd20,j=5.6hz);13cnmr(100mhz,cdcl3)δ24.2,37.7,38.6,39.2,42.8,53.1,53.3,56.2,66.5,66.5,100.1,119.8,121.9,123.5,123.6,124.5,126.2,127.9,129.5,131.3,136.8,149.7,151.9,152.2,157.8,163.0,163.1,163.4,166.1。hrms(es+)(m+h)+c35h40n6o6计算值641.3090,实测值641.3088。2,7-双(3-吗啉代丙基)-4-((4-(吡咯烷-1-基)丁基)氨基)苯并[lmn][3,8]菲罗啉-1,3,6,8(2h,7h)-四酮(4e):将化合物3a和3b(100mg,0.17mmol)、4-(1-吡咯烷基)-1-丁胺(48mg,51μl,0.33mmol)和nmp(1.5ml)的混合物混悬于微波容器中。将管用橡皮杯密封并在微波辐照下加热至125℃持续30分钟。在去除溶剂后,将粗混合物通过制备型hplc纯化以得到呈红色固体的甲酸盐4e。产量4e(17mg,0.024mmol,14%)。1hnmr(400mhz,cdcl3)δ1.94-2.11(m,12h),2.59-2.77(m,12h),3.14(t,2h,j=7.8hz),3.28-3.29(m,4h),3.63-3.70(m,6h),3.76(t,4h,j=4.6hz),4.23-4.27(m,4h),8.17(s,1h),8.34(d,1h,j=8hz),8.64(d,1h,j=8hz),10.10(t,1h,exchd20,j=5.4hz);13cnmr(100mhz,cdcl3)δ23.2,23.4,23.7,23.8,26.7,38.3,38.9,42.4,52.7,52.8,53.3,54.7,55.8,55.9,65.7,65.9,100.2,119.5,119.5,123.6,124.7,126.3,128.1,129.5,131.4,152.3,162.9,163.1,163.4,166.2。hrms(es+)(m+h)+c36h48n6o6计算值661.3713,实测值661.3713。2,7-双(3-吗啉代丙基)-4-((2-(哌啶-1-基)乙基)氨基)苯并[lmn][3,8]菲罗啉-1,3,6,8(2h,7h)-四酮(4f):将化合物3a和3b(100mg,0.17mmol)、1-(2-氨基乙基)哌啶(42mg,47μl,0.33mmol)和nmp(1.5ml)的混合物混悬于微波容器中。将管用橡皮杯密封并在微波辐照下加热至125℃持续30分钟。在去除溶剂后,将粗混合物通过制备型hplc纯化以得到呈红色固体的甲酸盐4f。产量4f(13mg,0.018mmol,11%)。1hnmr(400mhz,cdcl3)δ1.57-1.60(m,2h),1.69-1.75(m,4h),1.95-2.00(m,4h),2.51-2.68(m,16h),2.87(t,2h,j=6.4hz),3.61(t,4h,j=4.6hz),3.67(t,4h,j=4.6hz),3.76-3.80(m,2h),4.22-4.28(m,4h),8.18(s,1h),8.31(d,1h,j=7.6hz),8.61(d,1h,j=8hz),10.24(t,1h,exchd20,j=5hz);13cnmr(100mhz,cdcl3)δ23.8,24.2,25.2,38.5,39.2,39.9,53.1,53.2,54.3,56.2,56.8,66.4,66.5,100.3,199.5,119.9,123.6,124.5,126.2,127.9,129.5,131.2,152.0,162.9,163.1,163.4,165.9。hrms(es+)(m+h)+c35h46n6o6计算值647.3566,实测值647.3557。4-((2-吗啉代乙基)氨基)-2,7-双(3-吗啉代丙基)苯并[lmn][3,8]菲罗啉-1,3,6,8(2h,7h)-四酮(4g):将化合物3a和3b(60mg,0.1mmol)、4-(2-氨基乙基)吗啉(26mg,30μl,0.2mmol)和nmp(1ml)的混合物混悬于微波容器中。将管用橡皮杯密封并在微波辐照下加热至125℃持续30分钟。在去除溶剂后,将粗混合物通过制备型hplc纯化以得到呈红色固体的甲酸盐4g。产量4g(15mg,0.022mmol,22%)。1hnmr(400mhz,cdcl3)δ1.92-1.96(m,4h),2.42-45(m,8h),2.49-2.54(m,4h),2.59(t,4h,j=4.4hz),2.80(t,2h,j=6hz),3.56(m,4h,j=4.4hz),3.62(t,4h,j=4.4hz),3.66-3.70(m,2h),3.78(t,4h,j=4.4hz),4.23-4.30(m,4h),8.21(s,1h),8.33(d,1h,j=7.6hz),8.64(d,1h,j=8hz),10.34(t,1h,exchd20,j=4.8hz);13cnmr(100mhz,cdcl3)δ24.4,24.6,38.7,39.4,40.1,53.5,53.6,56.4,56.5,56.8,66.93,66.96,66.99,100.3,119.5,120.0,123.7,124.5,126.2,127.9,129.5,131.2,152.1,163.1,163.5,165.9。hrms(es+)(m+h)+c34h44n6o7计算值649.3376,实测值649.3350。4-((四氢呋喃基甲基)氨基)-2,7-双(3-吗啉代propyl)苯并[lmn][3,8]菲罗啉-1,3,6,8(2h,7h)-四酮(4h):将化合物3a和3b(60mg,0.1mmol)、四氢糠胺(21mg,21μl,0.2mmol)和nmp(1ml)的混合物混悬于微波容器中。将管用橡皮杯密封并在微波辐照下加热至125℃持续30分钟。在去除溶剂后,将粗混合物通过制备型hplc纯化以得到呈红色固体的甲酸盐4h。产量4h(10mg,0.015mmol,15%)。1hnmr(400mhz,cdcl3)δ1.26-1.28(m,4h),1.71-1.78(m,6h),1.94-2.05(m,2h),2.12-2.51(m,10h),3.56-3.57(m,4h),3.62-3.64(m,4h),3.74-3.78(m,1h),3.84-3.87(m,1h),3.97-4.00(m,1h),4.24-4.28(m,4h),8.27(s,1h),8.34(d,1h,j=7.6hz),8.65(d,1h,j=7.6hz),10.34(t,1h,exchd20,j=4.8hz);13cnmr(100mhz,cdcl3)δ24.4,24.6,25.9,38.8,39.4,47.3,53.6,56.4,56.5,66.94,66.97,68.6,100.2,119.5,120.0,123.7,124.5,126.2,127.9,129.5,131.2,152.5,163.1,163.4,166.2。hrms(es+)(m+h)+c33h41n5o7计算值620.3096,实测值620.3084。4-((2-(二乙基氨基)乙基)氨基)-2,7-双(3-吗啉代丙基)苯并[lmn][3,8]菲罗啉-1,3,6,8(2h,7h)-四酮(4i):将化合物3a(24mg,0.04mmol)、n,n-二乙基乙二胺(9mg,11μl,0.08mmol)和nmp(0.8ml)混悬于微波容器中。将管用橡皮杯密封并在微波辐照下加热至125℃持续30分钟。在去除溶剂后,将粗混合物通过柱色谱使用ch2cl2/meoh/nh337%9.5/0.5/0.03的混合物作为洗脱液纯化以得到呈红色固体的化合物4i。产量4i(15mg,0.022mmol,59%)。1hnmr(400mhz,cdcl3)δ1.11(t,6h,j=7hz),1.46-1.47(m,4h),1.90-2.53(m,12h),2.64-2.69(m,4h),2.86(t,2h,j=6.2hz),3.56-3.57(m,4h),3.61-3.65(m,6h),4.23-4.30(m,4h),8.21(s,1h),8.31(d,1h,j=7.6hz),8.62(d,1h,j=8hz),10.27(t,1h,exchd20,j=5hz);13cnmr(100mhz,cdcl3)δ11.8,24.4,24.5,38.6,39.3,47.1,51.6,53.5,53.6,56.5,66.9,100.1,119.4,120.2,123.7,124.3,126.1,127.8,129.6,131.2,152.1,163.12,163.16,163.5,165.9。hrms(es+)(m+h)+c34h46n6o6计算值635.3552,实测值635.3557。4-((4-羟基苯乙基)氨基)-2,7-双(3-吗啉代丙基)苯并[lmn][3,8]菲罗啉-1,3,6,8(2h,7h)-四酮(4j):将化合物3a和3b(139mg,0.23mmol)、酪胺盐酸盐(80mg,0.46mmol)、三乙胺(47mg,65μl,0.46mmol)和nmp(2ml)的混合物混悬于微波容器中。将管用橡皮杯密封并在微波辐照下加热至125℃持续30分钟。在去除溶剂后,将粗混合物通过使用ch2cl2/meoh9.5/0.5的混合物作为洗脱液的柱色谱纯化以得到呈红色固体的化合物4j。产量4j(11mg,0.016mmol,7%)。1hnmr(400mhz,cdcl3)δ1.75-1.82(m,4h),2.31-2.43(m,12h),2.91(t,2h,j=6.6hz),3.41-3.44(m,8h),3.9-3.71(m,2h),4.02(m,4h),6.72(d,2h,j=8.4hz),7.15(d,2h,j=8hz),7.84(s,1h),8.02(d,1h,j=8hz),8.30(d,1h,j=8hz),9.86(brs,1h,exchd20).13cnmr(100mhz,cdcl3),δ29.9,30.9,38.4,38.9,44.5,53.4,53.5,56.2,56.4,66.5,66.7,94.4,100.1,116.1,119.4,120.2,123.5,124.5,126.2,129.0,130.2,131.3,155.4,162.9,163.1,163.5,165.8。hrms(es+)(m+h)+c36h41n5o7计算值656.3074,实测值656.3084。参考方法2方案2–i)5,5-二甲基-1,3-二溴海因,h2so4,80℃,72h;ii)3-吗啉代丙胺,乙酸,微波,130℃,25min;iii)胺,nmp,微波,125℃,30min根据方案2优化4c的合成。该工艺涉及步骤ii的产物混合物的纯化,以分离出3a和3b和无溴副产物。这使得通过柱色谱更容易纯化产物4。这还允许进行放大,但不能用如方法1中所用的制备型hplc。使用不同的反应条件来获得化合物3a。所有使用的条件和化合物3a和3b的相对产量列于表1中。表1:测试的用于合成中间体3a的反应条件。2-溴-1,4,5,8-萘四羧酸二酸酐(2a)和2,6-二溴-1,4,5,8-萘四羧酸二酸酐(2b):将萘二酸酐(1)(150mg,0.56mmol)在硫酸(1.5ml)中浆化,并将获得的混悬液在室温下搅拌5min以使其完全溶解。在1h的时间段内缓慢添加5,5-二甲基-1,3-二溴海因(88mg,0.308mmol),并紧紧塞住圆底烧瓶以避免溴从反应混合物中逸出。将溶液在80℃下搅拌72h,然后倾倒至冰(30ml)上。将形成的黄色固体过滤,用水(2x10ml)洗涤并真空干燥,得到混合物2a和2b。由于溶解度问题没有获得nmr数据。所得的混合物没有进一步纯化就用于下一步中。n,n’-双(3-(吗啉代)丙基氨基)-2-溴-1,4,5,8-萘四羧酸二酰亚胺(3a).将化合物2a和2b(194mg,0.56mmol)用超声处理混悬于微波反应容器内的冰乙酸(1.5ml)中。将3-吗啉代丙胺(242mg,245μl,1.68mmol)逐滴加入至搅拌的混合物中。将反应管密封并在125℃下在微波中处理30min。然后将溶液用碳酸钾碱化并用氯仿(3x5ml)萃取。将有机相收集、经硫酸钠干燥并蒸发。使用ch2cl2/meoh96/40的混合物作为洗脱液,通过柱色谱纯化获得的残余物。产量3a(107mg,0.18mmol,35%)1hnmr(400mhz,cdcl3)δ1.95-1.97(m,4h),2.40-2.43(m,8h),2.52-2.53(m,4h),3.51-3.54(m,8h),4.28-4.33(q,4h),8.77(d,1h,j=8hz),8.82(d,1h,j=7.6hz),8.935(s,1h).13cnmr(100mhz,cdcl3,tms)δ29.3,29.7,31.2,38.1,53.5,56.4,56.5,59.5,66.9,126.0,126.1,126.7,126.9,128.6,130.6,130.8,131.6,138.4,161.1,161.8,161.9,162.5。hrms(es+)c28h31brn4o6[m+h]+计算值:600.1543。实测值:600.1536。2,7-双(3-吗啉代丙基)-4-((2-(吡咯烷-1-基)乙基)氨基)苯并[lmn][3,8]菲罗啉-1,3,6,8(2h,7h)-四酮(4c):将化合物3a(0.15g,0.25mmol)、1-(2-氨基乙基)吡咯烷(0.06ml,0.51mmol)和nmp(1ml)混悬于微波反应容器中。将反应管密封并在130℃于微波中处理25min。在已冷却至室温后,将溶剂浓缩并将粗混合物通过柱色谱(使用ch2cl2/meoh/nh395/5/0.4的混合物作为洗脱液)纯化。获得呈红色油状物的化合物4c(118mg,0.186mmol,75%产量)。1hnmr(400mhz,cdcl3,tms)δ2.019-2.040(m,8h),2.683-2.782(m,12h),3.195(t,4h,j=6.4hz),3.32(t,2h,j=6.6hz),3.708(t,4h,j=4.8hz),3.764(t,4h,j=4.6hz),4.009-4.024(m,2h),4.22(t,4h,j=7hz),8.237(s,1h),8.28(d,1h,j=7.6hz),8.56(d,1h,j=8hz),10.174(t,1h,exchd20,j=4.2hz).13cnmr(100mhz,cdcl3)δ23.3,23.6,23.7,38.2,38.9,40.1,52.6,52.8,53.9,53.9,55.8,65.6,65.8,100.7,119.1,119.5,123.5,124.9,126.1,128.0,129.2,131.4,151.8,162.8,163.2,165.9,166.5。hrms(es+)(m+h)+c34h44n6o6计算值633.3389,实测值633.3401。盐形成然后将4c转化成盐形式以提高其水溶解度。获得不同的盐,评价它们在5mg/ml的溶解度和所得的水溶液的ph。结果报告在表2中。表2:不同的4c盐的溶解度数据。生物物理学数据和细胞生物学数据fret测定使用经修改用作以96-孔形式进行高通量筛选的荧光共振能量转移(fret)分析来研究萘二酰亚胺化合物稳定四联体dna序列的能力。研究了若干条四联体序列(包括人端粒g-四联体dna序列5'-fam-d(ggg[ttaggg]3)-tamra-3'和双链体序列5'-fam-dtatagctata-heg-tatagctata-tamra-3'(heg接头:[(-ch2-ch2-o-)6。标记的寡核苷酸已将它们附接至供体荧光团fam:6-羧基荧光素和受体荧光团tamra:6-羧基四甲基罗丹明。将fret探针序列在60mm二甲胂酸钾缓冲液(ph7.4)中自储液稀释至正确的浓度(400nm),然后通过加热至85℃退火10min,然后在加热块中冷却至室温。将化合物以于dmso中的10mm储备溶液储存;使用10mm的hcl以初始1:10的稀释度制备最终溶液(在2×浓度下),然后60mm的二甲胂酸钾缓冲液(ph7.4)用于所有后续步骤中。因此,反应体积中的最大hcl浓度(在20μm的配体浓度下)为200μm,正好处于所用缓冲液的范围内。还进行了相关对照以检查对分析的干扰。通过将50μl退火的dna等分到每个孔中,然后添加50μl化合物溶液来准备96孔板(mjresearch,waltham,ma)。在dnaengineopticonopticon(mjresearch)上进行测量,其中激发在450-495nm且检测在515-545nm。以0.5℃的间隔在30-100℃的范围中获得荧光,在每次读取前维持30s恒温以确保稳定的值。使用于程序origin7.0(originlabcorp.,northampton,ma)中编写的脚本进行最终的数据分析。origin7.0中的高级曲线拟合功能用于计算δtm值。所有测定一式三份或更好地进行。δtm的esd为±0.1℃。表3:用一系列富含g的序列进行1μm浓度的化合物4a-j的fret分析的δtm值(℃):人端粒(f21t),两条来自hsp90启动子hsp90a、hsp90b(热激蛋白90)的序列,两条来自kras基因的启动子区的序列,一条来自bcl-2基因的启动子区,以及t环(tloop)(代表双链体dna)。esd来自一式三份测量并且平均0.3℃。细胞培养和细胞毒性测试人癌细胞系和人体细胞系wi-38(肺成纤维细胞)购自美国典型培养物保藏中心(americantypeculturecollection)。将细胞系维持在补充有10%胎牛血清(invitrogen,uk)、2mml-谷氨酰胺(invitrogen,netherlands)和如供应商指定的其它组分的适当培养基中。将细胞系维持在37℃,5%co2下并常规传代。将药物溶解在dmso中,并通过0.22μm孔径的过滤器单元过滤,然后添加至细胞系适当的培养基。在96孔板中使用磺酰罗丹明b(srb)测量细胞生长抑制。通过取得表示为未处理的对照孔的吸光度的百分比的每种药物浓度在540nm处的平均吸光度来确定50%抑制浓度(ic50)。对于qrt-pcr分析,将mia-paca2细胞接种至与用于在t75培养烧瓶中的ic50测定所用的密度相等的密度,并在10ml的dmem中生长24h以允许在将化合物3d添加至培养基之前附着。在化合物暴露后,将细胞在pbs中洗涤两次,通过胰蛋白酶消化收获,通过离心(300g,5min,4℃)收集并重悬于rlt缓冲液(qiagen)中。使用qiashredder离心柱将样品均质化并储存在-80℃下,之后进行rna提取。在不同的日期进行三次生物重复实验。a549mcf7miapaca2panc1altwi384a0.0670.3570.0590.0450.2241.834b0.0860.3160.0480.0460.0851.494c0.0240.1590.0120.0220.0931.194d0.1301.0700.2200.3401.292.244e0.0260.2220.0360.0330.0891.224f0.1981.1100.1080.0840.5355.334g2.18>250.2060.22010.87417.654h0.1461.030.1390.8080.711.884i0.8253.331.0850.9092.675.534j0.0921.5380.0590.1630.4511.65mm410.0190.0700.0110.0030.0630.230表4:在癌细胞系组中的化合物4a-j和mm41的短期96hr的ic50值(以μm计),所述癌细胞系组包括mcf7(乳腺)、a549(肺癌)、mia-paca2/panc1(胰腺癌)、alt(端粒的替代延伸)和wi38(肺成纤维细胞)细胞系。esd平均为0.25μm。(*)表示5μm而不是1μm的浓度。mm41是在micco等人jmedchem,2013中强调的二-吗啉代-二-n-甲基-哌嗪萘二酰亚胺衍生物。抗肿瘤活性方案在携带用基质胶生长的胰腺肿瘤细胞系miapaca-2(在右侧腹中5×106个细胞)的皮下异种移植物的cd1裸小鼠中,研究化合物4c(mm41-tri)与mm41和吉西他滨相比的治疗效果。实验概要1在右侧腹中sc接种5×106个miapaca-2细胞(未补充的rpmi+基质胶)。2等待肿瘤皮下建立(约11天)。3每周测量肿瘤的尺寸,直至它们达到0.1cm3的平均尺寸。在肿瘤发展的这个阶段,小鼠准备开始疗法研究。4对小鼠重新分组,以形成5个治疗组,每组由8只小鼠组成,每组携带大约平均0.1cm3的肿瘤尺寸。5通过iv施用相应剂量的mm41或化合物4c或吉西他滨。组1:每周两次,用15mg/kg剂量的吉西他滨处理8只小鼠。组2:每周两次,用15mg/kg剂量的mm41(四取代的nd化合物)处理8只小鼠。组3:每周两次,用15mg/kg剂量的化合物4c处理8只小鼠。组4:每周两次,用10mg/kg剂量的化合物4c处理8只小鼠。组5:每周两次,仅用盐水处理8只对照小鼠。肿瘤生长曲线示于图1中。15mg/kg的化合物4c(标记为mm41-tri)显示出非常显著的抗肿瘤响应。mm41是在micco等人jmedchem).2013中强调的二-吗啉代-二-n-甲基-哌嗪萘二酰亚胺衍生物。动物在治疗后没有任何显著的重量减轻。基因表达方法细胞培养细胞系购自美国典型培养物保藏中心(atcc)并在液氮下储存。将细胞维持在37℃的潮湿5%co2气氛中的单层培养t75(75cm2烧瓶)中。根据(atcc)将细胞维持在适当的培养基中,其必要的补充物包括l-谷氨酰胺(thermofisherscientific目录号25030024)、青霉素-链霉素溶液hybrid-max(sigma目录号p7539)和胎牛血清(fbs,thermofisherscientific目录号10270106)。将细胞在80-90%汇合下传代。rna提取将mia-paca2细胞在孵育前48小时接种在mattek35mm玻璃底皿中。检查了化合物4c的三个孵育时间:0、6和24h。对于rna提取,将细胞(最多1×107个细胞)通过在1×pbs(不含氯化钙和氯化镁,购自sigma目录号d1408)中冲洗细胞两次后进行胰蛋白酶消化来收获。在用与胰蛋白酶等体积的培养基中和培养基后,将细胞转移到无菌离心管,在室温下以400rpm离心4分钟以沉淀细胞。在离心后去除培养基和胰蛋白酶后,使用qiagen的rneasyminikit(目录号74104)从细胞沉淀中提取rna。通过使用nanodrop2000分光光度计检查rna质量,并验证每个重复样品在>25ng/μl浓度下的最小量为约500ng的rna。由uclgenomicsfacility使用illuminanextseq500仪器进行rna-seq测序研究,并且对0hr暴露时相对于对照细胞,所分析的细胞基因组中的所有单个基因的rna表达水平获得数据。每个时间点使用四次重复样品,因此总共对12个单个基因组进行分析并获得每个时间点的统计学数据。基因表达分析-用化合物4c处理后基因表达的变化数据取自来自处理的mia-paca2细胞的rna-seq数据,其显示出选自示出最显著表达下调(如通过rna水平所测量)的那些基因的对数倍表达变化。表5:选择最下调的基因(显示出相对于t=0hr即未处理的细胞的对数倍变化)表6:所选的最下调的基因的功能表7:选择一些最上调的基因(显示出相对于t=0小时,即未处理的细胞的对数倍变化)基因表达变化的分析显示出在两个时间点的一致性的变化模式,其中经下调的基因(表5)通常编码参与转录激活、gtp酶活性和癌症途径(包括dna损伤响应)的蛋白。令人惊讶的是,最强烈下调的基因倾向于在其启动子区域内具有推定的dna四联体序列。相比之下,上调基因(表7)倾向于编码参与膜和细胞外基质结构和功能的蛋白,并且在其启动子中推定的四联体序列显著表达不足。已使用ensembl基因组浏览器(版本86,http://www.ensembl.org/index.html)从一级序列估计每个基因启动子中推定的四联体序列的数目。先前已报道了上述分析中最下调的若干基因(例如,prdm16、cbfa2t3和shank2),当发现被甲基化时,与提高的胰腺癌患者存活有关(thompsonmj,rubbil,dawsondw,donahuetr,pellegrinim(2015)pancreaticcancerpatientsurvivalcorrelateswithdnamethylationofpancreasdevelopmentgenes.plosone10(6):e0128814)。trex1和tp73基因的基因产物参与dna修复:据报道,这些基因中的多态性也与胰腺癌患者存活有关(dongx,liy,hesskr,abbruzzesejl,lid.(2011)dnamismatchrepairgenepolymorphismsaffectsurvivalinpancreaticcancer.oncologist16(1),61-70)。以前的生物信息学研究表明,人类基因组含有300,000至700,000条潜在的四联体形成序列(toddak,johnstonm,neidles.(2005)highlyprevalentputativequadruplexsequencemotifsinhumandna.nucleicacidsresearch33(9),2901-2907;huppertjl,balasubramanians.(2005)prevalenceofquadruplexesinthehumangenome.nucleicacidsresearch33(9),2908-2916;kwokck,marsicog,sahakyanab,chambersvs,balasubramanians.(2016)rg4-seqrevealswidespreadformationofg-quadruplexstructuresinthehumantranscriptome.naturemethods13(10),841-844)。然而,最近的关于细胞内活性染色质中四联体形成的研究(-hertschr,beraldid,lensingsv,marsicog,zynerk,parrya,diantoniom,pikej,kimurah,naritam,tannahilld,balasubramanians.(2016)g-quadruplexstructuresmarkhumanregulatorychromatin.naturegenetics48(10),1267-1272)已经显示,在人染色质中仅存在约10,000个四联体,并且富集于主要是参与人类癌症的基因的启动子区域中。这与仅在真核细胞中存在少量折叠rna四联体的发现(guoju,barteldp.(2016)rnag-quadruplexesaregloballyunfoldedineukaryoticcellsanddepletedinbacteria.science353(6306).pii:aaf5371)支持了四联体-选择性靶向在人类癌症治疗中可行的假设。令人惊讶的是,已经发现mia-paca2胰腺癌细胞当用化合物4c处理时显示基因表达在相对较少的基因中有变化,所述基因的大部分在其启动子中具有推定的四联体-形成序列,这与该化合物选择性靶向这些核基因的概念一致。令人惊讶的是,许多这些靶向基因与提高的患者存活相关,其中它们在人类中的表达被下调,这支持了本公开中的化合物诸如化合物4c可在人类癌症的治疗中具有效用的概念。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1